Suppression of NF-κB Activity: A Viral Immune Evasion Mechanism

 , , , ,
, , , ,
Abstract
:1. Introduction
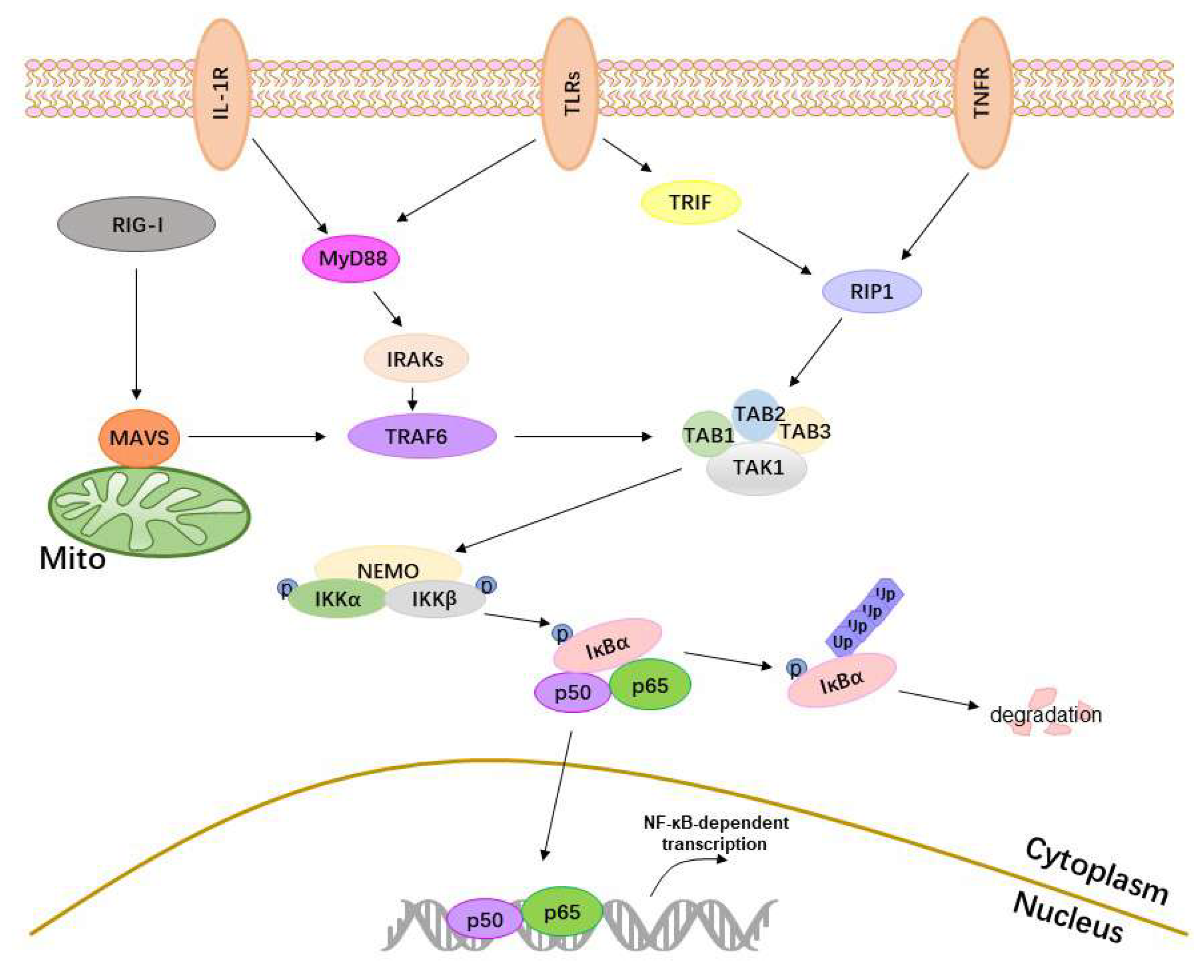
2. The Activation of the NF-κB
2.1. Receptors
2.2. Adaptor Proteins
2.3. IKKs, IκBα and p50/p65
3. Viruses Suppress NF-κB Activation
3.1. Targeting Receptors and Adaptor Proteins
3.2. Targeting IKKs
3.3. Targeting IκBα
3.4. Targeting p50/p65 and Reducing NF-κB Transcriptional Activity
4. The Specific NF-κB Inhibitors from Viruses
4.1. Proteases Encoded by Viruses
4.1.1. NS3/4A
4.1.2. The 3C and 3C-Like Proteases
4.2. DUBs Encoded by Viruses
4.2.1. Papain-Like Protease (PLP)
4.2.2. Other Viral DUBs
4.3. PDL Motifs Encoded by Viruses
4.4. PPase-Binding Proteins Encoded by Viruses
4.5. SH Protein Encoded by Viruses
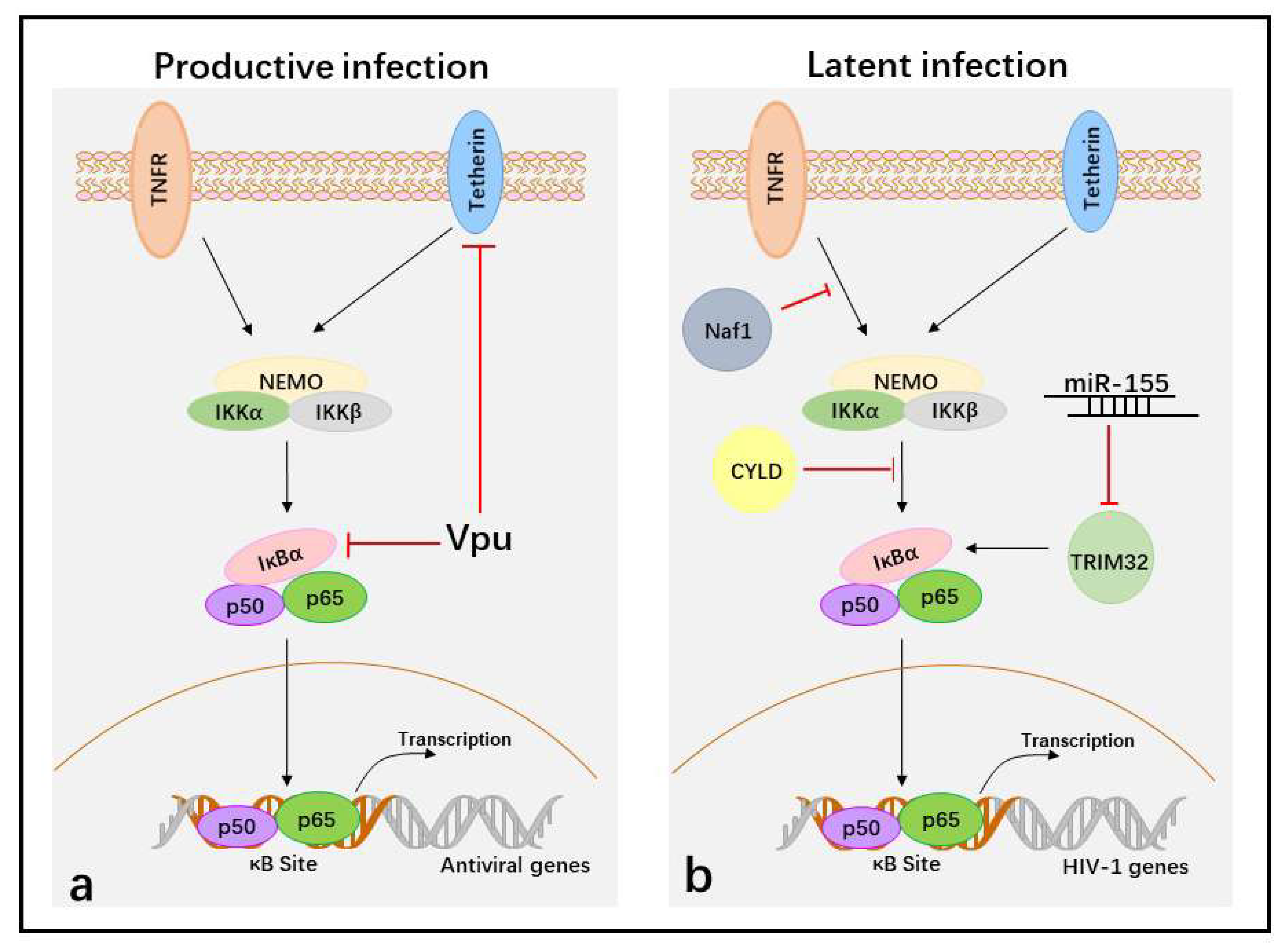
5. Suppression of NF-κB Activity to Facilitate HIV-1 Immune Evasion
6. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Lester, S.N.; Li, K. Toll-like receptors in antiviral innate immunity. J. Mol. Biol. 2014, 426, 1246–1264. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. SnapShot: Pattern-Recognition Receptors. Cell 2007, 129, 1024. [Google Scholar] [CrossRef] [PubMed]
- Quicke, K.M.; Diamond, M.S.; Suthar, M.S. Negative regulators of the RIG-I-like receptor signaling pathway. Eur. J. Immunol. 2017, 47, 615–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, M.; Kouwaki, T.; Fukushima, Y.; Oshiumi, H. Regulation of RIG-I Activation by K63-Linked Polyubiquitination. Front. Immunol. 2017, 8, 1942. [Google Scholar] [CrossRef] [PubMed]
- Ting, A.T.; Bertrand, M.J.M. More to life than NF-κB in TNFR1 signaling. Trends Immunol. 2016, 37, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Palomo, J.; Dietrich, D.; Martin, P.; Palmer, G.; Gabay, C. The interleukin (IL)-1 cytokine family--Balance between agonists and antagonists in inflammatory diseases. Cytokine 2015, 76, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Prestonhurlburt, P.; Kopp, E.; Stadlen, A.; Chen, C.; Ghosh, S.; Janeway, A.C. MyD88 Is an Adaptor Protein in the hToll/IL-1 Receptor Family Signaling Pathways. Mol. Cell 1998, 2, 253–258. [Google Scholar] [CrossRef]
- Yamamoto, M.; Sato, S.; Mori, K.; Hoshino, K.; Takeuchi, O.; Takeda, K.; Akira, S. Cutting edge: A novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J. Immunol. 2002, 169, 6668–6672. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Ea, C.K. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar]
- Dixit, E.; Boulant, S.; Zhang, Y.; Lee, A.S.Y.; Odendall, C.; Shum, B.; Hacohen, N.; Chen, Z.J.; Whelan, S.P.; Fransen, M. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 2010, 141, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Horner, S.M.; Liu, H.M.; Park, H.S.; Briley, J.; Gale, M., Jr. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. USA 2011, 108, 14590–14595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 2011, 146, 448–461. [Google Scholar]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA Is an Adapter Protein Required for Virus-Triggered IFN-β Signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Peng, Z.; Liu, Y.F.; Cheng, G.H.; Wang, Y.Y.; Peng, Z.; Liu, Y.F.; Cheng, G.H. TRAF-mediated regulation of immune and inflammatory responses. Sci. China Life Sci. 2010, 53, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Humphries, F.; Yang, S.; Wang, B.; Moynagh, P.N. RIP kinases: Key decision makers in cell death and innate immunity. Cell Death Differ. 2015, 22, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.; Nanda, S.; Cohen, P. Molecular control of the NEMO family of ubiquitin-binding proteins. Nat. Rev. Mol. Cell Biol. 2013, 14, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Xia, Y.; Parker, A.S.; Verma, I.M. IKK Biology. Immunol. Rev. 2012, 246, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Yang, X.D.; Lamb, A.; Chen, L.F. Posttranslational modifications of NF-κB: Another layer of regulation for NF-κB signaling pathway. Cell. Signal. 2010, 22, 1282–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Ke, H.; Han, M.; Chen, N.; Fang, W.; Yoo, D. Nonstructural Protein 11 of Porcine Reproductive and Respiratory Syndrome Virus Suppresses Both MAVS and RIG-I Expression as One of the Mechanisms to Antagonize Type I Interferon Production. PLoS ONE 2016, 11, e0168314. [Google Scholar] [CrossRef] [PubMed]
