The Microtubule-Associated Innate Immune Sensor GEF-H1 Does Not Influence Mouse Norovirus Replication in Murine Macrophages



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Virus Infection
2.2. Plasmids and Antibodies
2.3. Transfection of Cell Lines
2.4. Immunofluorescence Staining and Confocal Imaging
2.5. Immunoblotting
2.6. Quantitative RT-PCR
2.7. Plaque Assay
2.8. Immunoprecipitation
2.9. GEF-H1 Silencing Via Sirna Treatment
3. Results
3.1. GEF-H1 Is Found within the MNV Replication Complex
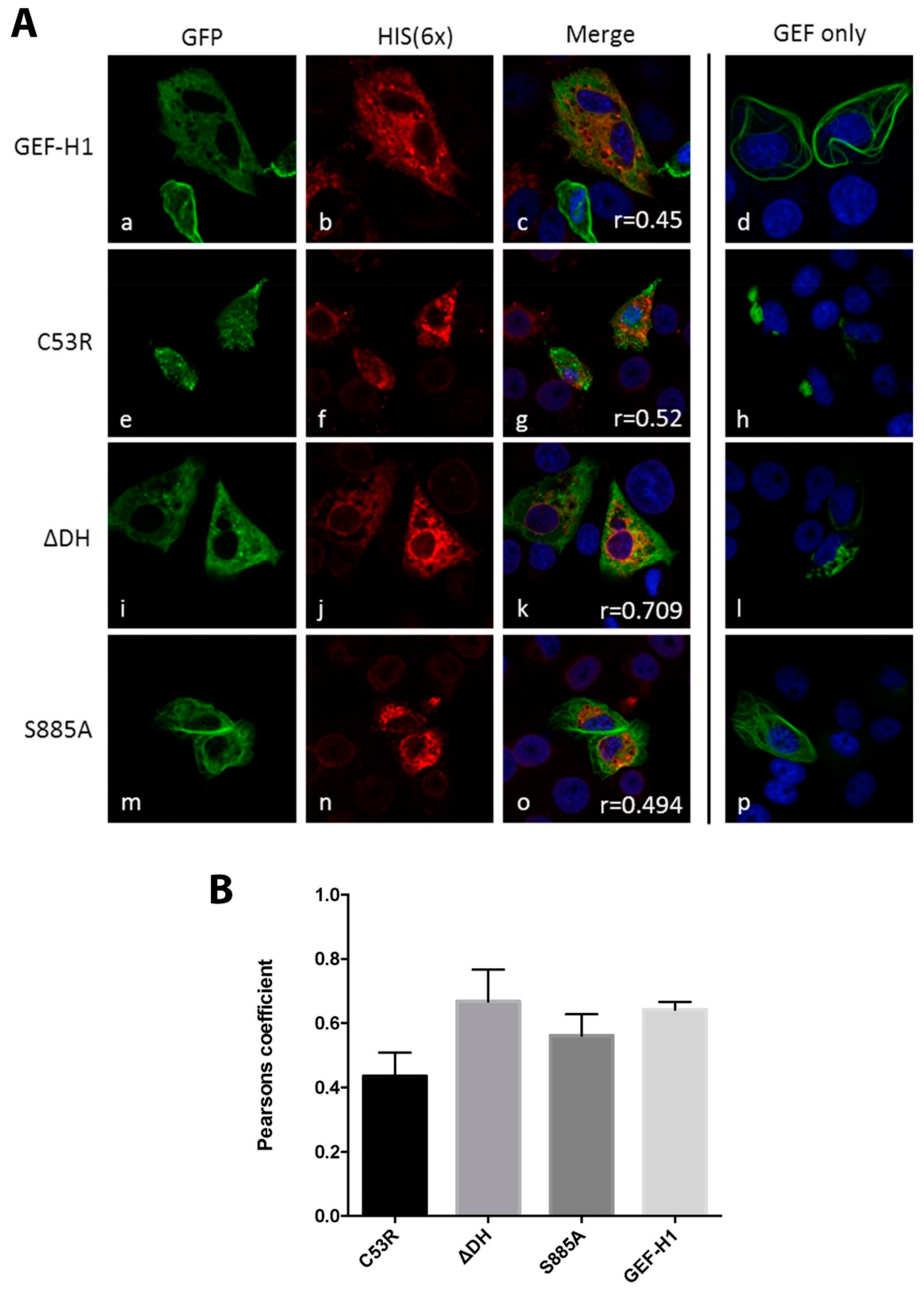
3.2. Expression of GEF-H1 Leads to Changes in the Localisation of the RC during MNV Infection
3.3. The MNV Protein NS3 Co-Localises with GEF-H1 and Alters Its Distribution
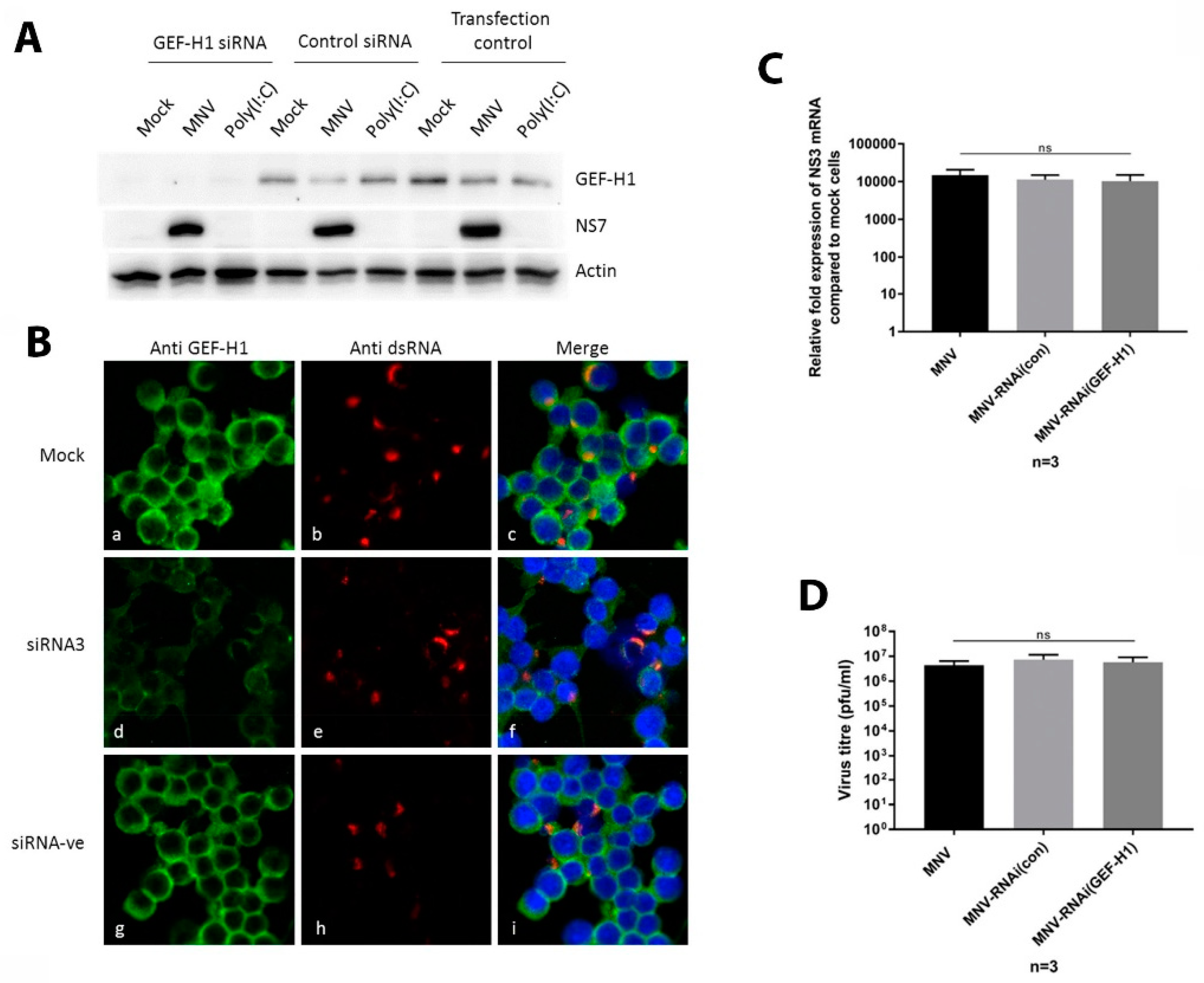
3.4. siRNA Knockdown of Endogenous GEF-H1 Does Not Affect MNV Replication Complex Formation or Localisation
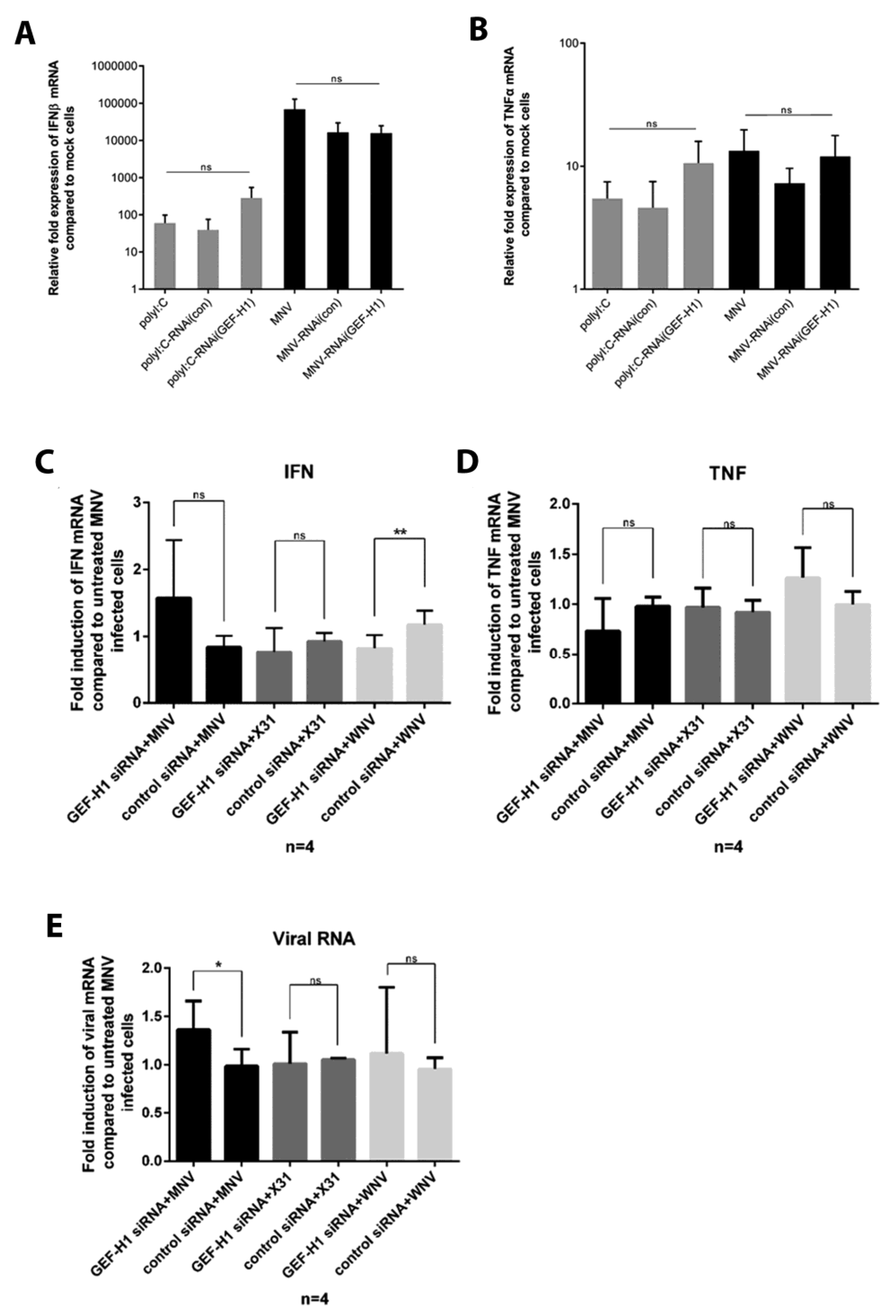
3.5. Suppression of the GEF-H1 Expression Does Not Affect the Production of Cytokines in Response to Poly(I:C) Stimulation or MNV Infection
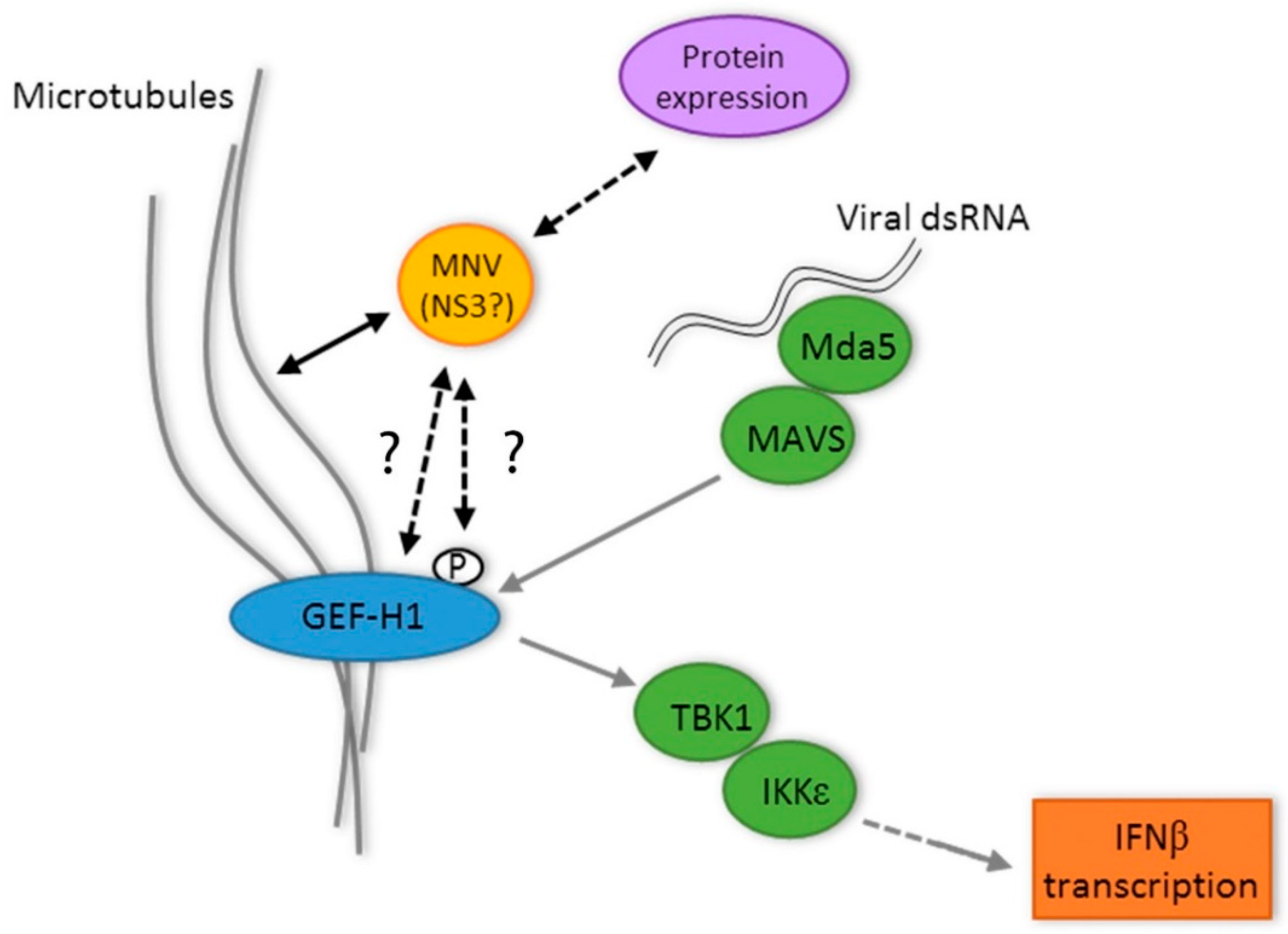
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Pires, S.M.; Fischer-Walker, C.L.; Lanata, C.F.; Devleesschauwer, B.; Hall, A.J.; Kirk, M.D.; Duarte, A.S.; Black, R.E.; Angulo, F.J. Aetiology-Specific Estimates of the Global and Regional Incidence and Mortality of Diarrhoeal Diseases Commonly Transmitted through Food. PLoS ONE 2015, 10, e0142927. [Google Scholar] [CrossRef] [Green Version]
