Phylodynamic Analysis of Ebola Virus Disease Transmission in Sierra Leone

 ,
,  , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Sample Processing, Sequencing, and Genome Assembly
2.3. Phylogenetic Analysis
2.4. Statistical Analyses
2.5. Ethical Statement
3. Results
3.1. Correlation Between Cases and Population Size
3.2. Sequencing of EBOV Genomes from SLE
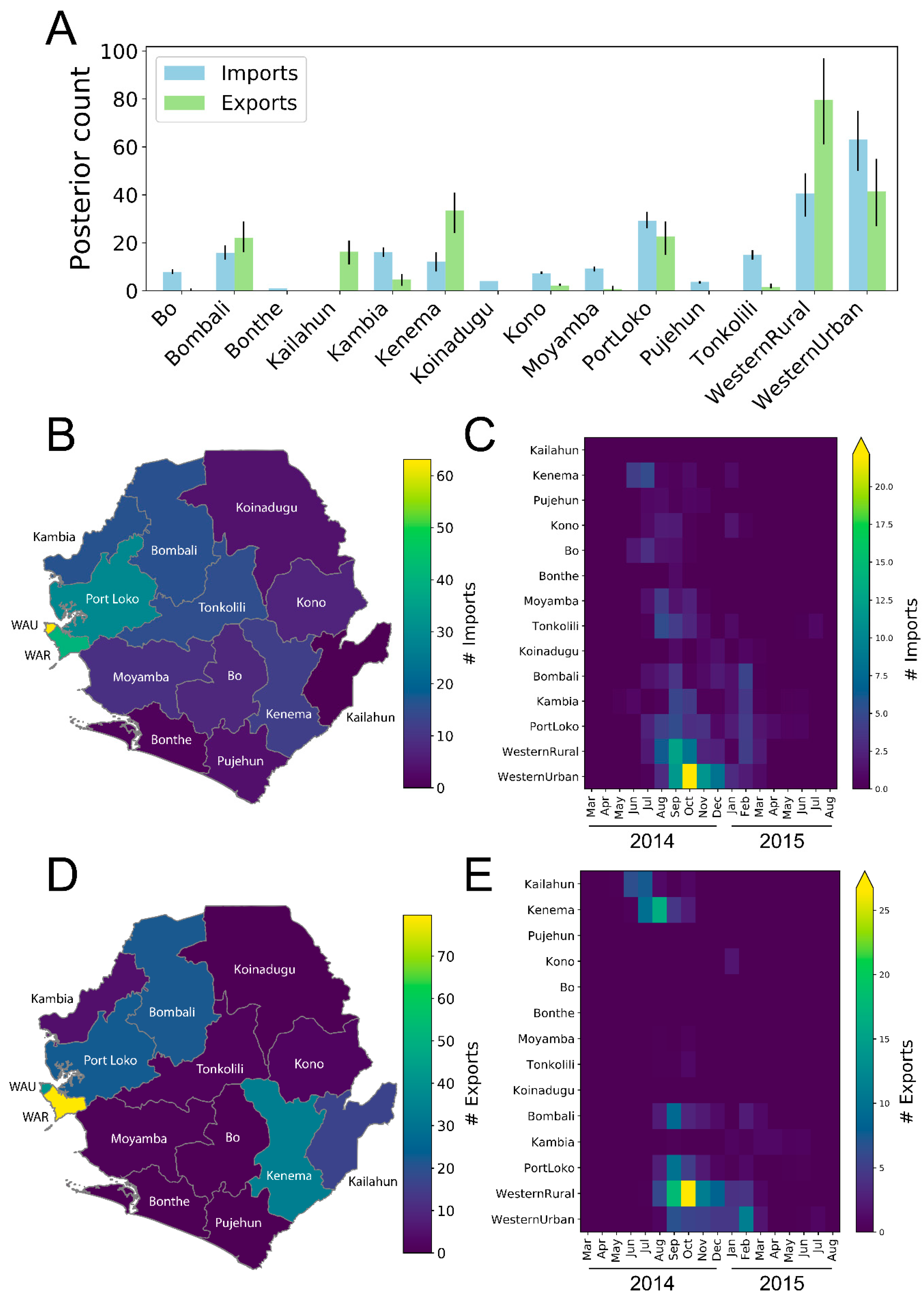
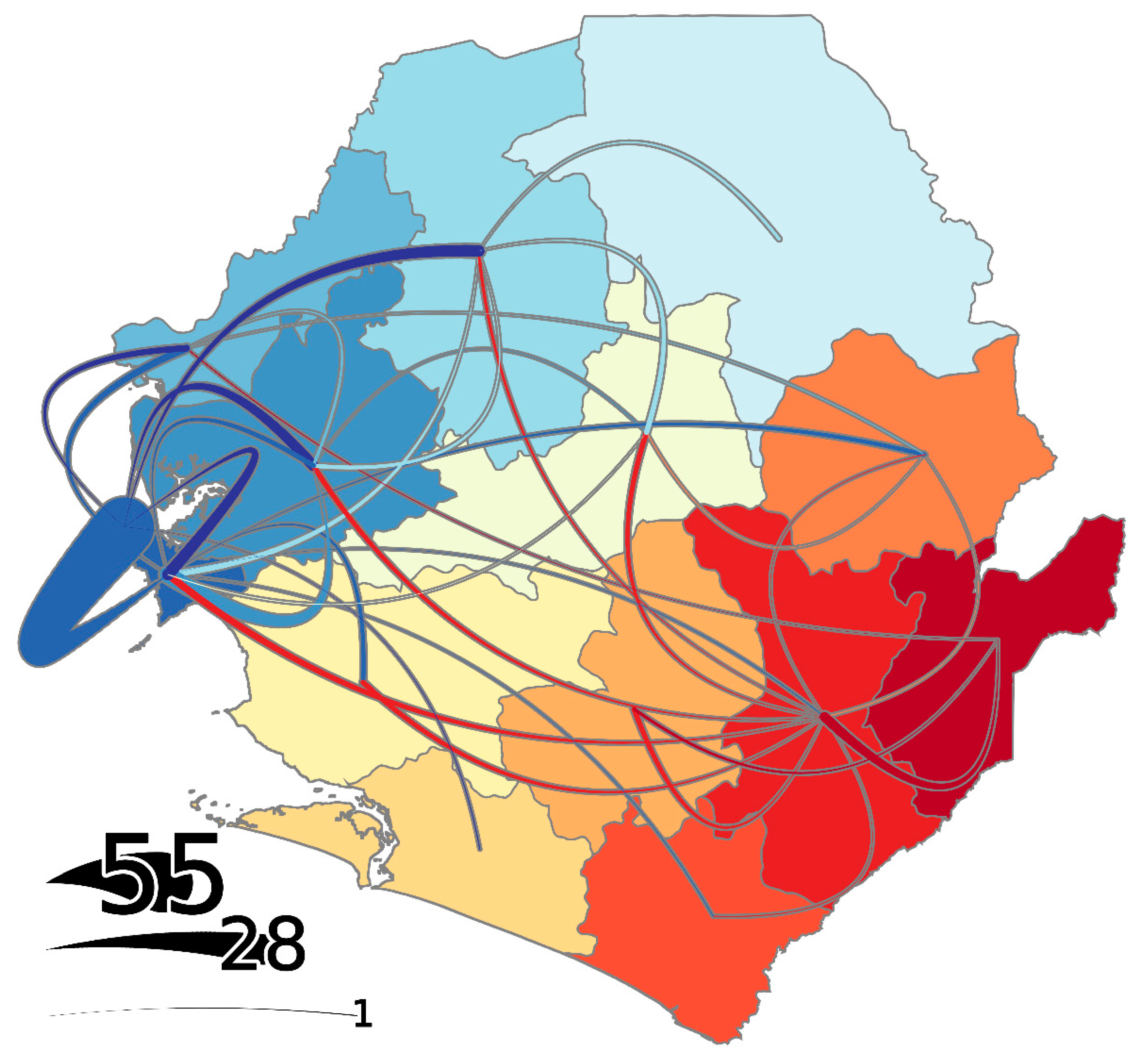
3.3. Districts as Sources, Sinks, or Hubs
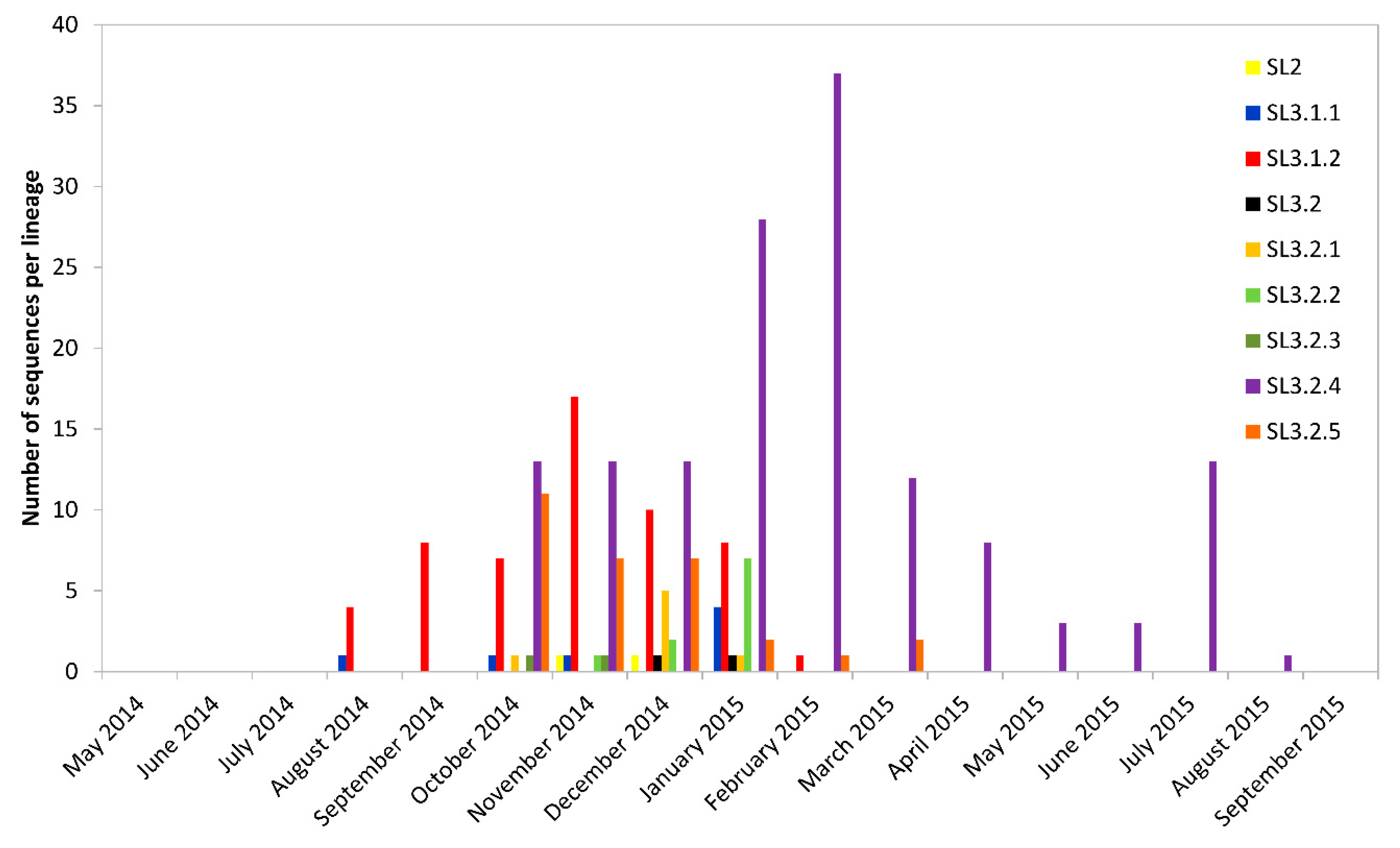
3.4. Temporal and Geographical Distribution of Lineages in SLE
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Ebola Situation Report—30 March 2016. Available online: http://apps.who.int/ebola/current-situation/ebola-situation-report-30-march-2016 (accessed on 3 May 2018).