- Lingel, A.; Ehlers, E.; Wang, Q.; Cao, M.; Wood, C.; Lin, R.; Zhang, L. Kaposi’s sarcoma-associated herpesvirus reduces cellular myeloid differentiation primary response gene-88 (MyD88) expression via modulation of its RNA. J. Virol. 2015, 90, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Lei, Q.; Li, L.; Cai, J.; Huang, W.; Qin, B.; Zhang, S. ORF3 of Hepatitis E Virus Inhibits the Expression of Proinflammatory Cytokines and Chemotactic Factors in LPS-Stimulated Human PMA-THP1 Cells by Inhibiting NF-κB Pathway. Viral Immunol. 2016, 29, 105–111. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Wang, M.; Huang, Y.; Peng, W.; Zheng, Z.; Xia, N.; Xu, J.; Tian, D. The ORF3 Protein of Genotype 1 Hepatitis E Virus Suppresses TLR3-induced NF-κB Signaling via TRADD and RIP1. Sci. Rep. 2016, 6, 27597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.; Liang, D.; Sun, R.; Jia, B.; Xia, T.; Xiao, H.; Lan, K. Kaposi’s sarcoma-associated herpesvirus-encoded replication and transcription activator impairs innate immunity via ubiquitin-mediated degradation of myeloid differentiation factor 88. J. Virol. 2015, 89, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, H.; Gubbels, R.; Ehlers, E.; Meyer, F.; Waterbury, T.; Lin, R.; Zhang, L. Kaposi sarcoma-associated herpesvirus degrades cellular Toll-interleukin-1 receptor domain-containing adaptor-inducing beta-interferon (TRIF). J. Biol. Chem. 2011, 286, 7865–7872. [Google Scholar] [CrossRef] [PubMed]
- van Lint, A.L.; Murawski, M.R.; Goodbody, R.E.; Severa, M.; Fitzgerald, K.A.; Finberg, R.W.; Knipe, D.M.; Kurt-Jones, E.A. Herpes simplex virus immediate-early ICP0 protein inhibits Toll-like receptor 2-dependent inflammatory responses and NF-κB signaling. J. Virol. 2010, 84, 10802–10811. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Dong, W.; Cao, Z.; Li, X.; Wang, J.; Qian, G.; Lv, Q.; Wang, C.; Guo, K.; Zhang, Y. TRAF6 is a novel NS3-interacting protein that inhibits classical swine fever virus replication. Sci. Rep. 2017, 7, 6737. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Ni, C.; Song, T.; Liu, Y.; Yang, X.; Zheng, Z.; Jia, Y.; Yuan, Y.; Guan, K.; Xu, Y. The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein. J. Immunol. 2010, 185, 1158–1168. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Qi, H.Y.; Boularan, C.; Huang, N.N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef] [PubMed]
- Bussey, K.A.; Reimer, E.; Todt, H.; Denker, B.; Gallo, A.; Konrad, A.; Ottinger, M.; Adler, H.; Stürzl, M.; Brune, W. The gammaherpesviruses Kaposi’s sarcoma-associated herpesvirus and murine gammaherpesvirus 68 modulate the Toll-like receptor-induced proinflammatory cytokine response. J. Virol. 2014, 88, 9245–9259. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, J.; Cristea, I.M. Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe 2013, 14, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Rebsamen, M.; Heinz, L.X.; Meylan, E.; Michallet, M.C.; Schroder, K.; Hofmann, K.; Vazquez, J.; Benedict, C.A.; Tschopp, J. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep. 2009, 10, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Mack, C.; Sickmann, A.; Lembo, D.; Brune, W. Inhibition of proinflammatory and innate immune signaling pathways by a cytomegalovirus RIP1-interacting protein. Proc. Natl. Acad. Sci. USA 2008, 105, 3094–3099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fliss, P.M.; Jowers, T.P.; Brinkmann, M.M.; Holstermann, B.; Mack, C.; Dickinson, P.; Hohenberg, H.; Ghazal, P.; Brune, W. Viral Mediated Redirection of NEMO/IKKγ to Autophagosomes Curtails the Inflammatory Cascade. PLoS Pathog. 2012, 8, e1002517. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Lan, P.; Hou, X.; Han, Q.; Lu, N.; Li, T.; Jiao, C.; Zhang, J.; Zhang, C.; Tian, Z. HBV inhibits LPS-induced NLRP3 inflammasome activation and IL-1β production via suppressing the NF-κB pathway and ROS production. J. Hepatol. 2016, 66, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Lang, T.; Lo, C.; Skinner, N.; Locarnini, S.; Visvanathan, K.; Mansell, A. The hepatitis B e antigen (HBeAg) targets and suppresses activation of the toll-like receptor signaling pathway. J. Hepatol. 2011, 55, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Kanda, T.; Imazeki, F.; Nakamoto, S.; Tanaka, T.; Arai, M.; Roger, T.; Shirasawa, H.; Nomura, F.; Yokosuka, O. Hepatitis B Viruse Antigen Physically Associates with Receptor-Interacting Serine/Threonine Protein Kinase 2 and Regulates IL-6 Gene Expression. J. Infect. Dis. 2017, 206, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Liu, G.; Go, J.; Kolli, D.; Zhang, G.; Bao, X. Human Metapneumovirus M2-2 Protein Inhibits Innate Immune Response in Monocyte-Derived Dendritic Cells. PLoS ONE 2014, 9, e91865. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Deng, X.; Deng, J.; Zhou, J.; Ren, Y.; Liu, S.; Prusak, D.J.; Wood, T.G.; Bao, X. Functional motifs responsible for human metapneumovirus M2-2-mediated innate immune evasion. Virology 2016, 499, 361–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; He, L.; Peng, Y.; Shi, X.; Chen, J.; Zhong, J.; Chen, X.; Cheng, G.; Deng, H. The hepatitis C virus protein NS3 suppresses TNF-α-stimulated activation of NF-κB by targeting LUBAC. Sci. Signal. 2015, 8, ra118. [Google Scholar] [CrossRef] [PubMed]