- Troeger, C.; Colombara, D.V.; Rao, P.C.; Khalil, I.A.; Brown, A.; Brewer, T.G.; Guerrant, R.L.; Houpt, E.R.; Kotloff, K.L.; Misra, K.; et al. Global disability-adjusted life-year estimates of long-term health burden and undernutrition attributable to diarrhoeal diseases in children younger than 5 years. Lancet Glob. Health 2018, 6, e255–e269. [Google Scholar] [CrossRef]
- Kotloff, K.L.; Nataro, J.P.; Blackwelder, W.C.; Nasrin, D.; Farag, T.H.; Panchalingam, S.; Wu, Y.; Sow, S.O.; Sur, D.; Breiman, R.F.; et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): A prospective, case-control study. Lancet 2013, 382, 209–222. [Google Scholar] [CrossRef]
- Bartsch, S.M.; Lopman, B.A.; Ozawa, S.; Hall, A.J.; Lee, B.Y. Global Economic Burden of Norovirus Gastroenteritis. PLoS ONE 2016, 11, e0151219. [Google Scholar] [CrossRef]
- Bartsch, S.M.; Lopman, B.A.; Hall, A.J.; Parashar, U.D.; Lee, B.Y. The potential economic value of a human norovirus vaccine for the United States. Vaccine 2012, 30, 7097–7104. [Google Scholar] [CrossRef] [Green Version]
- Kroneman, A.; Vega, E.; Vennema, H.; Vinje, J.; White, P.A.; Hansman, G.; Green, K.; Martella, V.; Katayama, K.; Koopmans, M. Proposal for a unified norovirus nomenclature and genotyping. Arch. Virol. 2013, 158, 2059–2068. [Google Scholar] [CrossRef] [Green Version]
- Martella, V.; Lorusso, E.; Decaro, N.; Elia, G.; Radogna, A.; D’Abramo, M.; Desario, C.; Cavalli, A.; Corrente, M.; Camero, M.; et al. Detection and molecular characterization of a canine norovirus. Emerg. Infect. Dis. 2008, 14, 1306–1308. [Google Scholar] [CrossRef]
