Robust Innate Immunity of Young Rabbits Mediates Resistance to Rabbit Hemorrhagic Disease Caused by Lagovirus Europaeus GI.1 But Not GI.2

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Treatments and Virus Inoculation
2.2. RNA Extraction
2.3. Virus Quantification
2.4. RNA Sequencing and Analysis
3. Results
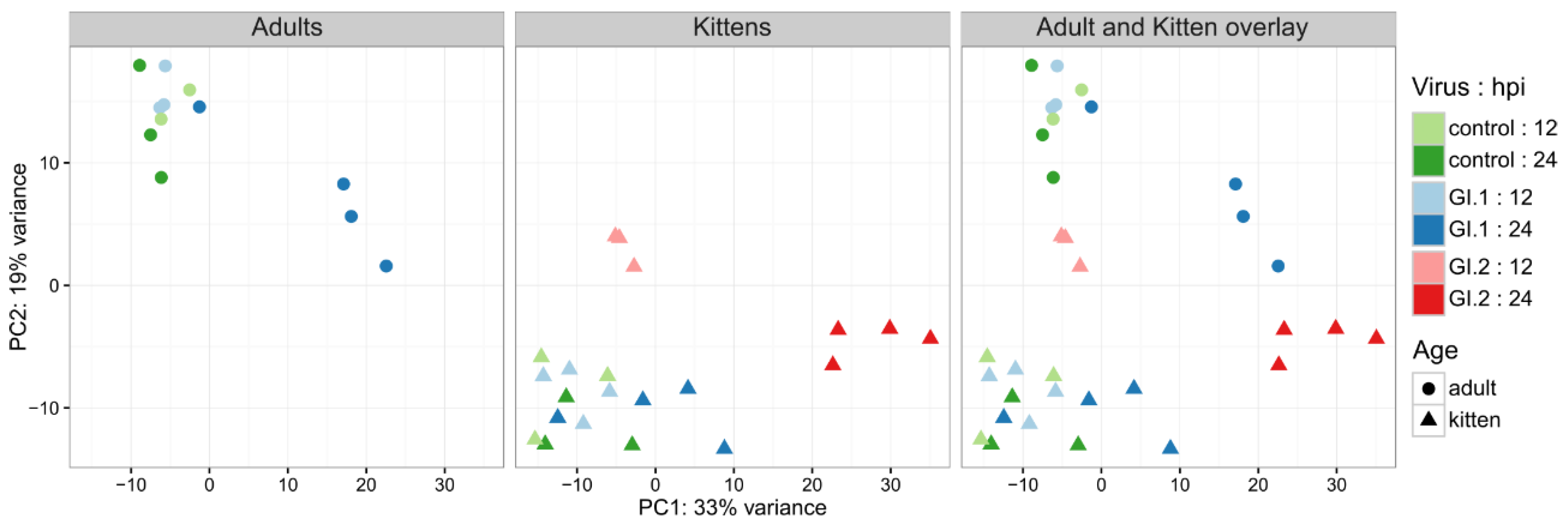
3.1. Genome Mapping and Significance Testing
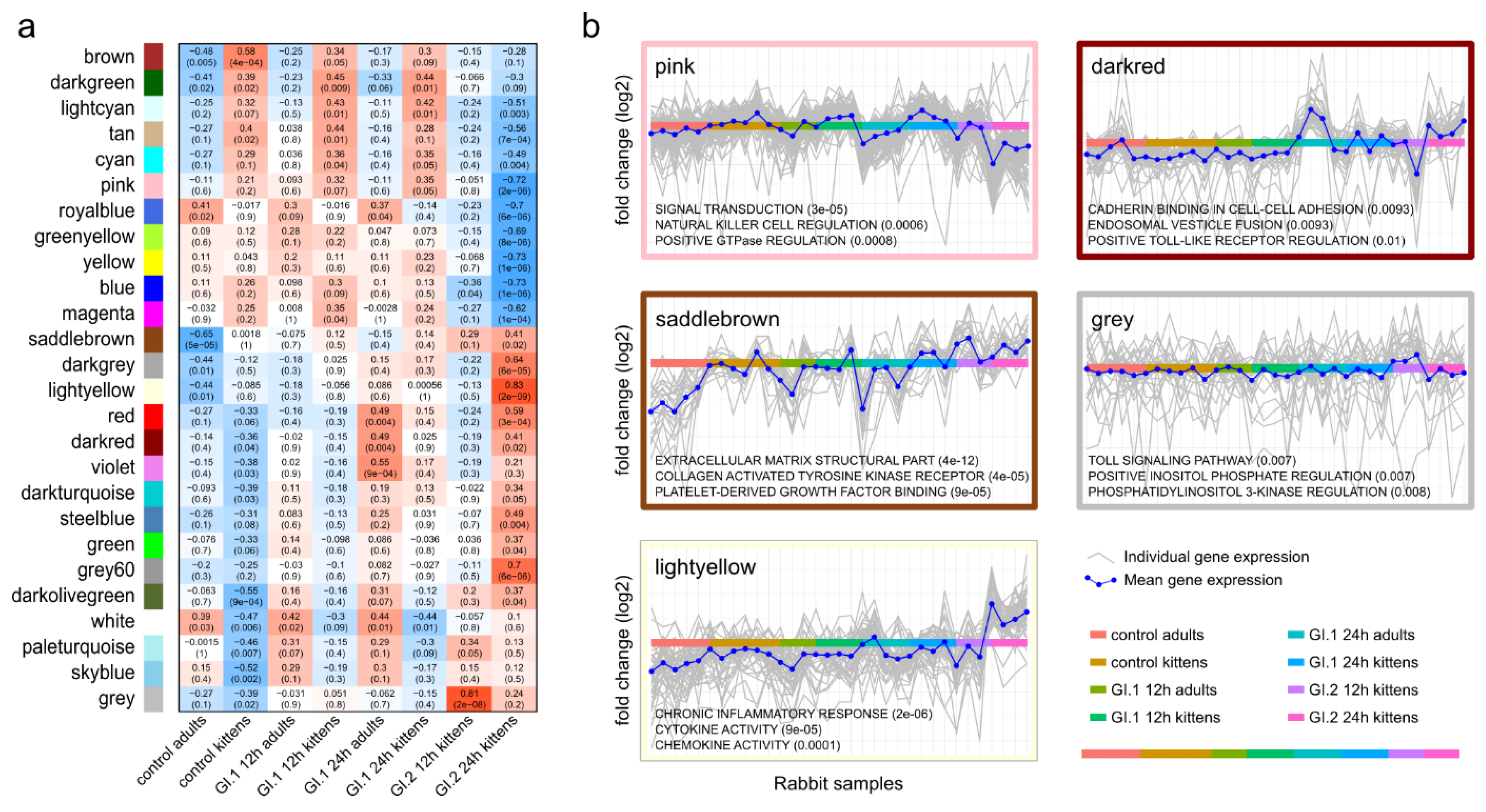
3.2. Differential Gene Expression and Pathway Enrichment in Adults and Kittens
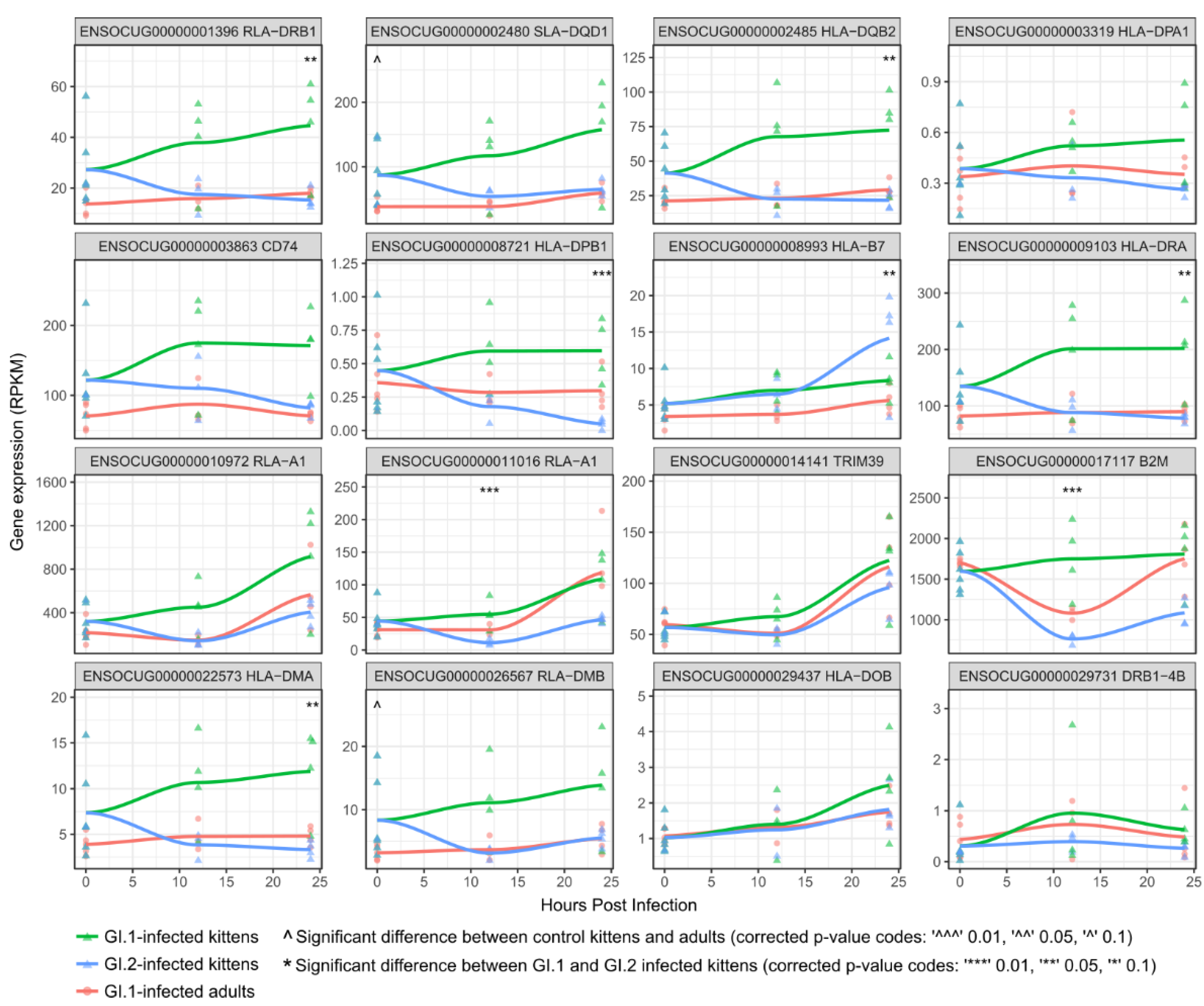
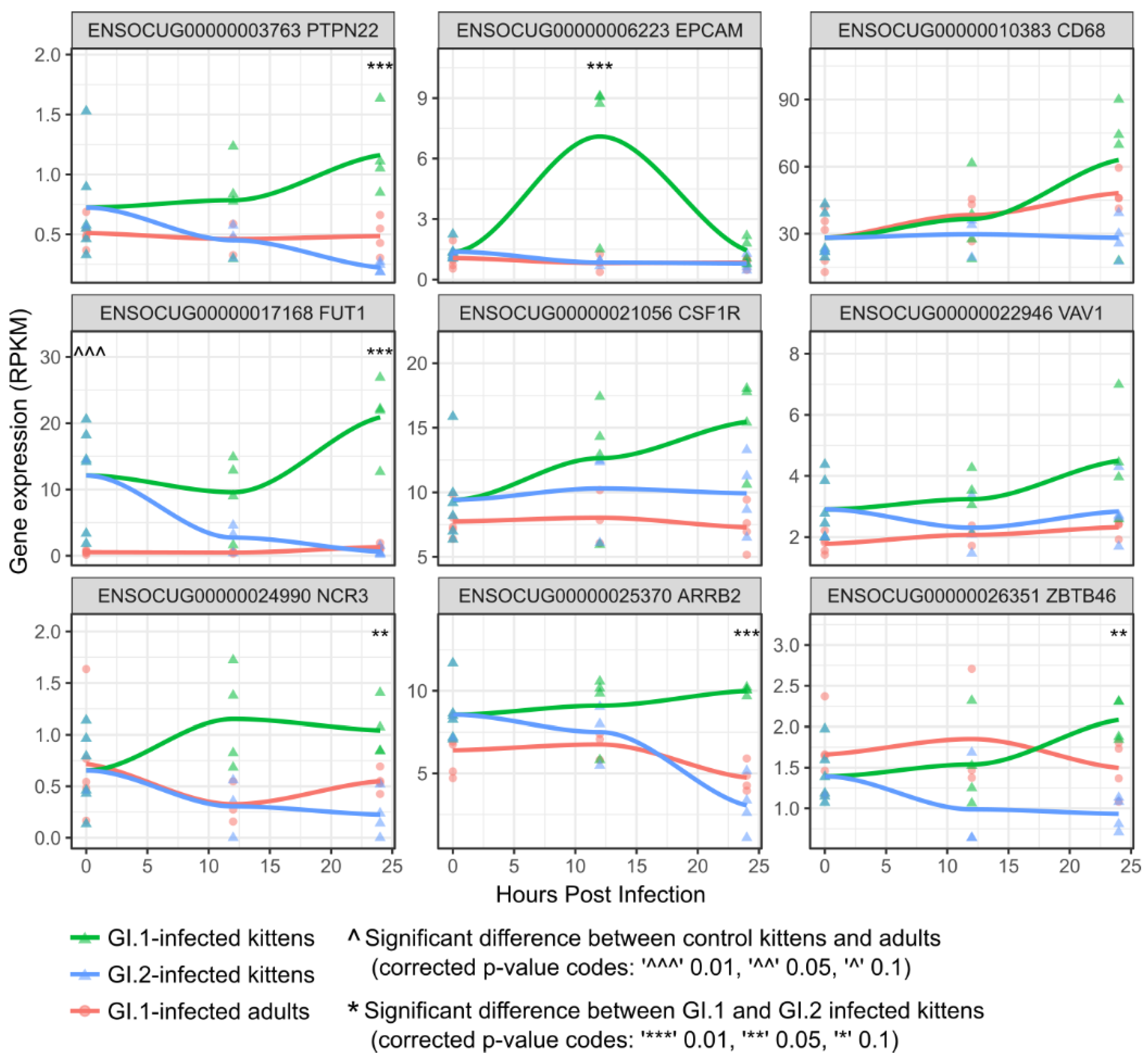
3.3. Kittens Upregulate Important Components of Innate Immunity Compared to Adults, Which May Limit GI.1-Induced Pathology
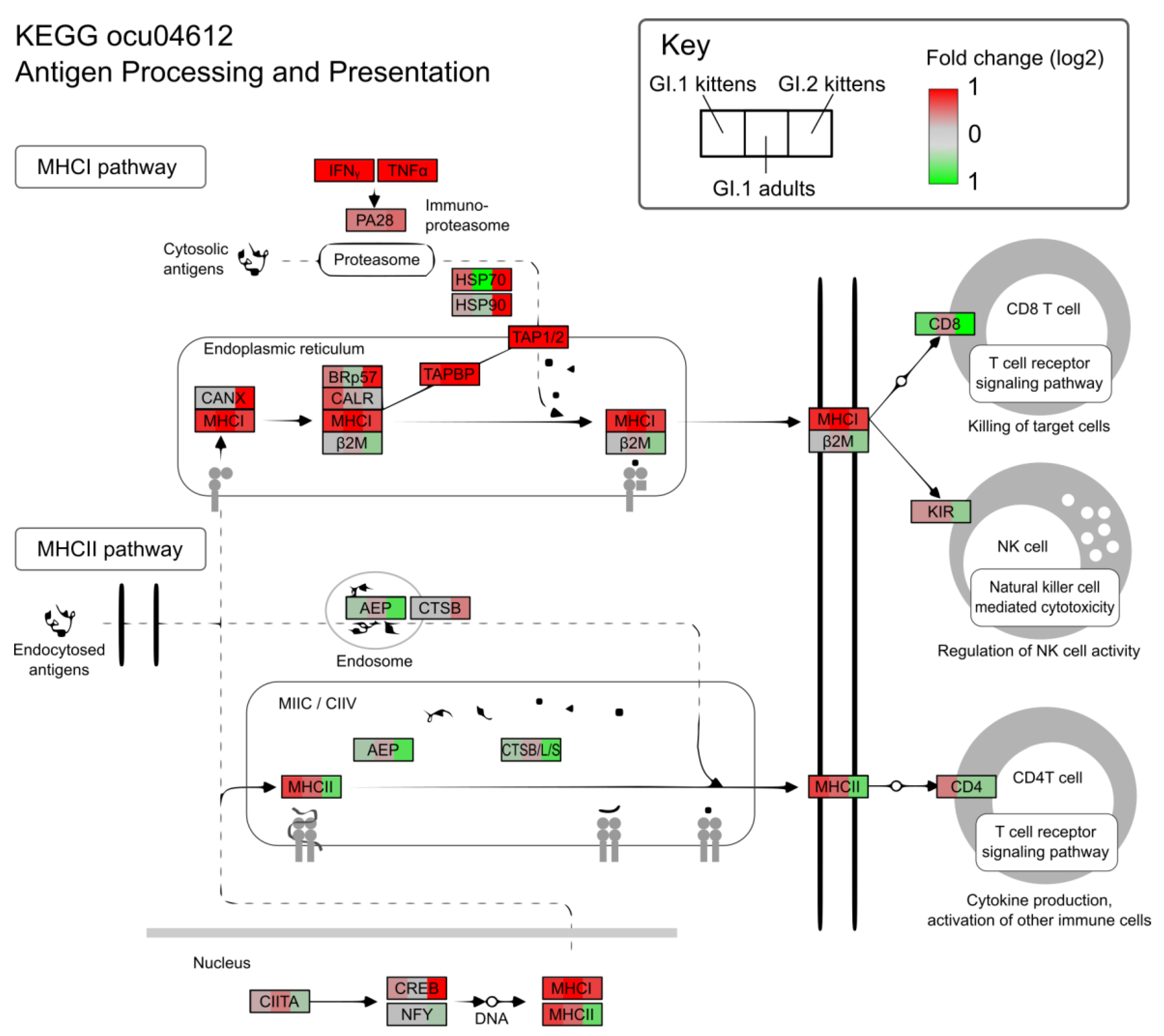
3.4. In Contrast to GI.1, GI.2 Infection Restricts the Activation of Several Innate Immune Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Data Availability
References
- Abrantes, J.; van der Loo, W.; Le Pendu, J.; Esteves, P.J. Rabbit haemorrhagic disease (RHD) and rabbit haemorrhagic disease virus (RHDV): A review. Vet. Res. 2012, 43, 12. [Google Scholar] [CrossRef] [PubMed]
- Le Gall-Recule, G.; Zwingelstein, F.; Boucher, S.; Le Normand, B.; Plassiart, G.; Portejoie, Y.; Decors, A.; Bertagnoli, S.; Guerin, J.L.; Marchandeau, S. Detection of a new variant of rabbit haemorrhagic disease virus in France. Vet. Rec. 2011, 168, 137–138. [Google Scholar] [CrossRef] [PubMed]
- Le Pendu, J.; Abrantes, J.; Bertagnoli, S.; Guitton, J.-S.; Le Gall-Reculé, G.; Lopes, A.M.; Marchandeau, S.; Alda, F.; Almeida, T.; Célio, A.P.; et al. Proposal for a unified classification system and nomenclature of lagoviruses. J. Gen. Virol. 2017, 98, 1658–1666. [Google Scholar] [CrossRef] [PubMed]
- Cooke, B.D.; Fenner, F. Rabbit haemorrhagic disease and the biological control of wild rabbits, Oryctolagus cuniculus, in Australia and New Zealand. Wildl. Res. 2002, 29, 689–706. [Google Scholar] [CrossRef]
- Dalton, K.P.; Nicieza, I.; Balseiro, A.; Muguerza, M.A.; Rosell, J.M.; Casais, R.; Alvarez, A.L.; Parra, F. Variant rabbit hemorrhagic disease virus in young rabbits, Spain. Emerg. Infect. Dis. 2012, 18, 2009–2012. [Google Scholar] [CrossRef] [PubMed]
- Abrantes, J.; Lopes, A.M.; Dalton, K.P.; Melo, P.; Correia, J.J.; Ramada, M.; Alves, P.C.; Parra, F.; Esteves, P.J. New variant of rabbit hemorrhagic disease virus, Portugal, 2012–2013. Emerg. Infect. Dis. 2013, 19, 1900–1902. [Google Scholar] [CrossRef] [PubMed]
- Westcott, D.G.; Frossard, J.P.; Everest, D.; Dastjerdi, A.; Duff, J.P.; Steinbach, F.; Choudhury, B. Incursion of RHDV2-like variant in Great Britain. Vet. Rec. 2014, 174, 333. [Google Scholar] [CrossRef] [PubMed]
- Baily, J.L.; Dagleish, M.P.; Graham, M.; Maley, M.; Rocchi, M.S. RHDV variant 2 presence detected in Scotland. Vet. Rec. 2014, 174, 411. [Google Scholar] [CrossRef] [PubMed]