- Brady, G.; Haas, D.A.; Farrell, P.J.; Pichlmair, A.; Bowie, A.G. Molluscum Contagiosum Virus protein MC005 inhibits NFκB activation by targeting NEMO-regulated IKK activation. J. Virol. 2017, 91, e00545-17. [Google Scholar] [CrossRef] [PubMed]
- Randall, C.M.H.; Jokela, J.A.; Shisler, J.L. The MC159 protein from the molluscum contagiosum poxvirus inhibits NF-κB activation by interacting with the IKK complex. J. Immunol. 2012, 188, 2371–2379. [Google Scholar] [CrossRef] [PubMed]
- Ember, S.W.J.; Ren, H.; Ferguson, B.J.; Smith, G.L. Vaccinia virus protein C4 inhibits NF-κB activation and promotes virus virulence. J. Gen. Virol. 2012, 93, 2098–2108. [Google Scholar] [CrossRef] [PubMed]
- Khatiwada, S.; Delhon, G.; Nagendraprabhu, P.; Chaulagain, S.; Luo, S.; Diel, D.G.; Flores, E.F.; Rock, D.L. A parapoxviral virion protein inhibits NF-κB signaling early in infection. PLoS Pathog. 2017, 13, e1006561. [Google Scholar] [CrossRef] [PubMed]
- Benfield, C.T.; Mansur, D.S.; Mccoy, L.E.; Ferguson, B.J.; Bahar, M.W.; Oldring, A.P.; Grimes, J.M.; Stuart, D.I.; Graham, S.C.; Smith, G.L. Mapping the IκB kinase beta (IKKβ)-binding interface of the B14 protein, a vaccinia virus inhibitor of IKKβ-mediated activation of nuclear factor κB. J. Biol. Chem. 2011, 286, 20727–20735. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.A.; Ryzhakov, G.; Cooray, S.; Randow, F.; Smith, G.L. Inhibition of IκB kinase by vaccinia virus virulence factor B14. PLoS Pathog. 2008, 4, e22. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.C.; Bahar, M.W.; Cooray, S.; Chen, R.A.; Whalen, D.M.; Abrescia, N.G.; Alderton, D.; Owens, R.J.; Stuart, D.I.; Smith, G.L.; et al. Vaccinia virus proteins A52 and B14 Share a Bcl-2-like fold but have evolved to inhibit NF-κB rather than apoptosis. PLoS Pathog. 2008, 4, e1000128. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Chakraborty, S.; Xu, G. Mechanism of vaccinia viral protein B14 mediated inhibition of IκB kinase β activation. J. Biol. Chem. 2018, RA118, 002817. [Google Scholar] [CrossRef] [PubMed]
- Mathers, C.; Schafer, X.; Martínez-Sobrido, L.; Munger, J. The human cytomegalovirus UL26 protein antagonizes NF-κB activation. J. Virol. 2014, 88, 14289–14300. [Google Scholar] [CrossRef] [PubMed]
- Diel, D.G.; Delhon, G.; Luo, S.; Flores, E.F.; Rock, D.L. A Novel Inhibitor of the NF-κB Signaling Pathway Encoded by the Parapoxvirus Orf Virus. J. Virol. 2010, 84, 3962–3973. [Google Scholar] [CrossRef] [PubMed]
- Nichols, D.B.; Shisler, J.L. The MC160 Protein Expressed by the Dermatotropic Poxvirus Molluscum Contagiosum Virus Prevents Tumor Necrosis Factor Alpha-Induced NF-κB Activation via Inhibition of I Kappa Kinase Complex Formation. J. Virol. 2006, 80, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Beaury, M.; Velagapudi, U.K.; Weber, S.; Soto, C.; Talele, T.T.; Nichols, D.B. The molluscum contagiosum virus death effector domain containing protein MC160 RxDL motifs are not required for its known viral immune evasion functions. Virus Genes 2017, 53, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Song, L.; Li, J.; Zhang, Z.; Peng, H.; Jiang, W.; Wang, Q.; Kang, T.; Chen, S.; Huang, W. Influenza A virus-encoded NS1 virulence factor protein inhibits innate immune response by targeting IKK. Cell. Microbiol. 2012, 14, 1849–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Ma, J.; Yoo, D. Inhibition of NF-κB activity by the porcine epidemic diarrhea virus nonstructural protein 1 for innate immune evasion. Virology 2017, 510, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Whitmer, T.; Malouli, D.; Uebelhoer, L.S.; Defilippis, V.R.; Früh, K.; Verweij, M.C. The ORF61 Protein Encoded by Simian Varicella Virus and Varicella-Zoster Virus Inhibits NF-κB Signaling by Interfering with IκBα Degradation. J. Virol. 2015, 89, 8687–8700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van, B.N.; Burles, K.; Schriewer, J.; Mehta, N.; Parker, S.; Buller, R.M.; Barry, M. EVM005: An Ectromelia-Encoded Protein with Dual Roles in NF-κB Inhibition and Virulence. PLoS Pathog. 2014, 10, e1004326. [Google Scholar]
- Kristin, B.; Nicholas, V.B.; Michele, B. Ectromelia virus encodes a family of Ankyrin/F-box proteins that regulate NFκB. Virology 2014, 470, 351–362. [Google Scholar]
- Wang, K.; Ni, L.; Wang, S.; Zheng, C. Herpes simplex virus 1 protein kinase US3 hyperphosphorylates p65/RelA and dampens NF-κB activation. J. Virol. 2014, 88, 7941–7951. [Google Scholar] [CrossRef] [PubMed]
