Viral Infections and Autoimmune Disease: Roles of LCMV in Delineating Mechanisms of Immune Tolerance

{kind=link}
Abstract
:1. Introduction
2. LCMV-Induced Mouse Models of Autoimmunity
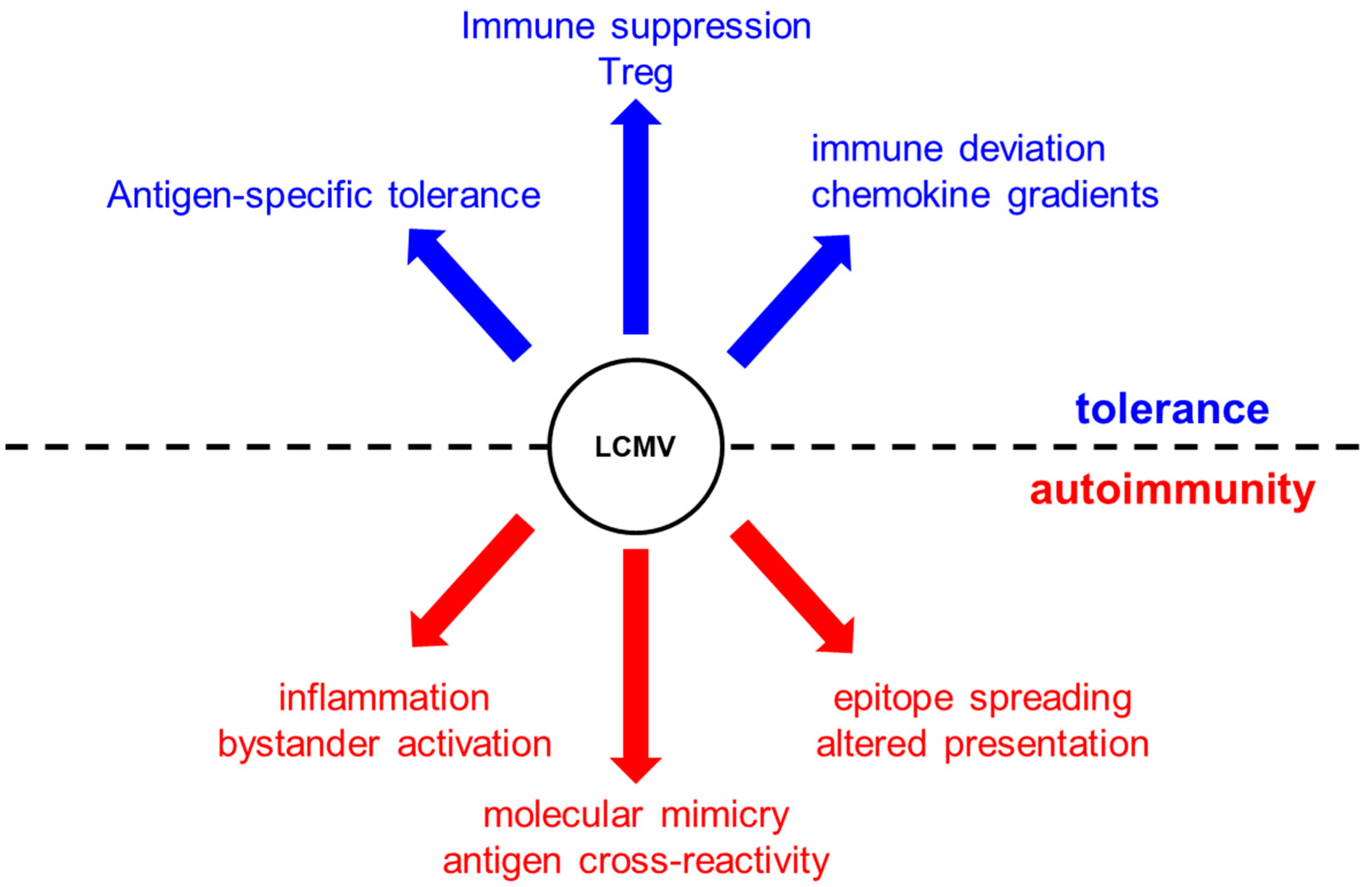
3. Mechanisms That Can Lead to Autoimmunity Following Viral Infection
3.1. Clonal Deletion, T-Cell Anergy, and Immune Ignorance
3.2. Molecular Mimicry, Epitope Spreading, and Bystander Activation
3.3. T-Cell Exhaustion and Immunopathology
4. The Hygiene Hypothesis and How Viral Infections Protect from Autoimmunity
4.1. Antigen-Specific Tolerance
4.2. Immune Suppression, Treg Invigoration, and Immune Deviation
5. Conclusions
Funding
Conflicts of Interest
References
- Filippi, C.; von Herrath, M. How viral infections affect the autoimmune process leading to type 1 diabetes. Cell. Immunol. 2005, 233, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Oldstone, M.B.; von Herrath, M.; Evans, C.F.; Horwitz, M.S. Virus-induced autoimmune disease: Transgenic approach to mimic insulin-dependent diabetes mellitus and multiple sclerosis. Curr. Top. Microbiol. Immunol. 1996, 206, 67–83. [Google Scholar] [PubMed]
- von Herrath, M.G.; Evans, C.F.; Horwitz, M.S.; Oldstone, M.B. Using transgenic mouse models to dissect the pathogenesis of virus-induced autoimmune disorders of the islets of Langerhans and the central nervous system. Immunol. Rev. 1996, 152, 111–143. [Google Scholar] [CrossRef] [PubMed]
- Christen, U.; Hintermann, E.; Holdener, M.; von Herrath, M.G. Viral triggers for autoimmunity: Is the ‘glass of molecular mimicry’ half full or half empty? J. Autoimmun. 2010, 34, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, R.S.; von Herrath, M.G.; Christen, U.; Whitton, J.L. Molecular mimicry, bystander activation, or viral persistence: Infections and autoimmune disease. Clin. Microbiol. Rev. 2006, 19, 80–94. [Google Scholar] [CrossRef]
- Buchmeier, M.J.; Welsh, R.M.; Dutko, F.J.; Oldstone, M.B. The virology and immunobiology of lymphocytic choriomeningitis virus infection. Adv. Immunol. 1980, 30, 275–331. [Google Scholar]
- Bonthius, D.J. Lymphocytic choriomeningitis virus: An underrecognized cause of neurologic disease in the fetus, child, and adult. Semin. Pediatr. Neurol. 2012, 19, 89–95. [Google Scholar] [CrossRef]
- Cole, G.A.; Nathanson, N.; Prendergast, R.A. Requirement for theta-bearing cells in lymphocytic choriomeningitis virus-induced central nervous system disease. Nature 1972, 238, 335–337. [Google Scholar] [CrossRef]
- Kim, J.V.; Kang, S.S.; Dustin, M.L.; McGavern, D.B. Myelomonocytic cell recruitment causes fatal CNS vascular injury during acute viral meningitis. Nature 2009, 457, 191–195. [Google Scholar] [CrossRef]