- Duarte, M.; Carvalho, C.; Bernardo, S.; Barros, S.V.; Benevides, S.; Flor, L.; Monteiro, M.; Marques, I.; Henriques, M.; Barros, S.C.; et al. Rabbit haemorrhagic disease virus 2 (RHDV2) outbreak in Azores: Disclosure of common genetic markers and phylogenetic segregation within the European strains. Infect. Genet. Evol. 2015, 35, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Martin-Alonso, A.; Martin-Carrillo, N.; Garcia-Livia, K.; Valladares, B.; Foronda, P. Emerging rabbit haemorrhagic disease virus 2 (RHDV2) at the gates of the African continent. Infect. Genet. Evol. 2016, 44, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Rocchi, M.; Maley, M.; Dagleish, M.; Vick, C.; Ryan, D.; Lee, A.; Jahns, H. RHDV-2 on the Isle of Man and in the Republic of Ireland. Vet. Rec. 2016, 179, 389–390. [Google Scholar] [CrossRef] [PubMed]
- Neimanis, A.S.; Ahola, H.; Zohari, S.; Larsson Pettersson, U.; Brojer, C.; Capucci, L.; Gavier-Widen, D. Arrival of rabbit haemorrhagic disease virus 2 to northern Europe: Emergence and outbreaks in wild and domestic rabbits (Oryctolagus cuniculus) in Sweden. Transbound. Emerg. Dis. 2017, 65, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Hall, R.N.; Mahar, J.E.; Haboury, S.; Stevens, V.; Holmes, E.C.; Strive, T. Emerging rabbit hemorrhagic disease virus 2 (RHDVb), Australia. Emerg. Infect. Dis. 2015, 21, 2276–2278. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.M.; Correia, J.; Abrantes, J.; Melo, P.; Ramada, M.; Magalhaes, M.J.; Alves, P.C.; Esteves, P.J. Is the new variant RHDV replacing genogroup 1 in Portuguese wild rabbit populations? Viruses 2014, 7, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Mahar, J.E.; Hall, R.N.; Peacock, D.; Kovaliski, J.; Piper, M.; Mourant, R.; Huang, N.; Campbell, S.; Gu, X.; Read, A.; et al. Rabbit haemorrhagic disease virus 2 (GI.2) is replacing endemic strains of RHDV in the Australian landscape within 18 months of its arrival. J. Virol. 2017, 92. [Google Scholar] [CrossRef] [PubMed]
- Le Gall-Recule, G.; Lavazza, A.; Marchandeau, S.; Bertagnoli, S.; Zwingelstein, F.; Cavadini, P.; Martinelli, N.; Lombardi, G.; Guerin, J.L.; Lemaitre, E.; et al. Emergence of a new lagovirus related to rabbit haemorrhagic disease virus. Vet. Res. 2013, 44, 81. [Google Scholar] [CrossRef] [PubMed]
- Puggioni, G.; Cavadini, P.; Maestrale, C.; Scivoli, R.; Botti, G.; Ligios, C.; Le Gall-Recule, G.; Lavazza, A.; Capucci, L. The new French 2010 rabbit hemorrhagic disease virus causes an RHD-like disease in the Sardinian Cape hare (Lepus capensis mediterraneus). Vet. Res. 2013, 44, 96. [Google Scholar] [CrossRef] [PubMed]
- Camarda, A.; Pugliese, N.; Cavadini, P.; Circella, E.; Capucci, L.; Caroli, A.; Legretto, M.; Mallia, E.; Lavazza, A. Detection of the new emerging rabbit haemorrhagic disease type 2 virus (RHDV2) in Sicily from rabbit (Oryctolagus cuniculus) and Italian hare (Lepus corsicanus). Res. Vet. Sci. 2014, 97, 642–645. [Google Scholar] [CrossRef] [PubMed]
- Velarde, R.; Cavadini, P.; Neimanis, A.; Cabezon, O.; Chiari, M.; Gaffuri, A.; Lavin, S.; Grilli, G.; Gavier-Widen, D.; Lavazza, A.; et al. Spillover events of infection of brown hares (Lepus europaeus) with rabbit haemorrhagic disease type 2 virus (RHDV2) caused sporadic cases of an European brown hare syndrome-like disease in Italy and Spain. Transbound. Emerg. Dis. 2016, 64, 1750–1761. [Google Scholar] [CrossRef] [PubMed]