- Sen, J.; Liu, X.; Roller, R.; Knipe, D.M. Herpes simplex virus US3 tegument protein inhibits Toll-like receptor 2 signaling at or before TRAF6 ubiquitination. Virology 2013, 439, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Su, C.; Pearson, A.; Mody, C.H.; Zheng, C. Herpes Simplex Virus 1 UL24 Abrogates the DNA Sensing Signal Pathway by Inhibiting NF-κB Activation. J. Virol. 2017, 91, e00025-17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, S.; Wang, K.; Zheng, C. Herpes simplex virus 1 DNA polymerase processivity factor UL42 inhibits TNF-α-induced NF-κB activation by interacting with p65/RelA and p50/NF-κB1. Med. Microbiol. Immunol. 2013, 202, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, K.; Wang, S.; Zheng, C. Herpes simplex virus 1 E3 ubiquitin ligase ICP0 protein inhibits tumor necrosis factor alpha-induced NF-κB activation by interacting with p65/RelA and p50/NF-κB1. J. Virol. 2013, 87, 12935–12948. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Yin, P.; Yang, X.; Zhang, L.; Jin, Q.; Zhu, G. Enterovirus 71 2C Protein Inhibits NF-κB Activation by Binding to RelA(p65). Sci. Rep. 2015, 5, 14302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diel, D.G.; Luo, S.; Delhon, G.; Peng, Y.; Flores, E.F.; Rock, D.L. Orf Virus ORFV121 Encodes a Novel Inhibitor of NF-κB That Contributes to Virus Virulence. J. Virol. 2011, 85, 2037–2049. [Google Scholar] [CrossRef] [PubMed]
- Brady, G.; Haas, D.A.; Farrell, P.J.; Pichlmair, A.; Bowie, A.G. Poxvirus Protein MC132 from Molluscum Contagiosum Virus Inhibits NF-κB Activation by Targeting p65 for Degradation. J. Virol. 2015, 89, 8406–8415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, L.; Filipe, J.; Seldon, M.P.; Fonseca, L.; Anrather, J.; Soares, M.P.; Simas, J.P. Termination of NF-κB activity through a gammaherpesvirus protein that assembles an EC 5 S ubiquitin-ligase. EMBO J. 2009, 28, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liang, D.; Lin, X.; Robertson, E.S.; Lan, K. Kaposi’s sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen reduces interleukin-8 expression in endothelial cells and impairs neutrophil chemotaxis by degrading nuclear p65. J. Virol. 2011, 85, 8606–8615. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Chen, Z.; Li, Y.; Zhao, Z.; He, W.; Zohaib, A.; Song, Y.; Deng, C.; Zhang, B.; Chen, H. Japanese Encephalitis Virus NS5 Inhibits Type I Interferon (IFN) Production by Blocking the Nuclear Translocation of IFN Regulatory Factor 3 and NF-κB. J. Virol. 2017, 91, e00039-17. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.L.; Friasstaheli, N.; Garcíasastre, A.; Schmaljohn, C.S. Hantaan virus nucleocapsid protein binds to importin alpha proteins and inhibits tumor necrosis factor alpha-induced activation of nuclear factor kappa B. J. Virol. 2009, 83, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, Z.; Zheng, Z.; Zheng, C.; Yan, L.; Hu, Q.; Ke, X.; Wang, H. Human Bocavirus NS1 and NS1-70 Proteins Inhibit TNF-α-Mediated Activation of NF-κB by targeting p65. Sci. Rep. 2016, 6, 28481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diel, D.G.; Luo, S.; Delhon, G.; Peng, Y.; Flores, E.F.; Rock, D.L. A Nuclear Inhibitor of NF-κB Encoded by a Poxvirus. J. Virol. 2011, 85, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Ni, L.; Wang, S.; Wang, K.; Lin, R.; Zheng, C. Herpes simplex virus 1-encoded tegument protein VP16 abrogates the production of beta interferon (IFN) by inhibiting NF-κB activation and blocking IFN regulatory factor 3 to recruit its coactivator CBP. J. Virol. 2013, 87, 9788–9801. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Foy, E.; Ferreon, J.C.; Nakamura, M.; Ferreon, A.C.; Ikeda, M.; Ray, S.C.; Gale, M., Jr.; Lemon, S.M. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. USA 2005, 102, 2992–2997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morikawa, K.; Lange, C.M.; Gouttenoire, J.; Meylan, E.; Brass, V.; Penin, F.; Moradpour, D. Nonstructural protein 3-4A: The Swiss army knife of hepatitis C virus. J. Viral Hepat. 2011, 18, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and Characterization of MAVS, a Mitochondrial Antiviral Signaling Protein that Activates NF-κB and IRF3. Cell 2015, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Horner, S.M.; Park, H.S.; Gale, M., Jr. Control of Innate Immune Signaling and Membrane Targeting by the Hepatitis C Virus NS3/4A Protease Are Governed by the NS3 Helix α0. J. Virol. 2012, 86, 3112–3120. [Google Scholar] [CrossRef] [PubMed]
- Romano, K.P.; Laine, J.M.; Deveau, L.M.; Cao, H.; Massi, F.; Schiffer, C.A. Molecular mechanisms of viral and host cell substrate recognition by hepatitis C virus NS3/4A protease. J. Virol. 2011, 85, 6106–6116. [Google Scholar] [CrossRef] [PubMed]