- Mesquita, J.R.; Barclay, L.; Nascimento, M.S.; Vinje, J. Novel norovirus in dogs with diarrhea. Emerg. Infect. Dis. 2010, 16, 980–982. [Google Scholar] [CrossRef]
- Jones, M.K.; Watanabe, M.; Zhu, S.; Graves, C.L.; Keyes, L.R.; Grau, K.R.; Gonzalez-Hernandez, M.B.; Iovine, N.M.; Wobus, C.E.; Vinje, J.; et al. Enteric bacteria promote human and mouse norovirus infection of B cells. Science 2014, 346, 755–759. [Google Scholar] [CrossRef] [Green Version]
- Ettayebi, K.; Crawford, S.E.; Murakami, K.; Broughman, J.R.; Karandikar, U.; Tenge, V.R.; Neill, F.H.; Blutt, S.E.; Zeng, X.L.; Qu, L.; et al. Replication of human noroviruses in stem cell-derived human enteroids. Science 2016, 353, 1387–1393. [Google Scholar] [CrossRef] [Green Version]
- Wobus, C.E.; Karst, S.M.; Thackray, L.B.; Chang, K.O.; Sosnovtsev, S.V.; Belliot, G.; Krug, A.; Mackenzie, J.M.; Green, K.Y.; Virgin, H.W. Replication of Norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS Biol. 2004, 2, e432. [Google Scholar] [CrossRef]
- Karst, S.M.; Wobus, C.E.; Lay, M.; Davidson, J.; Virgin, H.W., IV. STAT1-Dependent Innate Immunity to a Norwalk-Like Virus. Science 2003, 299, 1575–1578. [Google Scholar] [CrossRef]
- McCartney, S.A.; Thackray, L.B.; Gitlin, L.; Gilfillan, S.; Virgin, H.W.; Colonna, M. MDA-5 recognition of a murine norovirus. PLoS Pathog. 2008, 4, e1000108. [Google Scholar] [CrossRef]