- Hall, R.N.; Peacock, D.E.; Kovaliski, J.; Mahar, J.E.; Mourant, R.; Piper, M.; Strive, T. Detection of RHDV2 in European brown hares (Lepus europaeus) in Australia. Vet. Rec. 2016, 180, 121. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.M.; Dalton, K.P.; Magalhaes, M.J.; Parra, F.; Esteves, P.J.; Holmes, E.C.; Abrantes, J. Full genomic analysis of new variant rabbit hemorrhagic disease virus revealed multiple recombination events. J. Gen. Virol. 2015, 96, 1309–1319. [Google Scholar] [CrossRef] [PubMed]
- Hall, R.N.; Mahar, J.E.; Read, A.J.; Mourant, R.; Piper, M.; Huang, N.; Strive, T. A strain-specific multiplex RT-PCR for Australian rabbit haemorrhagic disease viruses uncovers a new recombinant virus variant in rabbits and hares. Transbound. Emerg. Dis. 2017, 65, e444–e456. [Google Scholar] [CrossRef] [PubMed]
- Morisse, J.P.; Le Gall, G.; Boilletot, E. Hepatitis of viral origin in Leporidae: Introduction and aetiological hypotheses. Rev. Sci. Tech. 1991, 10, 269–310. [Google Scholar] [CrossRef] [PubMed]
- Cooke, B.D.; Berman, D. Effect of inoculation route and ambient temperature on the survival time of rabbits, Oryctolagus cuniculus, infected with Rabbit Haemorrhagic Disease Virus. Wildl. Res. 2000, 27, 137–142. [Google Scholar] [CrossRef]
- Capucci, L.; Cavadini, P.; Schiavitto, M.; Lombardi, G.; Lavazza, A. Increased pathogenicity in rabbit haemorrhagic disease virus type 2 (RHDV2). Vet. Rec. 2017, 180, 426. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wang, F.; Fan, Z.; Hu, B.; Liu, X.; Wei, H.; Xue, J.; Xu, W.; Qiu, R. Identification of novel rabbit hemorrhagic disease virus B-cell epitopes and their interaction with host histo-blood group antigens. J. Gen. Virol. 2016, 97, 356–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leuthold, M.M.; Dalton, K.P.; Hansman, G.S. Structural analysis of a rabbit hemorrhagic disease virus binding to histo-blood group antigens. J. Virol. 2015, 89, 2378–2387. [Google Scholar] [CrossRef] [PubMed]
- Nystrom, K.; Le Gall-Recule, G.; Grassi, P.; Abrantes, J.; Ruvoen-Clouet, N.; Le Moullac-Vaidye, B.; Lopes, A.M.; Esteves, P.J.; Strive, T.; Marchandeau, S.; et al. Histo-blood group antigens act as attachment factors of rabbit hemorrhagic disease virus infection in a virus strain-dependent manner. PLoS Pathog. 2011, 7, e1002188. [Google Scholar] [CrossRef] [PubMed]
- Guillon, P.; Ruvoen-Clouet, N.; Le Moullac-Vaidye, B.; Marchandeau, S.; Le Pendu, J. Association between expression of the H histo-blood group antigen, alpha1,2fucosyltransferases polymorphism of wild rabbits, and sensitivity to rabbit hemorrhagic disease virus. Glycobiology 2009, 19, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Ruvoen-Clouet, N.; Ganiere, J.P.; Andre-Fontaine, G.; Blanchard, D.; Le Pendu, J. Binding of rabbit hemorrhagic disease virus to antigens of the ABH histo-blood group family. J. Virol. 2000, 74, 11950–11954. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.M.; Breiman, A.; Lora, M.; Le Moullac-Vaidye, B.; Galanina, O.; Nyström, K.; Marchandeau, S.; Le Gall-Reculé, G.; Strive, T.; Neimanis, A.; et al. Host specific glycans are correlated with susceptibility to infection by lagoviruses, but not with their virulence. J. Virol. 