- Goba, A.; Khan, S.H.; Fonnie, M.; Fullah, M.; Moigboi, A.; Kovoma, A.; Sinnah, V.; Yoko, N.; Rogers, H.; Safai, S.; et al. An outbreak of Ebola virus disease in the Lassa fever zone. J. Infect. Dis. 2016, 214, S110–S121. [Google Scholar] [CrossRef]
- Gire, S.K.; Goba, A.; Andersen, K.G.; Sealfon, R.S.; Park, D.J.; Kanneh, L.; Jalloh, S.; Momoh, M.; Fullah, M.; Dudas, G.; et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 2014, 345, 1369–1372. [Google Scholar] [CrossRef] [Green Version]
- Park, J.P.; Dudas, G.; Wohl, S.; Goba, A.; Whitmer, S.L.; Andersen, K.G.; Sealfon, R.S.; Ladner, J.T.; Kugelman, J.R.; Matranga, C.B.; et al. Ebola virus epidemiology, transmission and evolution during seven months in Sierra Leone. Cell 2015, 161, 1516–1526. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Shi, W.F.; Liu, D.; Qian, J.; Liang, L.; Bo, X.C.; Liu, J.; Ren, H.G.; Fan, H.; Ni, M.; et al. Genetic diversity and evolutionary dynamics of Ebola virus in Sierra Leone. Nature 2015, 524, 93–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudas, G.; Carvalho, M.; Bedford, T.; Tattem, A.J.; Baele, G.; Faria, N.R.; Park, D.J.; Ladner, J.T.; Arias, A.; Asogun, D.; et al. Virus genomes reveal factors that spread and sustained the Ebola epidemic. Nature 2017, 544, 309–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Yao, H.; Liu, D.; Ren, H.; Hu, Y.; Kargbo, D.; Teng, Y.; Deng, Y.; Lu, H.; Liu, X.; et al. Mapping the clinical outcomes and genetic evolution of Ebola virus in Sierra Leone. JCI Insight 2017, 2, e88333. [Google Scholar] [CrossRef] [PubMed]
- Paweska, J.T.; Jansen van Vuren, P.; Meier, G.H.; Le Roux, C.; Conteh, O.S.; Kemp, A.; Fourie, C.; Naidoo, P.; Naicker, S.; Ohaebosim, P.; et al. South African Ebola diagnostic response in Sierra Leone: A modular high biosafety field laboratory. PLoS Negl. Trop. Dis. 2017, 11, e0005665. [Google Scholar] [CrossRef]
- Morlan, J.D.; Qu, K.; Sinicropi, D.V. Selective depletion of rRNA enables whole transcriptome profiling of archival fixed tissue. PLoS ONE 2012, 7, e42882. [Google Scholar] [CrossRef]
- Djikeng, A.; Halpin, R.; Kuzmickas, R.; Depasse, J.; Feldblyum, J.; Sengamalay, N.; Afonso, C.; Zhang, X.; Anderson, N.G.; Ghedin, E.; et al. Viral genome sequencing by random priming methods. BMC Genom. 2008, 9, 5. [Google Scholar] [CrossRef]
- Ladner, J.T.; Wiley, M.R.; Mate, S.; Dudas, G.; Prieto, K.; Lovett, S.; Nagle, E.R.; Beitzel, B.; Gilbert, M.L.; Fakoli, L.; et al. Evolution and spread of Ebola virus in Liberia, 2014–2015. Cell Host Microbe 2015, 18, 659–669. [Google Scholar] [CrossRef]
- Mate, S.E.; Kugelman, J.R.; Nyenswah, T.G.; Ladner, J.T.; Wiley, M.R.; Cordier-Lassalle, T.; Christie, A.; Schroth, G.P.; Gross, S.M.; Davies-Wayne, G.J.; et al. Molecular evidence of sexual transmission of ebola virus. N. Engl. J. Med. 2015, 373, 2448–2454. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality control and processing of metagenomics datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Jansen van Vuren, P.; Grobbelaar, A.; Storm, N.; Conteh, O.; Konneh, K.; Kamara, A.; Sanne, I.; Paweska, J.T. Comparative evaluation of the diagnostic performance of the prototype Cepheid GeneXpert Ebola assay. J. Clin. Microbiol. 2016, 54, 359–367. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUTi and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Ho, S.Y.; Phillips, M.J.; Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef] [PubMed]