- Ohashi, P.S.; Oehen, S.; Buerki, K.; Pircher, H.; Ohashi, C.T.; Odermatt, B.; Malissen, B.; Zinkernagel, R.M.; Hengartner, H. Ablation of “tolerance” and induction of diabetes by virus infection in viral antigen transgenic mice. Cell 1991, 65, 305–317. [Google Scholar] [CrossRef]
- Oldstone, M.B.; Nerenberg, M.; Southern, P.; Price, J.; Lewicki, H. Virus infection triggers insulin-dependent diabetes mellitus in a transgenic model: Role of anti-self (virus) immune response. Cell 1991, 65, 319–331. [Google Scholar] [CrossRef]
- Holdener, M.; Hintermann, E.; Bayer, M.; Rhode, A.; Rodrigo, E.; Hintereder, G.; Johnson, E.F.; Gonzalez, F.J.; Pfeilschifter, J.; Manns, M.P.; et al. Breaking tolerance to the natural human liver autoantigen cytochrome P450 2D6 by virus infection. J. Exp. Med. 2008, 205, 1409–1422. [Google Scholar] [CrossRef] [PubMed]
- Christen, U. Animal models of autoimmune hepatitis. Biochim. Biophys. Acta. Mol. Basis Dis. 2019, 1865, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Christen, U.; Hintermann, E. Pathogens and autoimmune hepatitis. Clin. Exp. Immunol. 2019, 195, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.F.; Horwitz, M.S.; Hobbs, M.V.; Oldstone, M.B. Viral infection of transgenic mice expressing a viral protein in oligodendrocytes leads to chronic central nervous system autoimmune disease. J. Exp. Med. 1996, 184, 2371–2384. [Google Scholar] [CrossRef] [PubMed]
- Wucherpfennig, K.W.; Strominger, J.L. Molecular mimicry in T cell-mediated autoimmunity: Viral peptides activate human T cell clones specific for myelin basic protein. Cell 1995, 80, 695–705. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Ramachandran, S.; Mann, M.; Popkin, D.L. Role of lymphocytic choriomeningitis virus (LCMV) in understanding viral immunology: Past, present and future. Viruses 2012, 4, 2650–2669. [Google Scholar] [CrossRef]
- Oldstone, M.B. Lessons learned and concepts formed from study of the pathogenesis of the two negative-strand viruses lymphocytic choriomeningitis and influenza. Proc. Natl. Acad. Sci. USA 2013, 110, 4180–4183. [Google Scholar] [CrossRef] [Green Version]
- Doyle, L.B.; Doyle, M.V.; Oldstone, M.B. Susceptibility of newborn mice with H-2k backgrounds to lymphocytic choriomeningitis virus infection. Immunology 1980, 40, 589–596. [Google Scholar]
- Oldstone, M.B.; Dixon, F.J. Change in susceptibility of C3H-HeJ mice to LCM virus infection. J. Immunol. 1973, 111, 1613–1615. [Google Scholar]
- Speiser, D.E.; Pircher, H.; Ohashi, P.S.; Kyburz, D.; Hengartner, H.; Zinkernagel, R.M. Clonal deletion induced by either radioresistant thymic host cells or lymphohemopoietic donor cells at different stages of class I-restricted T cell ontogeny. J. Exp. Med. 1992, 175, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Elson, C.J.; Barker, R.N.; Thompson, S.J.; Williams, N.A. Immunologically ignorant autoreactive T cells, epitope spreading and repertoire limitation. Immunol. Today 1995, 16, 71–76. [Google Scholar] [CrossRef]