2017, 92. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.Y.; Lee, B.J.; Tai, J.H.; Park, J.H.; Lee, Y.S. Apoptosis in rabbit haemorrhagic disease. J. Comp. Pathol. 2000, 123, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Prieto, J.M.; Fernandez, F.; Alvarez, V.; Espi, A.; Garcia Marin, J.F.; Alvarez, M.; Martin, J.M.; Parra, F. Immunohistochemical localisation of rabbit haemorrhagic disease virus VP-60 antigen in early infection of young and adult rabbits. Res. Vet. Sci. 2000, 68, 181–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcato, P.S.; Benazzi, C.; Vecchi, G.; Galeotti, M.; Della Salda, L.; Sarli, G.; Lucidi, P. Clinical and pathological features of viral haemorrhagic disease of rabbits and the European brown hare syndrome. Rev. Sci. Tech. 1991, 10, 371–392. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, M.; Peshev, R.; Yanchev, I.; Bozhkov, S.; Doumanova, L.; Dimitrov, T.; Zacharieva, S. Immunohistochemical localization of the rabbit haemorrhagic disease viral antigen. Arch. Virol. 1992, 127, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Stoerckle-Berger, N.; Keller-Berger, B.; Ackermann, M.; Ehrensperger, F. Immunohistological diagnosis of rabbit haemorrhagic disease (RHD). Zbl. Veterinarmed. B 1992, 39, 237–245. [Google Scholar] [CrossRef]
- Park, J.H.; Itakura, C. Detection of rabbit haemorrhagic disease virus antigen in tissues by immunohistochemistry. Res. Vet. Sci. 1992, 52, 299–306. [Google Scholar] [CrossRef]
- Ramiro-Ibanez, F.; Martin-Alonso, J.M.; Garcia Palencia, P.; Parra, F.; Alonso, C. Macrophage tropism of rabbit hemorrhagic disease virus is associated with vascular pathology. Virus Res. 1999, 60, 21–28. [Google Scholar] [CrossRef]
- Gelmetti, D.; Grieco, V.; Rossi, C.; Capucci, L.; Lavazza, A. Detection of rabbit haemorrhagic disease virus (RHDV) by in situ hybridisation with a digoxigenin labelled RNA probe. J. Virol. Methods 1998, 72, 219–226. [Google Scholar] [CrossRef]
- Fuchs, A.; Weissenbock, H. Comparative histopathological study of rabbit haemorrhagic disease (RHD) and European brown hare syndrome (EBHS). J. Comp. Pathol. 1992, 107, 103–113. [Google Scholar] [CrossRef]
- Ferreira, P.G.; Costa-e-Silva, A.; Monteiro, E.; Oliveira, M.J.; Aguas, A.P. Transient decrease in blood heterophils and sustained liver damage caused by calicivirus infection of young rabbits that are naturally resistant to rabbit haemorrhagic disease. Res. Vet. Sci. 2004, 76, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, P.G.; Costa, E.S.A.; Monteiro, E.; Oliveira, M.J.; Aguas, A.P. Liver enzymes and ultrastructure in rabbit haemorrhagic disease (RHD). Vet. Res. Commun. 2006, 30, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Tunon, M.J.; Sanchez-Campos, S.; Garcia-Ferreras, J.; Alvarez, M.; Jorquera, F.; Gonzalez-Gallego, J. Rabbit hemorrhagic viral disease: Characterization of a new animal model of fulminant liver failure. J. Lab. Clin. Med. 2003, 141, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, P.G.; Costa-e-Silva, A.; Oliveira, M.J.; Monteiro, E.; Cunha, E.M.; Aguas, A.P. Severe leukopenia and liver biochemistry changes in adult rabbits after calicivirus infection. Res. Vet. Sci. 2006, 80, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Plassiart, G.; Guelfi, J.F.; Ganiere, J.P.; Wang, B.; Andre-Fontaine, G.; Wyers, M. Hematological parameters and visceral lesions relationships in rabbit viral hemorrhagic disease. Zbl. Veterinarmed. B 1992, 39, 443–453. [Google Scholar] [CrossRef]