- Enosi Tuipulotu, D.; Netzler, N.E.; Lun, J.H.; Mackenzie, J.M.; White, P.A. RNA Sequencing of Murine Norovirus-Infected Cells Reveals Transcriptional Alteration of Genes Important to Viral Recognition and Antigen Presentation. Front. Immunol. 2017, 8, 959. [Google Scholar] [CrossRef]
- Cotton, B.T.; Hyde, J.L.; Sarvestani, S.T.; Sosnovtsev, S.V.; Green, K.Y.; White, P.A.; Mackenzie, J.M. The Norovirus NS3 Protein Is a Dynamic Lipid- and Microtubule-Associated Protein Involved in Viral RNA Replication. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Hyde, J.L.; Gillespie, L.K.; Mackenzie, J.M. Mouse norovirus 1 utilizes the cytoskeleton network to establish localization of the replication complex proximal to the microtubule organizing center. J. Virol. 2012, 86, 4110–4122. [Google Scholar] [CrossRef]
- Hyde, J.L.; Mackenzie, J.M. Subcellular localization of the MNV-1 ORF1 proteins and their potential roles in the formation of the MNV-1 replication complex. Virology 2010, 406, 138–148. [Google Scholar] [CrossRef] [Green Version]
- Hyde, J.L.; Sosnovtsev, S.V.; Green, K.Y.; Wobus, C.; Virgin, H.W.; Mackenzie, J.M. Mouse norovirus replication is associated with virus-induced vesicle clusters originating from membranes derived from the secretory pathway. J. Virol. 2009, 83, 9709–9719. [Google Scholar] [CrossRef]
- Krendel, M.; Zenke, F.T.; Bokoch, G.M. Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat. Cell Biol. 2002, 4, 294–301. [Google Scholar] [CrossRef]