- Lang, K.S.; Recher, M.; Junt, T.; Navarini, A.A.; Harris, N.L.; Freigang, S.; Odermatt, B.; Conrad, C.; Ittner, L.M.; Bauer, S.; et al. Toll-like receptor engagement converts T-cell autoreactivity into overt autoimmune disease. Nat. Med. 2005, 11, 138–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmer, B.; Fleckenstein, B.T.; Vergelli, M.; Jung, G.; McFarland, H.; Martin, R.; Wiesmuller, K.H. Identification of high potency microbial and self ligands for a human autoreactive class II-restricted T cell clone. J. Exp. Med. 1997, 185, 1651–1659. [Google Scholar] [CrossRef] [PubMed]
- Pfizenmaier, K.; Trostmann, H.; Rollinghoff, M.; Wagner, H. Temporary presence of self-reactive cytotoxic T lymphocytes during murine lymphocytic choriomeningitis. Nature 1975, 258, 238–240. [Google Scholar] [CrossRef]
- Serreze, D.V.; Ottendorfer, E.W.; Ellis, T.M.; Gauntt, C.J.; Atkinson, M.A. Acceleration of type 1 diabetes by a coxsackievirus infection requires a preexisting critical mass of autoreactive T-cells in pancreatic islets. Diabetes 2000, 49, 708–711. [Google Scholar] [CrossRef]
- Donermeyer, D.L.; Beisel, K.W.; Allen, P.M.; Smith, S.C. Myocarditis-inducing epitope of myosin binds constitutively and stably to I-Ak on antigen-presenting cells in the heart. J. Exp. Med. 1995, 182, 1291–1300. [Google Scholar] [CrossRef]
- Huber, S.A.; Lodge, P.A. Coxsackievirus B-3 myocarditis in Balb/c mice. Evidence for autoimmunity to myocyte antigens. Am. J. Pathol. 1984, 116, 21–29. [Google Scholar]
- Fujinami, R.S.; Oldstone, M.B. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: Mechanism for autoimmunity. Science 1985, 230, 1043–1045. [Google Scholar] [CrossRef]
- Horwitz, M.S.; Bradley, L.M.; Harbertson, J.; Krahl, T.; Lee, J.; Sarvetnick, N. Diabetes induced by Coxsackie virus: Initiation by bystander damage and not molecular mimicry. Nat. Med. 1998, 4, 781–785. [Google Scholar] [CrossRef]
- Murali-Krishna, K.; Altman, J.D.; Suresh, M.; Sourdive, D.J.; Zajac, A.J.; Miller, J.D.; Slansky, J.; Ahmed, R. Counting antigen-specific CD8 T cells: A reevaluation of bystander activation during viral infection. Immunity 1998, 8, 177–187. [Google Scholar] [CrossRef]
- Duke, R.C. Self recognition by T cells. I. Bystander killing of target cells bearing syngeneic MHC antigens. J. Exp. Med. 1989, 170, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Sevilla, N.; Homann, D.; von Herrath, M.; Rodriguez, F.; Harkins, S.; Whitton, J.L.; Oldstone, M.B. Virus-induced diabetes in a transgenic model: Role of cross-reacting viruses and quantitation of effector T cells needed to cause disease. J. Virol. 2000, 74, 3284–3292. [Google Scholar] [CrossRef] [PubMed]