- Ueda, K.; Park, J.H.; Ochiai, K.; Itakura, C. Disseminated intravascular coagulation (DIC) in rabbit haemorrhagic disease. Jpn. J. Vet. Res. 1992, 40, 133–141. [Google Scholar] [PubMed]
- Robinson, A.J.; So, P.T.M.; Müller, W.J.; Cooke, B.D.; Capucci, L. Statistical models for the effect of age and maternal antibodies on the development of rabbit haemorrhagic disease in Australian wild rabbits. Wildl. Res. 2002, 29, 663–671. [Google Scholar] [CrossRef]
- Ferreira, P.G.; Dinis, M.; Costa, E.S.A.; Aguas, A.P. Adult rabbits acquire resistance to lethal calicivirus infection by adoptive transfer of sera from infected young rabbits. Vet. Immunol. Immunopathol. 2008, 121, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Matthaei, M.; Kerr, P.J.; Read, A.J.; Hick, P.; Haboury, S.; Wright, J.D.; Strive, T. Comparative quantitative monitoring of rabbit haemorrhagic disease viruses in rabbit kittens. Virol. J. 2014, 11, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikami, O.; Park, J.H.; Kimura, T.; Ochiai, K.; Itakura, C. Hepatic lesions in young rabbits experimentally infected with rabbit haemorrhagic disease virus. Res. Vet. Sci. 1999, 66, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Marques, R.M.; Costa, E.S.A.; Aguas, A.P.; Teixeira, L.; Ferreira, P.G. Early inflammatory response of young rabbits attending natural resistance to calicivirus (RHDV) infection. Vet. Immunol. Immunopathol. 2012, 150, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Marques, R.M.; Teixeira, L.; Aguas, A.P.; Ribeiro, J.C.; Costa-e-Silva, A.; Ferreira, P.G. Immunosuppression abrogates resistance of young rabbits to rabbit haemorrhagic disease (RHD). Vet. Res. 2014, 45, 14. [Google Scholar] [CrossRef] [PubMed]
- Ruvoen-Clouet, N.; Blanchard, D.; Andre-Fontaine, G.; Ganiere, J.P. Partial characterization of the human erythrocyte receptor for rabbit haemorrhagic disease virus. Res. Virol. 1995, 146, 33–41. [Google Scholar] [CrossRef]
- Liu, J.; Kerr, P.J.; Wright, J.D.; Strive, T. Serological assays to discriminate rabbit haemorrhagic disease virus from Australian non-pathogenic rabbit calicivirus. Vet. Microbiol. 2012, 157, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Neimanis, A.; Pettersson, U.L.; Huang, N.; Gavier-Widen, D.; Strive, T. Elucidation of the pathology and tissue distribution of Lagovirus europaeus GI.2/RHDV2 (rabbit haemorrhagic disease virus 2) in young and adult rabbits (Oryctolagus cuniculus). Vet. Res. 2018, 49, 46. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Rahnenfuhrer, J. topGO: Enrichment Analysis for Gene Ontology. Bioconductor. Available online: http://www.bioconductor.org/packages//2.10/bioc/html/topGO.html (accessed on 1 October 2010).