- Gagné, B.; Tremblay, N.; Park, A.Y.; Baril, M.; Lamarre, D. Importin β1 targeting by Hepatitis C Virus NS3/4A Protein Restricts IRF3 and NF-κB Signaling of IFNB1 Antiviral Response. Traffic 2017, 18, 362–377. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Morosky, S.A.; Delormeaxford, E.; Dybdahlsissoko, N.; Oberste, M.S.; Wang, T.; Coyne, C.B. The Coxsackievirus B 3Cpro Protease Cleaves MAVS and TRIF to Attenuate Host Type I Interferon and Apoptotic Signaling. PLoS Pathog. 2011, 7, e1001311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Z.; Li, L.; Lei, X.; Zhou, H.; Zhou, Z.; He, B.; Wang, J. Enterovirus 68 3C Protease Cleaves TRIF To Attenuate Antiviral Responses Mediated by Toll-Like Receptor 3. J. Virol. 2014, 88, 6650–6659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, X.; Han, N.; Xiao, X.; Jin, Q.; He, B.; Wang, J. Enterovirus 71 3C inhibits cytokine expression through cleavage of the TAK1/TAB1/TAB2/TAB3 complex. J. Virol. 2014, 88, 9830–9841. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.; Su, J.; Wang, H.; Chang, J.; Wang, S.; Zheng, W.; Cai, Y.; Wei, W.; Gordy, J.T.; Markham, R. Disruption of MDA5 mediated innate immune responses by the 3C proteins of Coxsackievirus A16, Coxsackievirus A6, and Enterovirus D68. J. Virol. 2017, 91, e00546-17. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fang, L.; Li, K.; Zhong, H.; Fan, J.; Ouyang, C.; Zhang, H.; Duan, E.; Luo, R.; Zhang, Z. Foot-and-mouth disease virus 3C protease cleaves NEMO to impair innate immune signaling. J. Virol. 2012, 86, 9311–9322. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fang, L.; Wei, D.; Zhang, H.; Luo, R.; Chen, H.; Li, K.; Xiao, S. Hepatitis A virus 3C protease cleaves NEMO to impair induction of beta interferon. J. Virol. 2014, 88, 10252–10258. [Google Scholar] [CrossRef] [PubMed]
- Blom, N.; Hansen, J.; Blaas, D.; Brunak, S. Cleavage site analysis in picornaviral polyproteins: Discovering cellular targets by neural networks. Protein Sci. 1996, 5, 2203–2216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, X.; Sun, Z.; Liu, X.; Jin, Q.; He, B.; Wang, J. Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll-like receptor 3. J. Virol. 2011, 85, 8811–8818. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Zhang, Q.; Guo, X.K.; Yu, Z.B.; Xu, A.T.; Tang, J.; Feng, W.H. Porcine Reproductive and Respiratory Syndrome Virus Nonstructural Protein 4 Antagonizes IFNβ Expression by Targeting NEMO. J. Virol. 2014, 88, 10934–10945. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fang, L.; Shi, Y.; Zhang, H.; Gao, L.; Peng, G.; Chen, H.; Li, K.; Xiao, S. Porcine Epidemic Diarrhoea Virus 3C-Like Protease Regulates its Interferon Antagonism by Cleaving NEMO. J. Virol. 2017, 90, 2090–2101. [Google Scholar] [CrossRef] [PubMed]
- Won, M.; Byun, H.S.; Park, K.A.; Gang, M.H. Post-translational control of NF-κB signaling by ubiquitination. Arch. Pharm. Res. 2016, 39, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Li, S.W.; Wang, C.Y.; Jou, Y.J.; Huang, S.H.; Hsiao, L.H.; Wan, L.; Lin, Y.J.; Kung, S.H.; Lin, C.W. SARS Coronavirus Papain-Like Protease Inhibits the TLR7 Signaling Pathway through Removing Lys63-Linked Polyubiquitination of TRAF3 and TRAF6. Int. J. Mol. Sci. 2016, 17, e678. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Tian, J.; Kang, H.; Guo, D.; Liu, J.; Liu, D.; Jiang, Q.; Li, Z.; Qu, J.; Qu, L. Transmissible Gastroenteritis Virus Papain-Like Protease 1 Antagonizes Production of Interferon-β through Its Deubiquitinase Activity. BioMed Res. Int. 2017, 2017, 7089091. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Chen, Z.H.; Lawson, S.R.; Fang, Y. The cysteine protease domain of porcine reproductive and respiratory syndrome virus nonstructural protein 2 possesses deubiquitinating and interferon antagonism functions. J. Virol. 2010, 84, 7832–7846. [Google Scholar] [CrossRef] [PubMed]
- van Kasteren, P.B.; Baileyelkin, B.A.; James, T.W.; Ninaber, D.K.; Beugeling, C.; Khajehpour, M.; Snijder, E.J.; Mark, B.L.; Kikkert, M. Deubiquitinase function of arterivirus papain-like protease 2 suppresses the innate immune response in infected host cells. Proc. Natl. Acad. Sci. USA 2013, 110, E838. [Google Scholar] [CrossRef] [PubMed]
- Kwon, K.M.; Oh, S.E.; Kim, Y.E.; Han, T.H.; Ahn, J.H. Cooperative inhibition of RIP1-mediated NF-κB signaling by cytomegalovirus-encoded deubiquitinase and inactive homolog of cellular ribonucleotide reductase large subunit. PLoS Pathog. 2017, 13, e1006423. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.; Su, C.; Xu, H.; Zheng, C. Herpes Simplex Virus Type 1 Ubiquitin-specific Protease UL36 abrogates NF-κB Activation in DNA Sensing Signal Pathway. J. Virol. 2016, 91, e02417-16. [Google Scholar]
- Inn, K.S.; Lee, S.H.; Rathbun, J.Y.; Wong, L.Y.; Toth, Z.; Machida, K.; Ou, J.H.J.; Jung, J.U. Inhibition of RIG-I-Mediated Signaling by Kaposi’s Sarcoma-Associated Herpesvirus-Encoded Deubiquitinase ORF64. J. Virol. 2011, 85, 10899–10904. [Google Scholar] [CrossRef] [PubMed]