- Murali-Krishna, K.; Altman, J.D.; Suresh, M.; Sourdive, D.; Zajac, A.; Ahmed, R. In vivo dynamics of anti-viral CD8 T cell responses to different epitopes. An evaluation of bystander activation in primary and secondary responses to viral infection. Adv. Exp. Med. Biol. 1998, 452, 123–142. [Google Scholar] [PubMed]
- McCoy, L.; Tsunoda, I.; Fujinami, R.S. Multiple sclerosis and virus induced immune responses: Autoimmunity can be primed by molecular mimicry and augmented by bystander activation. Autoimmunity 2006, 39, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Vanderlugt, C.J.; Miller, S.D. Epitope spreading. Curr. Opin. Immunol. 1996, 8, 831–836. [Google Scholar] [CrossRef]
- Brehm, M.A.; Selin, L.K.; Welsh, R.M. CD8 T cell responses to viral infections in sequence. Cell. Microbiol. 2004, 6, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Blattman, J.N.; Murali-Krishna, K.; van der Most, R.; Ahmed, R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 2003, 77, 4911–4927. [Google Scholar] [CrossRef]
- Godkin, A.; Smith, K.A. Chronic infections with viruses or parasites: Breaking bad to make good. Immunology 2017, 150, 389–396. [Google Scholar] [CrossRef]
- Cornberg, M.; Clute, S.C.; Watkin, L.B.; Saccoccio, F.M.; Kim, S.K.; Naumov, Y.N.; Brehm, M.A.; Aslan, N.; Welsh, R.M.; Selin, L.K. CD8 T cell cross-reactivity networks mediate heterologous immunity in human EBV and murine vaccinia virus infections. J. Immunol. 2010, 184, 2825–2838. [Google Scholar] [CrossRef]
- Nie, S.; Lin, S.J.; Kim, S.K.; Welsh, R.M.; Selin, L.K. Pathological features of heterologous immunity are regulated by the private specificities of the immune repertoire. Am. J. Pathol. 2010, 176, 2107–2112. [Google Scholar] [CrossRef] [PubMed]
- Selin, L.K.; Nahill, S.R.; Welsh, R.M. Cross-reactivities in memory cytotoxic T lymphocyte recognition of heterologous viruses. J. Exp. Med. 1994, 179, 1933–1943. [Google Scholar] [CrossRef] [PubMed]
- Welsh, R.M.; Che, J.W.; Brehm, M.A.; Selin, L.K. Heterologous immunity between viruses. Immunol. Rev. 2010, 235, 244–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brehm, M.A.; Pinto, A.K.; Daniels, K.A.; Schneck, J.P.; Welsh, R.M.; Selin, L.K. T cell immunodominance and maintenance of memory regulated by unexpectedly cross-reactive pathogens. Nat. Immunol. 2002, 3, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Adams, A.B.; Pearson, T.C.; Larsen, C.P. Heterologous immunity: An overlooked barrier to tolerance. Immunol. Rev. 2003, 196, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Adams, A.B.; Williams, M.A.; Jones, T.R.; Shirasugi, N.; Durham, M.M.; Kaech, S.M.; Wherry, E.J.; Onami, T.; Lanier, J.G.; Kokko, K.E.; et al. Heterologous immunity provides a potent barrier to transplantation tolerance. J. Clin. Investig. 2003, 111, 1887–1895. [Google Scholar] [CrossRef]