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer Publishing Company: New York, NY, USA, 2009; p. 216. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahne, F.; Ivanek, R. Visualizing genomic data using Gviz and Bioconductor. Methods Mol. Biol. 2016, 1418, 335–351. [Google Scholar] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Meyers, G.; Wirblich, C.; Thiel, H.-J. Genomic and subgenomic RNAs of rabbit hemorrhagic disease virus are both protein-linked and packaged into particles. Virology 1991, 184, 677–686. [Google Scholar] [CrossRef]
- Van den Broeke, C.; Jacob, T.; Favoreel, H.W. Rho’ing in and out of cells: Viral interactions with Rho GTPase signaling. Small GTPases 2014, 5, e28318. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Takeda, K.; Kawano, M.; Takai, T.; Ishii, N.; Ogasawara, K. Natural killer (NK)–dendritic cell interactions generate MHC class II-dressed NK cells that regulate CD4(+) T cells. Proc. Natl. Acad. Sci. USA 2011, 108, 18360–18365. [Google Scholar] [CrossRef] [PubMed]
- Roche, P.A.; Furuta, K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat. Rev. Immunol. 2015, 15, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Syal, G.; Fausther, M.; Dranoff, J.A. Advances in cholangiocyte immunobiology. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G1077–G1086. [Google Scholar] [CrossRef] [PubMed]
- Hattoum, A.; Rubin, E.; Orr, A.; Michalopoulos, G.K. Expression of hepatocyte epidermal growth factor receptor, FAS and glypican 3 in EpCAM-positive regenerative clusters of hepatocytes, cholangiocytes, and progenitor cells in human liver failure. Hum. Pathol. 2013, 44, 743–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neefjes, J.; Jongsma, M.L.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Cruickshank, S.M.; Southgate, J.; Selby, P.J.; Trejdosiewicz, L.K. Expression and cytokine regulation of immune recognition elements by normal human biliary epithelial and established liver cell lines in vitro. J. Hepatol. 1998, 29, 550–558. [Google Scholar] [CrossRef]
- Biron, C.A.; Byron, K.S.; Sullivan, J.L. Severe herpesvirus infections in an adolescent without natural killer cells. N. Engl. J. Med. 1989, 320, 1731–1735. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.K.; Issekutz, A.C.; Fraser, R.; Schmit, P.; Morash, B.; Monaco-Shawver, L.; Orange, J.S.; Fernandez, C.V. Bilateral adrenal EBV-associated smooth muscle tumors in a child with a natural killer cell deficiency. Blood 2012, 119, 4009–4012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, N.C.; Mathew, P.A. NKp44 and natural cytotoxicity receptors as damage-associated molecular pattern recognition receptors. Front. Immunol. 2015, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Burn, G.L.; Svensson, L.; Sanchez-Blanco, C.; Saini, M.; Cope, A.P. Why is PTPN22 a good candidate susceptibility gene for autoimmune disease? FEBS Lett. 2011, 585, 3689–3698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Begovich, A.B.; Carlton, V.E.H.; Honigberg, L.A.; Schrodi, S.J.; Chokkalingam, A.P.; Alexander, H.C.; Ardlie, K.G.; Huang, Q.; Smith, A.M.; Spoerke, J.M.; et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am. J. Hum. Genet. 2004, 75, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Pisegna, S.; Zingoni, A.; Pirozzi, G.; Cinque, B.; Cifone, M.G.; Morrone, S.; Piccoli, M.; Frati, L.; Palmieri, G.; Santoni, A. Src-dependent Syk activation controls CD69-mediated signaling and function on human NK cells. J. Immunol. 2002, 169, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Malorni, W.; Quaranta, M.G.; Straface, E.; Falzano, L.; Fabbri, A.; Viora, M.; Fiorentini, C. The Rac-activating toxin cytotoxic necrotizing factor 1 oversees NK cell-mediated activity by regulating the actin/microtubule interplay. J. Immunol. 2003, 171, 4195–4202. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445. [Google Scholar] [CrossRef] [PubMed]
- Satpathy, A.T.; Wu, X.; Albring, J.C.; Murphy, K.M. Re(de)fining the dendritic cell lineage. Nat. Immunol. 2012, 13, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Holness, C.L.; Simmons, D.L. Molecular cloning of CD68, a human macrophage marker related to lysosomal glycoproteins. Blood 1993, 81, 1607. [Google Scholar] [PubMed]
- Satpathy, A.T.; Kc, W.; Albring, J.C.; Edelson, B.T.; Kretzer, N.M.; Bhattacharya, D.; Murphy, T.L.; Murphy, K.M. Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J. Exp. Med. 2012, 209, 1135–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, A.; Eck, S.L. EpCAM: A new therapeutic target for an old cancer antigen. Cancer Biol. Ther. 2003, 2, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Haller, O.; Kochs, G. Interferon-induced Mx proteins: Dynamin-like GTPases with antiviral activity. Traffic 2002, 3, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; DeFranco, R.; Grappone, C.; Milani, S.; Pastacaldi, S.; Pinzani, M.; Romanelli, R.G.; Laffi, G.; Gentilini, P. Increased expression of monocyte chemotactic protein-1 during active hepatic fibrogenesis: Correlation with monocyte infiltration. Am. J. Pathol. 1998, 152, 423–430. [Google Scholar] [PubMed]
- Otogawa, K.; Kinoshita, K.; Fujii, H.; Sakabe, M.; Shiga, R.; Nakatani, K.; Ikeda, K.; Nakajima, Y.; Ikura, Y.; Ueda, M.; et al. Erythrophagocytosis by liver macrophages (Kupffer cells) promotes oxidative stress, inflammation, and fibrosis in a rabbit model of steatohepatitis. Am. J. Pathol. 2007, 170, 967–980. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.B. Vaccination against and immune response to viral haemorrhagic disease of rabbits: A review of research in the People’s Republic of China. Rev. Sci. Tech. 1991, 10, 481–498. [Google Scholar] [PubMed]