- Pathak, R.; Dermardirossian, C. GEF-H1: Orchestrating the interplay between cytoskeleton and vesicle trafficking. Small GTPases 2013, 4, 174–179. [Google Scholar] [CrossRef]
- Waheed, F.; Dan, Q.; Amoozadeh, Y.; Zhang, Y.; Tanimura, S.; Speight, P.; Kapus, A.; Szaszi, K. Central role of the exchange factor GEF-H1 in TNF-alpha-induced sequential activation of Rac, ADAM17/TACE, and RhoA in tubular epithelial cells. Mol. Biol. Cell 2013, 24, 1068–1082. [Google Scholar] [CrossRef]
- Ren, Y.; Li, R.; Zheng, Y.; Busch, H. Cloning and characterization of GEF-H1, a microtubule-associated guanine nucleotide exchange factor for Rac and Rho GTPases. J. Biol. Chem. 1998, 273, 34954–34960. [Google Scholar] [CrossRef]
- Pathak, R.; Delorme-Walker, V.D.; Howell, M.C.; Anselmo, A.N.; White, M.A.; Bokoch, G.M.; Dermardirossian, C. The microtubule-associated Rho activating factor GEF-H1 interacts with exocyst complex to regulate vesicle traffic. Dev. Cell 2012, 23, 397–411. [Google Scholar] [CrossRef]
- Birkenfeld, J.; Nalbant, P.; Yoon, S.H.; Bokoch, G.M. Cellular functions of GEF-H1, a microtubule-regulated Rho-GEF: Is altered GEF-H1 activity a crucial determinant of disease pathogenesis? Trends Cell Biol. 2008, 18, 210–219. [Google Scholar] [CrossRef]
- Chiang, H.S.; Zhao, Y.; Song, J.H.; Liu, S.; Wang, N.; Terhorst, C.; Sharpe, A.H.; Basavappa, M.; Jeffrey, K.L.; Reinecker, H.C. GEF-H1 controls microtubule-dependent sensing of nucleic acids for antiviral host defenses. Nat. Immunol. 2014, 15, 63–71. [Google Scholar] [CrossRef]