- Welsh, R.M.; Selin, L.K. No one is naive: The significance of heterologous T-cell immunity. Nat. Rev. Immunol. 2002, 2, 417–426. [Google Scholar] [CrossRef]
- Selin, L.K.; Cornberg, M.; Brehm, M.A.; Kim, S.K.; Calcagno, C.; Ghersi, D.; Puzone, R.; Celada, F.; Welsh, R.M. CD8 memory T cells: Cross-reactivity and heterologous immunity. Semin Immunol 2004, 16, 335–347. [Google Scholar] [CrossRef]
- Jofra, T.; Di Fonte, R.; Galvani, G.; Kuka, M.; Iannacone, M.; Battaglia, M.; Fousteri, G. Tr1 cell immunotherapy promotes transplant tolerance via de novo Tr1 cell induction in mice and is safe and effective during acute viral infection. Eur. J. Immunol. 2018, 48, 1389–1399. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.B.; Pagni, P.P.; Fousteri, G.; Sachithanantham, S.; Dave, A.; Rodriguez-Calvo, T.; Miller, J.; von Herrath, M. Regulatory T cells control diabetes without compromising acute anti-viral defense. Clin. Immunol. 2014, 153, 298–307. [Google Scholar] [CrossRef] [Green Version]
- Kahan, S.M.; Zajac, A.J. Immune Exhaustion: Past Lessons and New Insights from Lymphocytic Choriomeningitis Virus. Viruses 2019, 11, 156. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, S.D.; Wherry, E.J. IL-10, T cell exhaustion and viral persistence. Trends Microbiol. 2007, 15, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Moskophidis, D.; Lechner, F.; Pircher, H.; Zinkernagel, R.M. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature 1993, 362, 758–761. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Wherry, E.J. CD8 T cell dysfunction during chronic viral infection. Curr. Opin. Immunol. 2007, 19, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Ha, S.J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef] [PubMed]
- Iwai, Y.; Terawaki, S.; Ikegawa, M.; Okazaki, T.; Honjo, T. PD-1 inhibits antiviral immunity at the effector phase in the liver. J. Exp. Med. 2003, 198, 39–50. [Google Scholar] [CrossRef]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.N.; Vanguri, V.K.; Ha, S.J.; West, E.E.; Keir, M.E.; Glickman, J.N.; Sharpe, A.H.; Ahmed, R. PD-L1 has distinct functions in hematopoietic and nonhematopoietic cells in regulating T cell responses during chronic infection in mice. J. Clin. Investig. 2010, 120, 2508–2515. [Google Scholar] [CrossRef]
- Okazaki, T.; Honjo, T. PD-1 and PD-1 ligands: From discovery to clinical application. Int. Immunol. 2007, 19, 813–824. [Google Scholar] [CrossRef]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Paluch, C.; Santos, A.M.; Anzilotti, C.; Cornall, R.J.; Davis, S.J. Immune Checkpoints as Therapeutic Targets in Autoimmunity. Front. Immunol. 2018, 9, 2306. [Google Scholar] [CrossRef] [PubMed]