- Tunon, M.J.; San Miguel, B.; Crespo, I.; Riezu-Boj, J.I.; Larrea, E.; Alvarez, M.; Gonzalez, I.; Bustos, M.; Gonzalez-Gallego, J.; Prieto, J. Cardiotrophin-1 promotes a high survival rate in rabbits with lethal fulminant hepatitis of viral origin. J. Virol. 2011, 85, 13124–13132. [Google Scholar] [CrossRef] [PubMed]
- Dalton, K.P.; Nicieza, I.; Abrantes, J.; Esteves, P.J.; Parra, F. Spread of new variant RHDV in domestic rabbits on the Iberian Peninsula. Vet. Microbiol. 2014, 169, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Alcami, A.; Koszinowski, U.H. Viral mechanisms of immune evasion. Immunol. Today 2000, 21, 447–455. [Google Scholar] [CrossRef]
- Karre, K.; Ljunggren, H.G.; Piontek, G.; Kiessling, R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 1986, 319, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Gumá, M.; Angulo, A.; López-Botet, M. NK Cell receptors involved in the response to human cytomegalovirus infection. In Immunobiology of Natural Killer Cell Receptors; Compans, R.W., Cooper, M.D., Honjo, T., Koprowski, H., Melchers, F., Oldstone, M.B.A., Olsnes, S., Potter, M., Vogt, P.K., Wagner, H., et al., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 207–223. [Google Scholar]
- McFadden, N.; Bailey, D.; Carrara, G.; Benson, A.; Chaudhry, Y.; Shortland, A.; Heeney, J.; Yarovinsky, F.; Simmonds, P.; Macdonald, A.; et al. Norovirus regulation of the innate immune response and apoptosis occurs via the product of the alternative open reading frame 4. PLoS Pathog. 2011, 7, e1002413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urakova, N.; Frese, M.; Hall, R.N.; Liu, J.; Matthaei, M.; Strive, T. Expression and partial characterisation of rabbit haemorrhagic disease virus non-structural proteins. Virology 2015, 484, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Schwensow, N.; Mazzoni, C.J.; Marmesat, E.; Fickel, J.; Peacock, D.; Kovaliski, J.; Sinclair, R.; Cassey, P.; Cooke, B.; Sommer, S. High adaptive variability and virus-driven selection on major histocompatibility complex (MHC) genes in invasive wild rabbits in Australia. Biol. Invasions 2017, 19, 1255–1271. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Virus | hpi | Age | Total Reads | Cleaned Reads | Mapped to Rabbit Genome * | Mapped to GI Genome ^ | RT-qPCR (Copies/mg Tissue) |
|---|---|---|---|---|---|---|---|---|
| A130 | cont | N/A ** | A | 36,608,811 | 33,019,057 | 30,372,424 (92.0%) | 0 (0.00%) | 55 |
| A440 | cont | 12 | A | 31,544,539 | 29,690,688 | 27,851,164 (93.8%) | 0 (0.00%) | 0 |
| A445 | cont | 12 | A | 33,024,742 | 30,839,504 | 28,557,666 (92.6%) | 0 (0.00%) | 0 |
| K11 | cont | 12 | K | 30,376,750 | 28,436,074 | 26,697,303 (93.9%) | 0 (0.00%) | 0 |
| K2 | cont | 12 | K | 26,466,336 | 24,621,221 | 23,121,670 (93.9%) | 0 (0.00%) | 0 |
| K9 | cont | 12 | K | 38,879,760 | 35,838,449 | 33,677,252 (94.0%) | 0 (0.00%) | 0 |
| A446 | cont | 24 | A | 26,875,056 | 25,190,314 | 23,565,537 (93.5%) | 0 (0.00%) | 0 |
| A447 | cont | 24 | A | 30,801,403 | 28,647,130 | 26,648,792 (93.0%) | 0 (0.00%) | 0 |
| K12 | cont | 24 | K | 21,522,682 | 19,510,404 | 18,264,437 (93.6%) | 0 (0.00%) | 0 |
| K14 | cont | 24 | K | 32,273,375 | 30,146,422 | 28,183,027 (93.5%) | 0 (0.00%) | 0 |
| K3 | cont | 24 | K | 34,270,680 | 31,856,522 | 29,771,558 (93.5%) | 0 (0.00%) | 0 |
| A444 | GI.1 | 12 | A | 41,554,574 | 38,521,840 | 35,819,416 (93.0%) | 0 (0.00%) | 94 |
| A451 | GI.1 | 12 | A | 34,269,029 | 31,413,217 | 29,237,190 (93.1%) | 0 (0.00%) | 28 |
| A452 | GI.1 | 12 | A | 30,744,544 | 28,741,202 | 26,469,612 (92.1%) | 0 (0.00%) | 0 |
| K1 | GI.1 | 12 | K | 35,648,932 | 33,188,895 | 31,053,518 (93.6%) | 0 (0.00%) | 0 |
| K13 | GI.1 | 12 | K | 28,834,630 | 26,852,299 | 25,283,903 (94.2%) | 0 (0.00%) | 0 |
| K5 | GI.1 | 12 | K | 32,247,351 | 30,065,559 | 28,089,931 (93.4%) | 0 (0.00%) | 0 |
| K7 | GI.1 | 12 | K | 34,983,905 | 32,474,265 | 30,104,239 (92.7%) | 0 (0.00%) | 85 |
| A443 | GI.1 | 24 | A | 32,025,317 | 29,266,843 | 27,437,173 (93.7%) | 4321 (0.01%) | 1,359,870 |
| A448 | GI.1 | 24 | A | 41,956,002 | 38,817,349 | 35,919,297 (92.5%) | 817 (0.00%) | 213,792 |
| A449 | GI.1 | 24 | A | 32,829,766 | 29,594,927 | 27,422,586 (92.7%) | 352 (0.00%) | 83,234 |
| A450 | GI.1 | 24 | A | 39,743,588 | 37,093,310 | 34,584,907 (93.2%) | 5 (0.00%) | 1205 |
| K10 | GI.1 | 24 | K | 26,707,074 | 24,485,698 | 22,779,842 (93.0%) | 2 (0.00%) | 307 |
| K4 | GI.1 | 24 | K | 35,102,440 | 32,387,798 | 30,179,957 (93.2%) | 49 (0.00%) | 18,929 |
| K6 | GI.1 | 24 | K | 30,314,748 | 27,870,816 | 25,865,930 (92.8%) | 19 (0.00%) | 6405 |
| K8 | GI.1 | 24 | K | 30,187,241 | 28,311,539 | 26,206,586 (92.6%) | 5 (0.00%) | 1490 |
| K379 | GI.2 | 12 | K | 32,179,571 | 30,150,297 | 28,485,814 (94.5%) | 0 (0.00%) | 81 |
| K380 | GI.2 | 12 | K | 30,937,894 | 29,113,196 | 27,110,731 (93.1%) | 0 (0.00%) | 66 |
| K381 | GI.2 | 12 | K | 24,208,541 | 22,521,030 | 20,774,527 (92.2%) | 0 (0.00%) | 0 |
| K375 | GI.2 | 24 | K | 31,299,465 | 29,544,132 | 27,913,303 (94.5%) | 26,558 (0.09%) | 20,966,939 |
| K376 | GI.2 | 24 | K | 30,626,983 | 28,656,602 | 26,667,507 (93.1%) | 1410 (0.00%) | 3,634,306 |
| K377 | GI.2 | 24 | K | 35,624,017 | 33,340,447 | 31,470,468 (94.4%) | 1947 (0.00%) | 323,906 |
| K378 | GI.2 | 24 | K | 26,510,091 | 24,505,534 | 22,803,925 (93.1%) | 22,528 (0.09%) | 4,857,896 |
| Comparison | Upregulated | Downregulated |
|---|---|---|
| Control adults vs. control kittens | 556 (up in adults) | 817 (up in kittens) |
| GI.1-infected kittens (12 hpi) * | 2 | 1 |
| GI.1-infected kittens (24 hpi) * | 60 | 2 |
| GI.1-infected adults (12 hpi) * | 2 | 0 |
| GI.1-infected adults (24 hpi) * | 296 | 26 |
| GI.2-infected kittens (12 hpi) * | 347 | 293 |
| GI.2-infected kittens (24 hpi) * | 2152 | 2059 |
| Ensembl Gene # | Annotation | Log Fold Change ^ | FDR * |
|---|---|---|---|
| ENSOCUG00000005528 | HAL (histidine ammonia-lyase) | 2.9 | 3.69 × 10−19 |