- Van, G.M.; Braem, S.G.; De, J.A.; Delagic, N.; Peeters, J.G.; Boer, I.G.; Moynagh, P.N.; Kremmer, E.; Wiertz, E.J.; Ovaa, H. Epstein-Barr virus large tegument protein BPLF1 contributes to innate immune evasion through interference with toll-like receptor signaling. PLoS Pathog. 2014, 10, e1003960. [Google Scholar]
- Kanarek, N.; London, N.; Schuelerfurman, O.; Benneriah, Y. Ubiquitination and degradation of the inhibitors of NF-kappaB. Cold Spring Harb. Perspect. Biol. 2010, 2, a000166. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.A.; Morelli, M.; Patton, J.T. Rotavirus NSP1 Requires Casein Kinase II-Mediated Phosphorylation for Hijacking of Cullin-RING Ligases. mBio 2017, 8, e01213-17. [Google Scholar] [CrossRef] [PubMed]
- Lutz, L.M.; Pace, C.R.; Arnold, M.M. Rotavirus NSP1 Associates with Components of the Cullin RING Ligase Family of E3 Ubiquitin Ligases. J. Virol. 2016, 90, 6036–6048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, S.; Mooney, N.; Li, B.; Kelly, M.R.; Feng, N.; Loktev, A.V.; Sen, A.; Patton, J.T.; Jackson, P.K.; Greenberg, H.B. Comparative Proteomics Reveals Strain-Specific β-TrCP Degradation via Rotavirus NSP1 Hijacking a Host Cullin-3-Rbx1 Complex. PLoS Pathog. 2016, 12, e1005929. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.; Dennis, A.F.; Patton, J.T. Putative E3 Ubiquitin Ligase of Human Rotavirus Inhibits NF-κB Activation by Using Molecular Mimicry to Target β-TrCP. mBio 2015, 6, e02490-14. [Google Scholar] [CrossRef] [PubMed]
- Mansur, D.S.; Motes, C.M.D.; Unterholzner, L.; Sumner, R.P.; Ferguson, B.J.; Ren, H.; Strnadova, P.; Bowie, A.G.; Smith, G.L. Poxvirus Targeting of E3 Ligase β-TrCP by Molecular Mimicry: A Mechanism to Inhibit NF-κB Activation and Promote Immune Evasion and Virulence. PLoS Pathog. 2013, 9, e1003183. [Google Scholar] [CrossRef] [PubMed]
- Bour, S.; Perrin, C.; Akari, H.; Strebel, K. The Human Immunodeficiency Virus Type 1 Vpu Protein Inhibits NF-κB Activation by Interfering with βTrCP-mediated Degradation of IκB. J. Biol. Chem. 2001, 276, 15920–15928. [Google Scholar] [CrossRef] [PubMed]
- Manganaro, L.; De, C.E.; Maestre, A.M.; Olivieri, K.; Garcíasastre, A.; Fernandezsesma, A.; Simon, V. HIV Vpu Interferes with NF-κB Activity but Not with Interferon Regulatory Factor 3. J. Virol. 2015, 89, 9781–9790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Liu, H.; Wang, C.; Zhang, J.; Man, J.; Gao, Y.; Zhang, P.; Li, W.; Zhao, J.; Pan, X.; et al. Deactivation of the kinase IKK by CUEDC2 through recruitment of the phosphatase PP1. Nat. Immunol. 2008, 9, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; Ji, Y.; Zhu, X.; Zheng, X. hCINAP negatively regulates NF-κB signaling by recruiting the phosphatase PP1 to deactivate IKK complex. J. Mol. Cell Biol. 2015, 7, 529–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Yan, Z.; Ma, Y.; Cao, Y.; He, B. A Herpesvirus Virulence Factor Inhibits Dendritic Cell Maturation through Protein Phosphatase 1 and IκB Kinase. J. Virol. 2011, 85, 3397–3407. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zheng, Z.; Liu, Y.; Zhang, Z.; Liu, Q.; Meng, J.; Ke, X.; Hu, Q.; Wang, H. 2C Proteins of Enteroviruses Suppress IKKβ Phosphorylation by Recruiting Protein Phosphatase 1. J. Virol. 2016, 90, 5141–5151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, D.A.; Abdulsada, H.; Knight, L.M.; Jackson, B.R.; Richards, K.; Prescott, E.L.; Peach, A.H.S.; Blair, G.E.; Macdonald, A.; Whitehouse, A. Merkel Cell Polyomavirus Small T Antigen Targets the NEMO Adaptor Protein to Disrupt Inflammatory Signaling. J. Virol. 2013, 87, 13853–13867. [Google Scholar] [CrossRef] [PubMed]
- Abdulsada, H.; Müller, M.; Mehta, R.; Toth, R.; Jsc, A.; Whitehouse, A.; Macdonald, A. The PP4R1 sub-unit of protein phosphatase PP4 is essential for inhibition of NF-κB by merkel polyomavirus small tumour antigen. Oncotarget 2017, 8, 25418–25432. [Google Scholar]
- Wilson, R.L.; Fuentes, S.P.; Taddeo, E.C.; Klatt, A.; Henderson, A.J.; He, B. Function of small hydrophobic proteins of paramyxovirus. J. Virol. 2006, 80, 1700–1709. [Google Scholar] [CrossRef] [PubMed]
- Franz, S.; Rennert, P.; Woznik, M.; Grützke, J.; Lüdde, A.; Arriero Pais, E.M.; Finsterbusch, T.; Geyer, H.; Mankertz, A.; Friedrich, N. Mumps Virus SH Protein Inhibits NF-κB Activation by Interacting with Tumor Necrosis Factor Receptor 1, Interleukin-1 Receptor 1, and Toll-Like Receptor 3 Complexes. J. Virol. 2017, 91, e01037-17. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, S.; Tran, K.C.; Luthra, P.; Teng, M.N.; He, B. Function of the respiratory syncytial virus small hydrophobic protein. J. Virol. 2007, 81, 8361–8366. [Google Scholar] [CrossRef] [PubMed]