- Frebel, H.; Nindl, V.; Schuepbach, R.A.; Braunschweiler, T.; Richter, K.; Vogel, J.; Wagner, C.A.; Loffing-Cueni, D.; Kurrer, M.; Ludewig, B.; et al. Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. J. Exp. Med. 2012, 209, 2485–2499. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, H.; Minato, N.; et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef]
- Wang, J.; Yoshida, T.; Nakaki, F.; Hiai, H.; Okazaki, T.; Honjo, T. Establishment of NOD-Pdcd1-/- mice as an efficient animal model of type I diabetes. Proc. Natl. Acad. Sci. USA 2005, 102, 11823–11828. [Google Scholar] [CrossRef]
- Ansari, M.J.; Salama, A.D.; Chitnis, T.; Smith, R.N.; Yagita, H.; Akiba, H.; Yamazaki, T.; Azuma, M.; Iwai, H.; Khoury, S.J.; et al. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J. Exp. Med. 2003, 198, 63–69. [Google Scholar] [CrossRef]
- Wang, J.; Okazaki, I.M.; Yoshida, T.; Chikuma, S.; Kato, Y.; Nakaki, F.; Hiai, H.; Honjo, T.; Okazaki, T. PD-1 deficiency results in the development of fatal myocarditis in MRL mice. Int. Immunol. 2010, 22, 443–452. [Google Scholar] [CrossRef]
- Bach, J.F. The hygiene hypothesis in autoimmunity: The role of pathogens and commensals. Nat. Rev. Immunol. 2018, 18, 105–120. [Google Scholar] [CrossRef]
- Dyrberg, T.; Schwimmbeck, P.L.; Oldstone, M.B. Inhibition of diabetes in BB rats by virus infection. J. Clin. Investig. 1988, 81, 928–931. [Google Scholar] [CrossRef] [PubMed]
- Oldstone, M.B. Prevention of type I diabetes in nonobese diabetic mice by virus infection. Science 1988, 239, 500–502. [Google Scholar] [CrossRef] [PubMed]
- Christen, U.; Benke, D.; Wolfe, T.; Rodrigo, E.; Rhode, A.; Hughes, A.C.; Oldstone, M.B.; von Herrath, M.G. Cure of prediabetic mice by viral infections involves lymphocyte recruitment along an IP-10 gradient. J. Clin. Investig. 2004, 113, 74–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippi, C.M.; Ehrhardt, K.; Estes, E.A.; Larsson, P.; Oldham, J.E.; von Herrath, M.G. TLR2 signaling improves immunoregulation to prevent type 1 diabetes. Eur. J. Immunol. 2011, 41, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Filippi, C.M.; Estes, E.A.; Oldham, J.E.; von Herrath, M.G. Immunoregulatory mechanisms triggered by viral infections protect from type 1 diabetes in mice. J. Clin. Investig. 2009, 119, 1515–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fousteri, G.; von Herrath, M.; Bresson, D. Mucosal exposure to antigen: Cause or cure of type 1 diabetes? Curr. Diab. Rep. 2007, 7, 91–98. [Google Scholar] [CrossRef]
- Barnett, L.A.; Whitton, J.L.; Wang, L.Y.; Fujinami, R.S. Virus encoding an encephalitogenic peptide protects mice from experimental allergic encephalomyelitis. J. Neuroimmunol. 1996, 64, 163–173. [Google Scholar] [CrossRef]