| ENSOCUG00000014137 | NAALAD2 (N-acetylated alpha-linked acidic dipeptidase 2) | −3.8 | 4.71 × 10−16 |
| ENSOCUG00000002950 | HN1 (hematological and neurological expressed 1) | −1.9 | 1.99 × 10−15 |
| ENSOCUG00000003663 | ACTG2 (actin, gamma 2, smooth muscle, enteric) | −2.3 | 7.09 × 10−13 |
| ENSOCUG00000014725 | N/A | 6.9 | 8.92 × 10−13 |
| ENSOCUG00000014317 | PIGR (polymeric immunoglobulin receptor) | 2.3 | 1.70 × 10−11 |
| ENSOCUG00000023455 | N/A | −3.0 | 1.70 × 10−11 |
| ENSOCUG00000002858 | CDK1 (cyclin dependent kinase 1) | −4.7 | 2.35 × 10−11 |
| ENSOCUG00000009882 | NCAPH (condensing complex subunit 2) | −2.9 | 2.35 × 10−11 |
| ENSOCUG00000012902 | N/A | −4.0 | 2.35 × 10−11 |
| Ensembl Gene # | Annotation | Log Fold Change | FDR * |
|---|---|---|---|
| Adults after 12 h | |||
| ENSOCUG00000023455 | N/A | 2.4 | 0.0003 |
| ENSOCUG00000025530 | N/A | 2.3 | 0.0350 |
| Adults after 24 h | |||
| ENSOCUG00000026233 | HRASLS2 (HRAS like suppressor 2) | 7.4 | 8.32 × 10−13 |
| ENSOCUG00000029154 | N/A | 5.4 | 3.30 × 10−12 |
| ENSOCUG00000002863 | IFIH1 (interferon induced with helicase C domain 1) | 3.4 | 1.31 × 10−11 |
| ENSOCUG00000009504 | CD80 (T-lymphocyte activation antigen CD80) | 3.5 | 1.31 × 10−11 |
| ENSOCUG00000020931 | SAMD9 (sterile alpha motif domain containing 9) | 4.6 | 5.12 × 10−11 |
| ENSOCUG00000021037 | MX2 (MX dynamin like GTPase 2) | 5.4 | 1.81 × 10−10 |
| ENSOCUG00000010311 | NXPE4 (neurexophilin and PC-esterase domain family member 4) | 3.7 | 3.05 × 10−10 |
| ENSOCUG00000009811 | ZNFX1 (zinc finger NFX1-type containing 1) | 2.6 | 3.61 × 10−10 |
| ENSOCUG00000000716 | EPSTI1 (epithelial stromal interaction 1) | 4.7 | 5.53 × 10−10 |
| ENSOCUG00000016280 | CXCL10 (C-X-C motif chemokine ligand 10) | 6.8 | 5.53 × 10−10 |
| Kittens after 12 h | |||
| ENSOCUG00000006223 | EPCAM (epithelial cell adhesion molecule) | 2.3 | 2.84 × 10−5 |
| ENSOCUG00000009559 | KIAA1841 | 1.3 | 0.0472 |
| ENSOCUG00000011988 | N/A | −3.8 | 0.0472 |
| Kittens after 24 h | |||
| ENSOCUG00000004501 | N/A | 4.1 | 4.62 × 10−6 |
| ENSOCUG00000006595 | UBA7 (ubiquitin like modifier activating enzyme 7) | 2.9 | 1.32 × 10−5 |
| ENSOCUG00000021924 | MX1 (MX dynamin like GTPase 1) | 4.2 | 1.84 × 10−5 |
| ENSOCUG00000006482 | IFI44 (interferon induced protein 44) | 2.9 | 0.0001 |
| ENSOCUG00000015823 | OAS2 (2’-5’-oligoadenylate synthetase 2) | 3.3 | 0.0001 |
| ENSOCUG00000013278 | DHX58 (DExH-box helicase 58) | 2.5 | 0.0002 |
| ENSOCUG00000027981 | ISG15 (ubiquitin-like modifier) | 5.4 | 0.0002 |
| ENSOCUG00000015872 | HERC5 (HECT and RLD domain containing E3 ubiquitin protein ligase 5) | 2.6 | 0.0002 |
| ENSOCUG00000024570 | IFIT5 (interferon induced protein with tetratricopeptide repeats 5) | 2.6 | 0.0002 |
| ENSOCUG00000024734 | N/A | 2.7 | 0.0002 |
| GO # | Term | Annotated | Significant | Expected | p-Value |
|---|---|---|---|---|---|
| Adults after 24 h | |||||
| GO:0051607 | defense response to virus | 106 | 29 | 1.72 | 2.40 × 10−19 |
| GO:0045071 | negative regulation of viral genome replication | 27 | 12 | 0.44 | 3.50 × 10−15 |
| GO:0006955 | immune response | 672 | 70 | 10.92 | 5.70 × 10−13 |
| GO:0071360 | cellular response to exogenous dsRNA | 11 | 5 | 0.18 | 4.60 × 10−7 |
| GO:0035455 | response to interferon-alpha | 12 | 5 | 0.19 | 7.80 × 10−7 |
| GO:0002474 | antigen processing and presentation of peptide antigen via MHC class 1 | 12 | 5 | 0.19 | 7.80 × 10−7 |
| GO:0060333 | interferon-gamma-mediated signaling pathway | 12 | 5 | 0.19 | 7.80 × 10−7 |
| GO:0034123 | positive regulation of toll-like receptor signaling pathway | 13 | 5 | 0.21 | 1.30 × 10−6 |
| GO:0050688 | regulation of defense response to virus | 38 | 9 | 0.62 | 2.10 × 10−6 |
| Kittens after 24 h | |||||
| GO:0051607 | defense response to virus | 106 | 17 | 0.37 | 4.60 × 10−17 |
| GO:0045071 | negative regulation of viral genome replication | 27 | 9 | 0.09 | 1.30 × 10−16 |
| GO:0035455 | response to interferon-alpha | 12 | 6 | 0.04 | 1.10 × 10−12 |
| GO:0032727 | positive regulation of interferon-alpha production | 11 | 4 | 0.04 | 4.00 × 10−8 |
| GO:0032728 | positive regulation of interferon-beta production | 22 | 4 | 0.08 | 8.60 × 10−7 |
| GO:0009615 | response to virus | 155 | 21 | 0.53 | 2.00 × 10−6 |
| GO:0039529 | RIG-I signaling pathway | 10 | 3 | 0.03 | 4.50 × 10−6 |
| GO:0006955 | immune response | 672 | 21 | 2.31 | 2.60 × 10−5 |
| GO:0035456 | response to interferon-beta | 14 | 3 | 0.05 | 6.60 × 10−5 |
| GO:0019941 | modification-dependent protein catabolic process | 354 | 4 | 1.22 | 0.00011 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neave, M.J.; Hall, R.N.; Huang, N.; McColl, K.A.; Kerr, P.; Hoehn, M.; Taylor, J.; Strive, T. Robust Innate Immunity of Young Rabbits Mediates Resistance to Rabbit Hemorrhagic Disease Caused by Lagovirus Europaeus GI.1 But Not GI.2. Viruses 2018, 10, 512. https://doi.org/10.3390/v10090512
Neave MJ, Hall RN, Huang N, McColl KA, Kerr P, Hoehn M, Taylor J, Strive T. Robust Innate Immunity of Young Rabbits Mediates Resistance to Rabbit Hemorrhagic Disease Caused by Lagovirus Europaeus GI.1 But Not GI.2. Viruses. 2018; 10(9):512. https://doi.org/10.3390/v10090512
Chicago/Turabian StyleNeave, Matthew J., Robyn N. Hall, Nina Huang, Kenneth A. McColl, Peter Kerr, Marion Hoehn, Jennifer Taylor, and Tanja Strive. 2018. "Robust Innate Immunity of Young Rabbits Mediates Resistance to Rabbit Hemorrhagic Disease Caused by Lagovirus Europaeus GI.1 But Not GI.2" Viruses 10, no. 9: 512. https://doi.org/10.3390/v10090512
APA StyleNeave, M. J., Hall, R. N., Huang, N., McColl, K. A., Kerr, P., Hoehn, M., Taylor, J., & Strive, T. (2018). Robust Innate Immunity of Young Rabbits Mediates Resistance to Rabbit Hemorrhagic Disease Caused by Lagovirus Europaeus GI.1 But Not GI.2. Viruses, 10(9), 512. https://doi.org/10.3390/v10090512