- Pollock, N.; Taylor, G.; Jobe, F.; Guzman, E. Modulation of the transcription factor NF-κB in antigen-presenting cells by bovine respiratory syncytial virus small hydrophobic protein. J. Gen. Virol. 2017, 98, 1587–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Xu, J.; Patel, J.; Fuentes, S.; Lin, Y.; Anderson, D.; Sakamoto, K.; Wang, L.F.; He, B. Function of the small hydrophobic protein of J paramyxovirus. J. Virol. 2011, 85, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Kolli, D.; Liu, T.; Shan, Y.; Garofalo, R.P.; Casola, A. Human Metapneumovirus Small Hydrophobic Protein Inhibits NF-κB Transcriptional Activity. J. Virol. 2008, 82, 8224–8229. [Google Scholar] [CrossRef] [PubMed]
- Sauter, D.; Hotter, D.; Van Driessche, B.; Sturzel, C.M.; Kluge, S.F.; Wildum, S.; Yu, H.; Baumann, B.; Wirth, T.; Plantier, J.; et al. Differential Regulation of NF-κB-Mediated Proviral and Antiviral Host Gene Expression by Primate Lentiviral Nef and Vpu Proteins. Cell Rep. 2015, 10, 586–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokarev, A.; Suarez, M.; Kwan, W.; Fitzpatrick, K.; Singh, R.K.; Guatelli, J.C. Stimulation of NF-κB activity by the HIV restriction factor BST2. J. Virol. 2013, 87, 2046–2057. [Google Scholar] [CrossRef] [PubMed]
- Shen, A.; Baker, J.J.; Scott, G.L.; Davis, Y.P.; Ho, Y.Y.; Siliciano, R.F. Endothelial Cell Stimulation Overcomes Restriction and Promotes Productive and Latent HIV-1 Infection of Resting CD4+ T Cells. J. Virol. 2013, 87, 9768–9779. [Google Scholar] [CrossRef] [PubMed]
- Cary, D.C.; Fujinaga, K.; Peterlin, B.M. Molecular mechanisms of HIV latency. J. Clin. Investig. 2016, 126, 448–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Wang, H.B.; Kuang, W.D.; Ren, X.X.; Song, S.T.; Zhu, H.Z.; Li, Q.; Xu, L.R.; Guo, H.J.; Wu, L. Naf1 Regulates HIV-1 Latency by Suppressing Viral Promoter-Driven Gene Expression in Primary CD4+ T Cells. J. Virol. 2017, 91, e01830-16. [Google Scholar] [CrossRef] [PubMed]
- Manganaro, L.; Pache, L.; Herrmann, T.; Marlett, J.M.; Hwang, Y.; Murry, J.P.; Miorin, L.; Ting, A.T.; Konig, R.; Garciasastre, A.; et al. Tumor Suppressor Cylindromatosis (CYLD) Controls HIV Transcription in an NF-κB-Dependent Manner. J. Virol. 2014, 88, 7528–7540. [Google Scholar] [CrossRef] [PubMed]
- Uchil, P.D.; Hinz, A.; Siegel, S.; Coenenstass, A.M.L.; Pertel, T.; Luban, J.; Mothes, W. TRIM protein mediated regulation of inflammatory and innate immune signaling and its association with antiretroviral activity. J. Virol. 2013, 87, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Ruelas, D.S.; Chan, J.K.L.; Oh, E.; Heidersbach, A.; Hebbeler, A.M.; Chavez, L.; Verdin, E.; Rape, M.; Greene, W.C. MicroRNA-155 Reinforces HIV Latency. J. Biol. Chem. 2015, 290, 13736–13748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Victoriano, A.F.; Okamoto, T. Transcriptional control of HIV replication by multiple modulators and their implication for a novel antiviral therapy. Aids Res. Hum. Retrovir. 2012, 28, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Li, J.; Cheng, L.; Liu, F.; Wu, N. Gp120 binding with DC-SIGN induces reactivation of HIV-1 provirus via the NF-κB signaling pathway. Acta Biochim. Biophys. Sin. 2016, 48, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Virus * | Viral Protein | Mechanisms of Modulation | Host Targets | References |
|---|---|---|---|---|
| HSV-1 | ICP0 | Promotes the degradation of MyD88 and TIRAP; binds to RHD of p50 and p65 | MyD88, TIRAP, p50, p65 | [26,62] |
| Us3 | Reduces TRAF6 polyubiquitination and hyperphosphorylates p65 | TRAF6, p65 | [58,59] | |
| UL24 | Binds to the RHD of p50 and p65 | p50, p65 | [60] | |
| UL42 | Binds to the RHD of p50 and p65 | p50, p65 | [61] | |
| VP16 | Binds to p65 and probably sequesters CBP | p65 | [72] | |
| UL36 | Cleaves polyubiquitin chains from IκBα | IκBα | [96] | |
| γ134.5 | Recruits both IKKα/β and PP1 to dephosphorylate IKKβ | IKKβ | [109] | |
| VZV | ORF61 | Prevents β-TrCP-mediated IκBα ubiquitination | IκBα | [55] |
| SVV | ORF61 | Binds to β-TrCP and interferes with IκBα ubiquitination | IκBα | [55] |
| HCMV | pUL83 | Blocks IFI16 pyrin aggregation | IFI16 | [31] |
| UL26 | Decreases phosphorylation of IKKα and IKKβ | IKKα, IKKβ | [49] | |
| UL48 | Cleaves K48- and K63-linked polyubiquitin chains of RIP1 | RIP1 | [95] | |
| MCMV | M45 | Disrupts DAI–RIP1 interactions or inhibits the ubiquitination of RIP1; induces degradation of NEMO | RIP1, NEMO | [32,33,34] |
| KSHV | RTA | Reduces levels of expressed TLR2, TLR4 and MyD88; degrades MyD88 and TRIF | TLR2, TLR4, MyD88, TRIF | [21,24,25,30] |
| LANA-1 | Causes p65 ubiquitination and degradation | p65 | [67] | |
| ORF64 | Reduces the ubiquitination of RIG-I | RIG-I | [97] | |
| EBV | BPLF1 | Removes ubiquitin chains from IκBα, TRAF6 and NEMO | IκBα, TRAF6, NEMO | [98] |
| MuHV-4 | ORF73 | Causes p65 ubiquitination and degradation | p65 | [66] |
| PV | 2C | Recruits both IKKα/β and PP1 to dephosphorylate IKKβ | IKKβ | [110] |