- Aichele, P.; Kyburz, D.; Ohashi, P.S.; Odermatt, B.; Zinkernagel, R.M.; Hengartner, H.; Pircher, H. Peptide-induced T-cell tolerance to prevent autoimmune diabetes in a transgenic mouse model. Proc. Natl. Acad. Sci. USA 1994, 91, 444–448. [Google Scholar] [CrossRef]
- Bielekova, B.; Goodwin, B.; Richert, N.; Cortese, I.; Kondo, T.; Afshar, G.; Gran, B.; Eaton, J.; Antel, J.; Frank, J.A.; et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83-99) in multiple sclerosis: Results of a phase II clinical trial with an altered peptide ligand. Nat. Med. 2000, 6, 1167–1175. [Google Scholar] [CrossRef]
- Kappos, L.; Comi, G.; Panitch, H.; Oger, J.; Antel, J.; Conlon, P.; Steinman, L. Induction of a non-encephalitogenic type 2 T helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. The Altered Peptide Ligand in Relapsing MS Study Group. Nat. Med. 2000, 6, 1176–1182. [Google Scholar] [CrossRef]
- Anderton, S.M. Peptide-based immunotherapy of autoimmunity: A path of puzzles, paradoxes and possibilities. Immunology 2001, 104, 367–376. [Google Scholar] [CrossRef]
- Fousteri, G.; Bresson, D.; von Herrath, M. Rational development of antigen-specific therapies for type 1 diabetes. Adv. Exp. Med. Biol. 2007, 601, 313–319. [Google Scholar]
- Hafler, D.A.; Weiner, H.L. Antigen-specific therapies for the treatment of autoimmune diseases. Springer Semin. Immunopathol. 1995, 17, 61–76. [Google Scholar] [CrossRef]
- Christen, U.; Von Herrath, M.G. IP-10 and type 1 diabetes: A question of time and location. Autoimmunity 2004, 37, 273–282. [Google Scholar] [CrossRef]
- McChesney, M.B.; Oldstone, M.B. Viruses perturb lymphocyte functions: Selected principles characterizing virus-induced immunosuppression. Annu. Rev. Immunol. 1987, 5, 279–304. [Google Scholar] [CrossRef]
- Mims, C.A.; Wainwright, S. The immunodepressive action of lymphocytic choriomeningitis virus in mice. J. Immunol. 1968, 101, 717–724. [Google Scholar]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fousteri, G.; Dave Jhatakia, A. Viral Infections and Autoimmune Disease: Roles of LCMV in Delineating Mechanisms of Immune Tolerance. Viruses 2019, 11, 885. https://doi.org/10.3390/v11100885
Fousteri G, Dave Jhatakia A. Viral Infections and Autoimmune Disease: Roles of LCMV in Delineating Mechanisms of Immune Tolerance. Viruses. 2019; 11(10):885. https://doi.org/10.3390/v11100885
Chicago/Turabian StyleFousteri, Georgia, and Amy Dave Jhatakia. 2019. "Viral Infections and Autoimmune Disease: Roles of LCMV in Delineating Mechanisms of Immune Tolerance" Viruses 11, no. 10: 885. https://doi.org/10.3390/v11100885
APA StyleFousteri, G., & Dave Jhatakia, A. (2019). Viral Infections and Autoimmune Disease: Roles of LCMV in Delineating Mechanisms of Immune Tolerance. Viruses, 11(10), 885. https://doi.org/10.3390/v11100885