- Gill, M.S.; Lemey, P.; Faria, N.R.; Rambaut, A.; Shapiro, B.; Suchard, M.A. Improving Bayesian population dynamics inference: A coalescent-based model for multiple loci. Mol. Biol. Evol. 2013, 30, 713–724. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef]
- Anonymous. Sierra Leone. Available online: https://www.citypopulation.de/php/sierraleone-admin.php?adm1id=42. (accessed on 6 June 2018).
- Rambaut, A.; Pybus, O.G.; Nelson, M.I.; Viboud, C.; Taubenberger, J.K.; Holmes, E.C. The genomic and epidemiological dynamics of human influenza A. virus. Nature 2008, 453, 615–619. [Google Scholar] [CrossRef]
- Gleason, B.; Redd, J.; Kilmarx, P.; Sesay, T.; Bayor, F.; Mozalevskis, A.; Connolly, A.; Akpablie, J.; Prybylski, D.; Moffett, D.; et al. Establishment of an Ebola Treatment Unit and Laboratory—Bombali district, Sierra Leone, July 2014–January 2015. MMWR Morb. Mortal. Wkly. Rep. 2015, 64, 1108–1111. [Google Scholar] [CrossRef] [PubMed]
- Neiderud, C. How urbanization affects the epidemiology of emerging infectious diseases. Infect. Ecol. Epidemiol. 2015, 5, 27060. [Google Scholar] [CrossRef] [PubMed]
- The Ebola Outbreak Epidemiology Team. Outbreak of Ebola virus disease in the Democratic Republic of the Congo, April–May 2018: An epidemiological study. Lancet 2018. [Google Scholar] [CrossRef]
- Green, A. Ebola outbreak in the DR Congo: Lessons learned. Lancet 2018, 391, 2096. [Google Scholar] [CrossRef]
- World Health Organization. Ebola virus disease, Democratic Republic of the Congo, External Situation Report 14. Available online: http://apps.who.int/iris/bitstream/handle/10665/272997/SITREP_EVD_DRC_20180701-eng.pdf?ua=1 (accessed on 9 July 2018).
- Mbala Kingebeni, P.; Villabona-Arenas, C.J.; Vidal, N.; Likofata, J.; Nsio-Mbeta, J.; Makiala-Mandanda, S.; Mukadi, D.; Mukadi, P.; Kumakamba, C.; Djokolo, B.; et al. Rapid confirmation of the Zaire Ebola virus in the outbreak of the Equateur Province in the Democratic Republic of the Congo: Implications for public health interventions. Clin. Infect. Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, C. Merck vaccine heads Ebola countermeasures. Nat. Biotechnol. 2018, 36, 563–565. [Google Scholar] [CrossRef]
- Nkengason, J.N.; Onyebujoh, P. Response to the Ebola virus disease outbreak in the Democratic Republic of the Congo. Lancet 2018, 391, 2395–2398. [Google Scholar] [CrossRef]
- Anonymous. Ebola Update (85): Democratic Republic of Congo (North Kivu) Case Update, Butembo. Promed Mail 2018. Available online: http://www.promedmail.org/direct.php?id=20180909.6017592 (accessed on 10 September 2018).