| CVA16 | 2C | Recruits both IKKα/β and PP1 to dephosphorylate IKKβ | IKKβ | [110] |
| CVB3 | 2C | Recruits both IKKα/β and PP1 to dephosphorylate IKKβ | IKKβ | [110] |
| 3C | Cleaves MAVS and TRIF | MAVS, TRIF | [80] | |
| EV71 | 2C | Interacts with the IPI domain of p65; recruits both IKKα/β and PP1 to dephosphorylate IKKβ | p65, IKKβ | [63,110] |
| 3C | Cleaves TRIF, TAK1, TAB1, TAB2, and TAB3 | TRIF, TAK1, TAB1, TAB2, TAB3 | [82,87] | |
| EV68 | 3C | Cleaves TRIF | TRIF | [81] |
| FMDV | 3C | Cleaves NEMO | NEMO | [84] |
| HAV | 3C | Cleaves NEMO | NEMO | [85] |
| HBV | HBX | Promotes the degradation of MAVS | MAVS | [28] |
| HBeAg | Interacts and colocalizes with Mal and TRAM; inhibits the expression of RIP2 | Mal, TRAM, RIP2 | [35,36,37] | |
| HCV | NS3 | Decreases LUBAC-mediated linear ubiquitylation of NEMO | NEMO | [40] |
| NS3/4A | Cleaves MAVS, TRIF, and Importin β1 | MAVS, TRIF, Importin β1 | [73,74,77,78,79] | |
| HEV | ORF3 | Reduces the mRNA levels of TLR4, TLR6, NOD2, and TRADD | TLR4, TLR6, NOD2, TRADD | [22,23] |
| HBoV | NS1-70 | Interacts with p65 RHD | p65 | [70] |
| NS1 | Interacts with p65 RHD and inhibits the phosphorylation of p65 | p65 | [70] | |
| JEV | NS5 | Blocks the interaction of importin α with p65 | importin α | [68] |
| IAV | NS1 | Decreases phosphorylation of IKKα and IKKβ | IKKα, IKKβ | [53] |
| CSFV | NS3 | Promotes the degradation of TRAF6 | TRAF6 | [27] |
| RV | NSP1 | Binds to β-TrCP and interferes with IκBα degradation | IκBα | [100,101,102,103] |
| PEDV | NSP1 | Inhibits the phosphorylation and degradation of IκBα | IκBα | [54] |
| NSP5 | Cleaves NEMO | NEMO | [89] | |
| PRRSV | NSP11 | Reduces the mRNA levels of both MAVS and RIG-I | MAVS, RIG-I | [20] |
| NSP4 | Cleaves NEMO | NEMO | [88] | |
| NSP2 | Interferes with the polyubiquitination of IκBα | IκBα | [93] | |
| HTNV | N protein | Blocks the interaction of importin α with p65 | importin α | [69] |
| CoV | ORF-9b | Promotes degradation of MAVS | MAVS | [29] |
| SARS-CoV | PLP | Removes Lys63-linked ubiquitin chains of TRAF3 and TRAF6 | TRAF3, TRAF6 | [91] |
| TGEV | PLP1 | Binds to and deubiquitinates RIG-I | RIG-I | [92] |
| hMPV | M2-2 | Prevents MAVS from recruiting downstream molecules and interacts with MyD88 | MAVS, MyD88 | [38,39] |
| SH | Unknown | Unknown | [118] | |
| MuV | SH | Interacts with TNFR1, IL-1R1, and TLR3 complexes | TNFR1, RIP1, IRAK1 | [113,114] |
| HIV-1 | Vpu | Binds to β-TrCP and diminishes degradation of IκBα, counteracts tetherin | IκBα, tetherin | [105,106,119,120] |
| MCV | MC005 | Inhibits the activity of the conformational state of NEMO | NEMO | [41] |
| MC159 | Interacts with NEMO | NEMO | [42] | |
| MC160 | Reduces IKKα protein levels and the phosphorylation of IKKα and IKKβ | IKKα, IKKβ | [51,52] | |
| MC132 | Causes p65 ubiquitination and degradation | p65 | [65] | |
| VACV | C4 | Interacts with NEMO and IKKβ | NEMO, IKKβ | [43] |
| B14 | Prevents IKKβ phosphorylation and activation | IKKβ | [45,46,47,48] | |
| A49 | Binds to β-TrCP and diminishes degradation of IκBα | IκBα | [104] | |
| ORFV | ORFV073 | Inhibits IKK activation, possibly by interacting with NEMO | Unknown | [44] |
| ORFV024 | Decreases phosphorylation of IKKα and IKKβ | IKKα, IKKβ | [50] | |
| ORFV121 | Binds to p65 and inhibits the phosphorylation of p65 | p65 | [64] | |
| ORFV002 | Decreases acetylation of p65 | p65 | [71] | |
| ECTV | EVM002 | Interacts with Skp1 via the F-box domain and diminishes the interaction between β-TrCP and the SCFβ-TrCP complex | Skp1 | [56,57] |
| EVM005 | ||||
| EVM154 | ||||
| EVM165 | ||||
| MCPyV | T antigen | Recruits a PP4R1/PP4C/PP2A Aβ phosphatase complex to dephosphorylate IKKs | NEMO | [111,112] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, L.; Zeng, Q.; Wang, M.; Cheng, A.; Jia, R.; Chen, S.; Zhu, D.; Liu, M.; Yang, Q.; Wu, Y.; et al. Suppression of NF-κB Activity: A Viral Immune Evasion Mechanism. Viruses 2018, 10, 409. https://doi.org/10.3390/v10080409
Deng L, Zeng Q, Wang M, Cheng A, Jia R, Chen S, Zhu D, Liu M, Yang Q, Wu Y, et al. Suppression of NF-κB Activity: A Viral Immune Evasion Mechanism. Viruses. 2018; 10(8):409. https://doi.org/10.3390/v10080409
Chicago/Turabian StyleDeng, Liyao, Qiurui Zeng, Mingshu Wang, Anchun Cheng, Renyong Jia, Shun Chen, Dekang Zhu, Mafeng Liu, Qiao Yang, Ying Wu, and et al. 2018. "Suppression of NF-κB Activity: A Viral Immune Evasion Mechanism" Viruses 10, no. 8: 409. https://doi.org/10.3390/v10080409
APA StyleDeng, L., Zeng, Q., Wang, M., Cheng, A., Jia, R., Chen, S., Zhu, D., Liu, M., Yang, Q., Wu, Y., Zhao, X., Zhang, S., Liu, Y., Yu, Y., Zhang, L., & Chen, X. (2018). Suppression of NF-κB Activity: A Viral Immune Evasion Mechanism. Viruses, 10(8), 409. https://doi.org/10.3390/v10080409