- Wang, H.; Wang, J.; Yang, J.; Yang, X.; He, J.; Wang, R.; Liu, S.; Zhou, L.; Ma, L. Guanine nucleotide exchange factor-H1 promotes inflammatory cytokine production and intracellular mycobacterial elimination in macrophages. Cell Cycle 2017, 16, 1695–1704. [Google Scholar] [CrossRef]
- Fritzlar, S.; Jegaskanda, S.; Aktepe, T.E.; Prier, J.E.; Holz, L.E.; White, P.A.; Mackenzie, J.M. Mouse Norovirus infection reduces the surface expression of MHC class I proteins and inhibits CD8+ T cell recognition and activation. J. Virol. 2018. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fritzlar, S.; White, P.A.; Mackenzie, J.M. The Microtubule-Associated Innate Immune Sensor GEF-H1 Does Not Influence Mouse Norovirus Replication in Murine Macrophages. Viruses 2019, 11, 47. https://doi.org/10.3390/v11010047
Fritzlar S, White PA, Mackenzie JM. The Microtubule-Associated Innate Immune Sensor GEF-H1 Does Not Influence Mouse Norovirus Replication in Murine Macrophages. Viruses. 2019; 11(1):47. https://doi.org/10.3390/v11010047
Chicago/Turabian StyleFritzlar, Svenja, Peter A. White, and Jason M. Mackenzie. 2019. "The Microtubule-Associated Innate Immune Sensor GEF-H1 Does Not Influence Mouse Norovirus Replication in Murine Macrophages" Viruses 11, no. 1: 47. https://doi.org/10.3390/v11010047
APA StyleFritzlar, S., White, P. A., & Mackenzie, J. M. (2019). The Microtubule-Associated Innate Immune Sensor GEF-H1 Does Not Influence Mouse Norovirus Replication in Murine Macrophages. Viruses, 11(1), 47. https://doi.org/10.3390/v11010047