- World Health Organization. Ebola Virus Disease, Democratic Republic of the Congo, External Situation Report 16. 21 November 2018. Available online: http://apps.who.int/iris/bitstream/handle/10665/275944/SITREP_EVD_DRC_20181121-eng.pdf?ua=1 (accessed on 23 November 2018).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| District | Median Import Events | Median Export Events | Ratio Exports/Imports | Source/Sink/Hub | Total #Cases | Cases/Import | Cases/Export |
|---|---|---|---|---|---|---|---|
| Bo | 8 | 0 | 0.02 | Sink | 358 | 46.31 | N/A |
| Bombali | 16 | 22 | 1.40 | Hub | 1064 | 67.56 | 48 |
| Bonthe | 1 | 0 | 0.00 | Sink | 1 | 1.00 | N/A |
| Kailahun | 0 | 16 | N/A | Source | 658 | N/A | 40 |
| Kambia | 16 | 5 | 0.29 | Sink | 277 | 17.11 | 58 |
| Kenema | 12 | 33 | 2.74 | Hub | 532 | 43.57 | 16 |
| Koinadugu | 4 | 0 | 0.00 | Sink | 155 | 38.65 | N/A |
| Kono | 7 | 2 | 0.30 | Sink | 450 | 62.59 | 210 |
| Moyamba | 9 | 1 | 0.08 | Sink | 276 | 29.68 | 394 |
| PortLoko | 29 | 23 | 0.77 | Hub | 1609 | 55.27 | 71 |
| Pujehun | 4 | 0 | 0.01 | Sink | 54 | 14.56 | N/A |
| Tonkolili | 15 | 2 | 0.10 | Sink | 505 | 33.53 | 326 |
| Western Area Rural | 41 | 80 | 1.97 | Hub | 1381 | 34.09 | 17 |
| Western Area Urban | 63 | 42 | 0.66 | Hub | 2520 | 39.92 | 61 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jansen van Vuren, P.; Ladner, J.T.; Grobbelaar, A.A.; Wiley, M.R.; Lovett, S.; Allam, M.; Ismail, A.; le Roux, C.; Weyer, J.; Moolla, N.; et al. Phylodynamic Analysis of Ebola Virus Disease Transmission in Sierra Leone. Viruses 2019, 11, 71. https://doi.org/10.3390/v11010071
Jansen van Vuren P, Ladner JT, Grobbelaar AA, Wiley MR, Lovett S, Allam M, Ismail A, le Roux C, Weyer J, Moolla N, et al. Phylodynamic Analysis of Ebola Virus Disease Transmission in Sierra Leone. Viruses. 2019; 11(1):71. https://doi.org/10.3390/v11010071
Chicago/Turabian StyleJansen van Vuren, Petrus, Jason T. Ladner, Antoinette A. Grobbelaar, Michael R. Wiley, Sean Lovett, Mushal Allam, Arshad Ismail, Chantel le Roux, Jacqueline Weyer, Naazneen Moolla, and et al. 2019. "Phylodynamic Analysis of Ebola Virus Disease Transmission in Sierra Leone" Viruses 11, no. 1: 71. https://doi.org/10.3390/v11010071
APA StyleJansen van Vuren, P., Ladner, J. T., Grobbelaar, A. A., Wiley, M. R., Lovett, S., Allam, M., Ismail, A., le Roux, C., Weyer, J., Moolla, N., Storm, N., Kgaladi, J., Sanchez-Lockhart, M., Conteh, O., Palacios, G., & Paweska, J. T. (2019). Phylodynamic Analysis of Ebola Virus Disease Transmission in Sierra Leone. Viruses, 11(1), 71. https://doi.org/10.3390/v11010071