Codon Usage in the Iflaviridae Family Is Not Diverse Though the Family Members Are Isolated from Diverse Host Taxa


Abstract
:1. Introduction
2. Materials and Methods
2.1. Genome Sequences
2.2. Nucleotide Composition
2.3. Relative Synonymous Codon Usage
2.4. Effective Number of Codons and ENc-Plot
2.5. Relative Neutrality Plot and Parity Rule 2 Bias Plot
2.6. Correspondence Analysis
2.7. Software and Calculation
3. Results
3.1. Nucleotide Composition
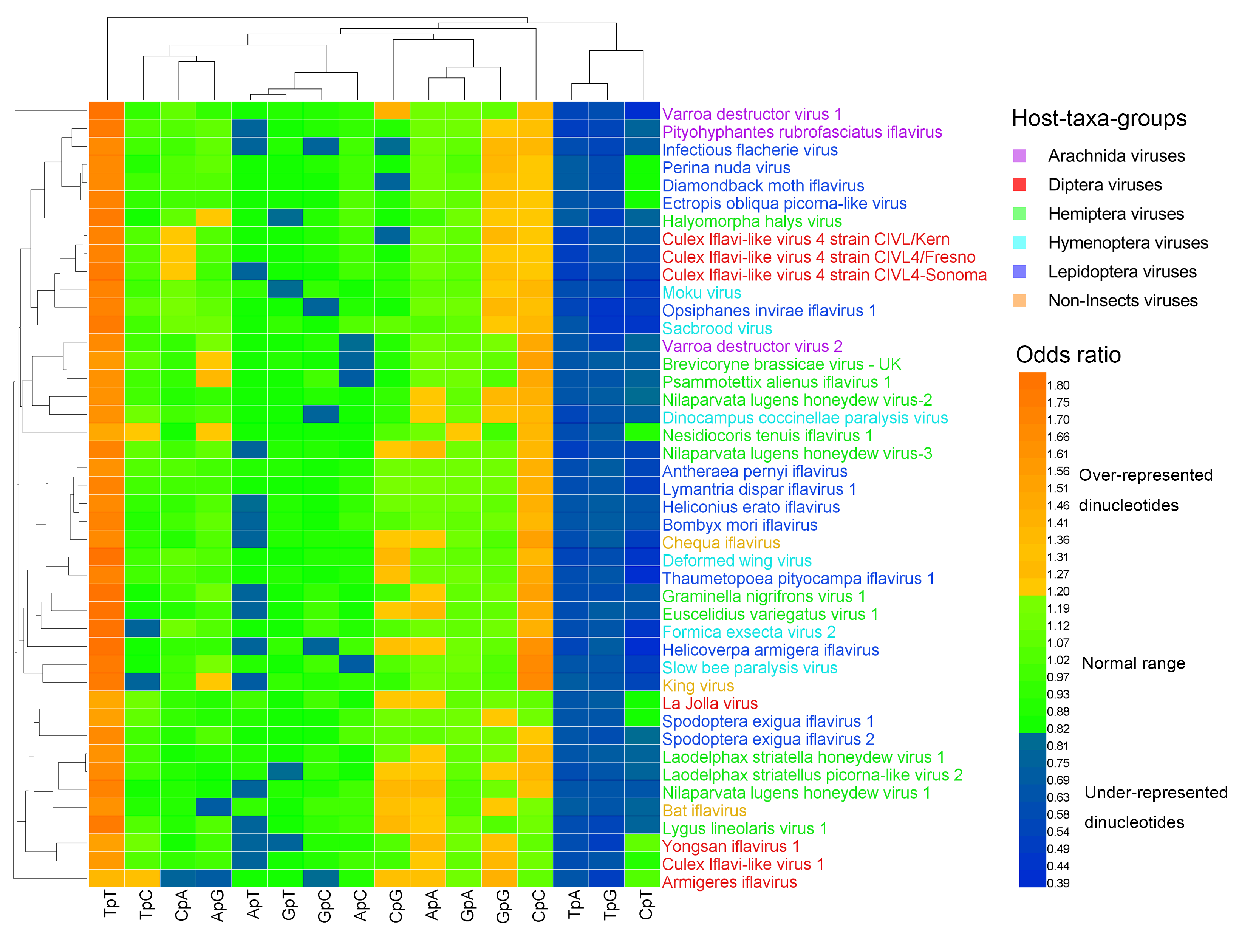
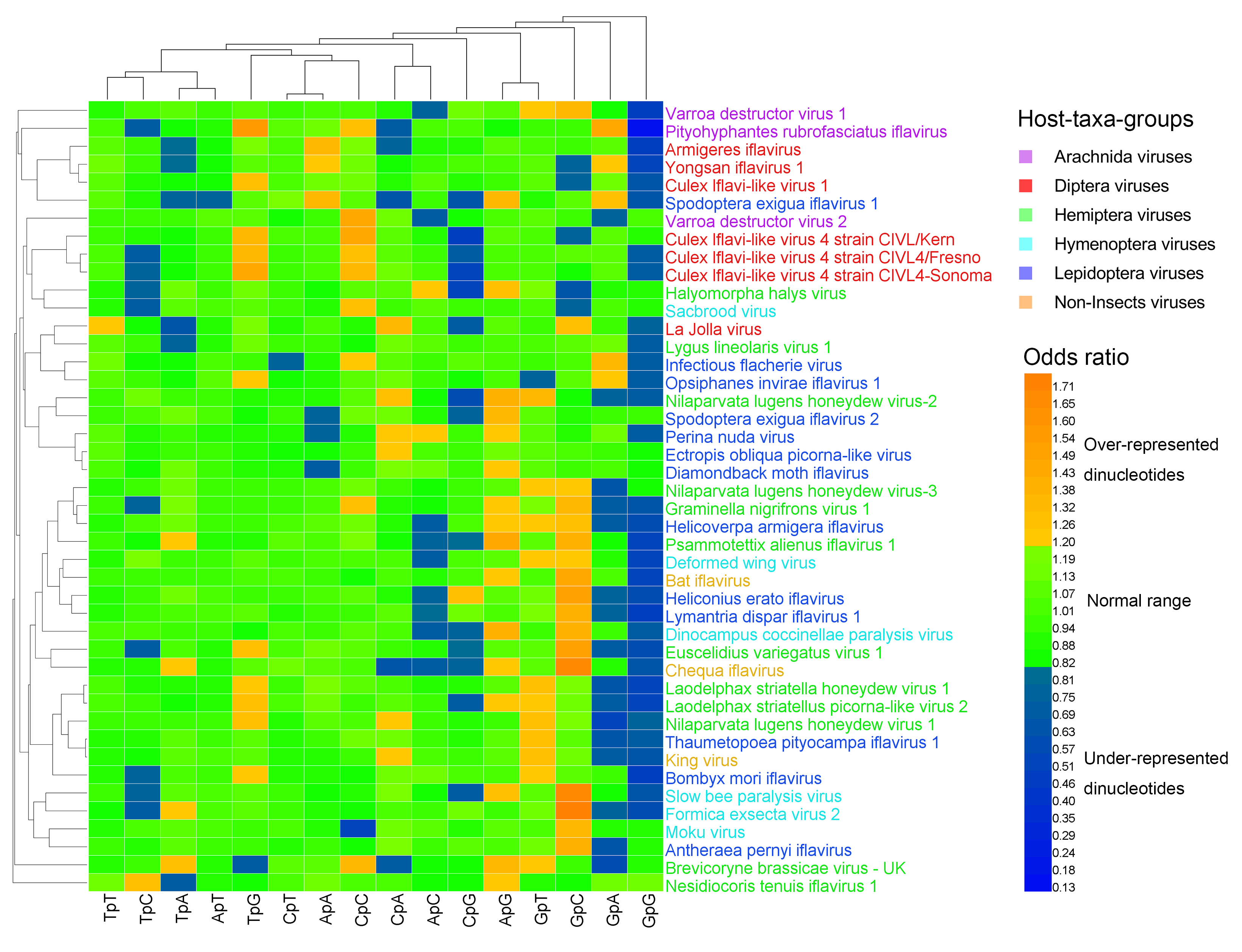
3.2. Dinucleotide Composition
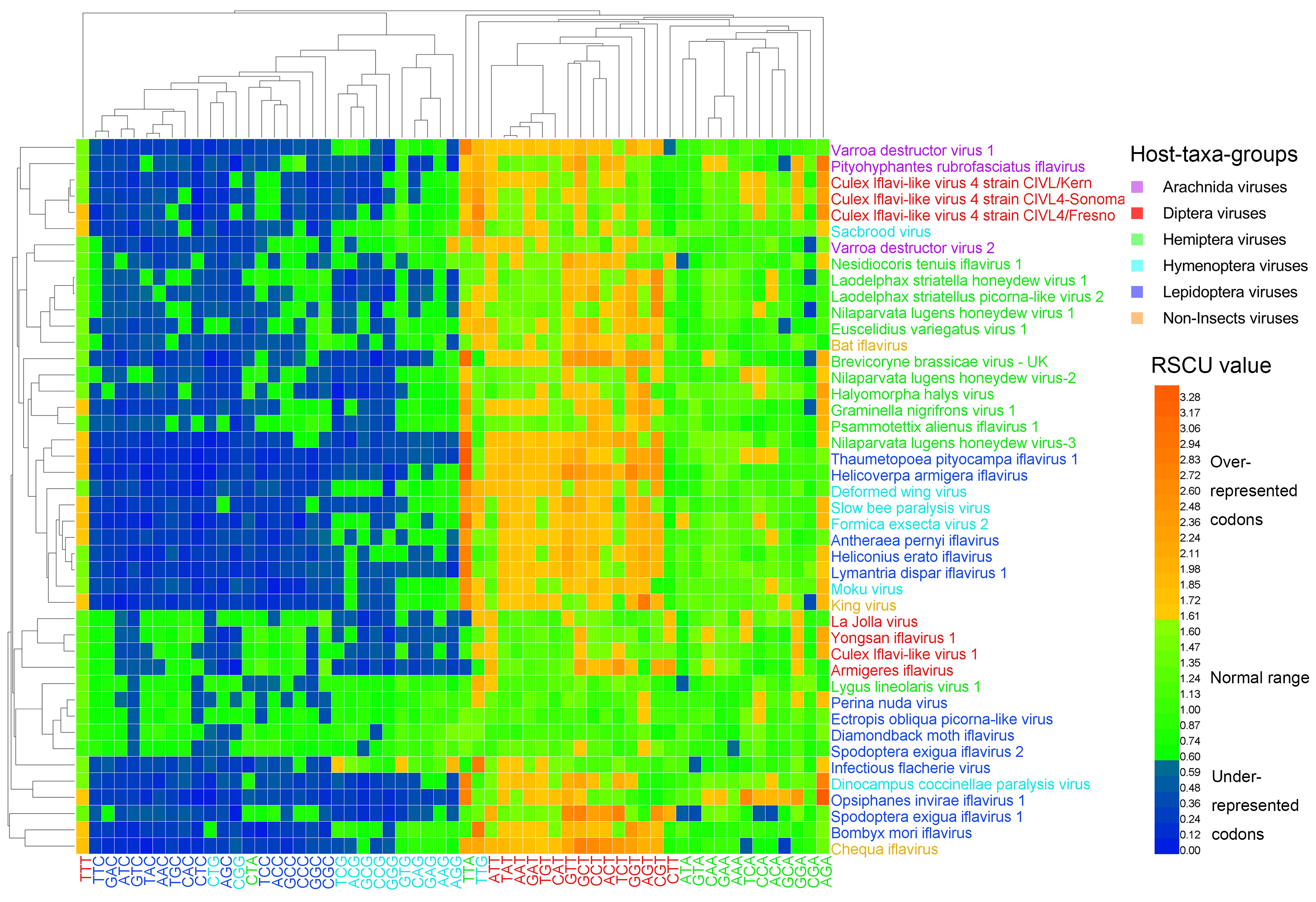
3.3. Synonymous Codon Usage
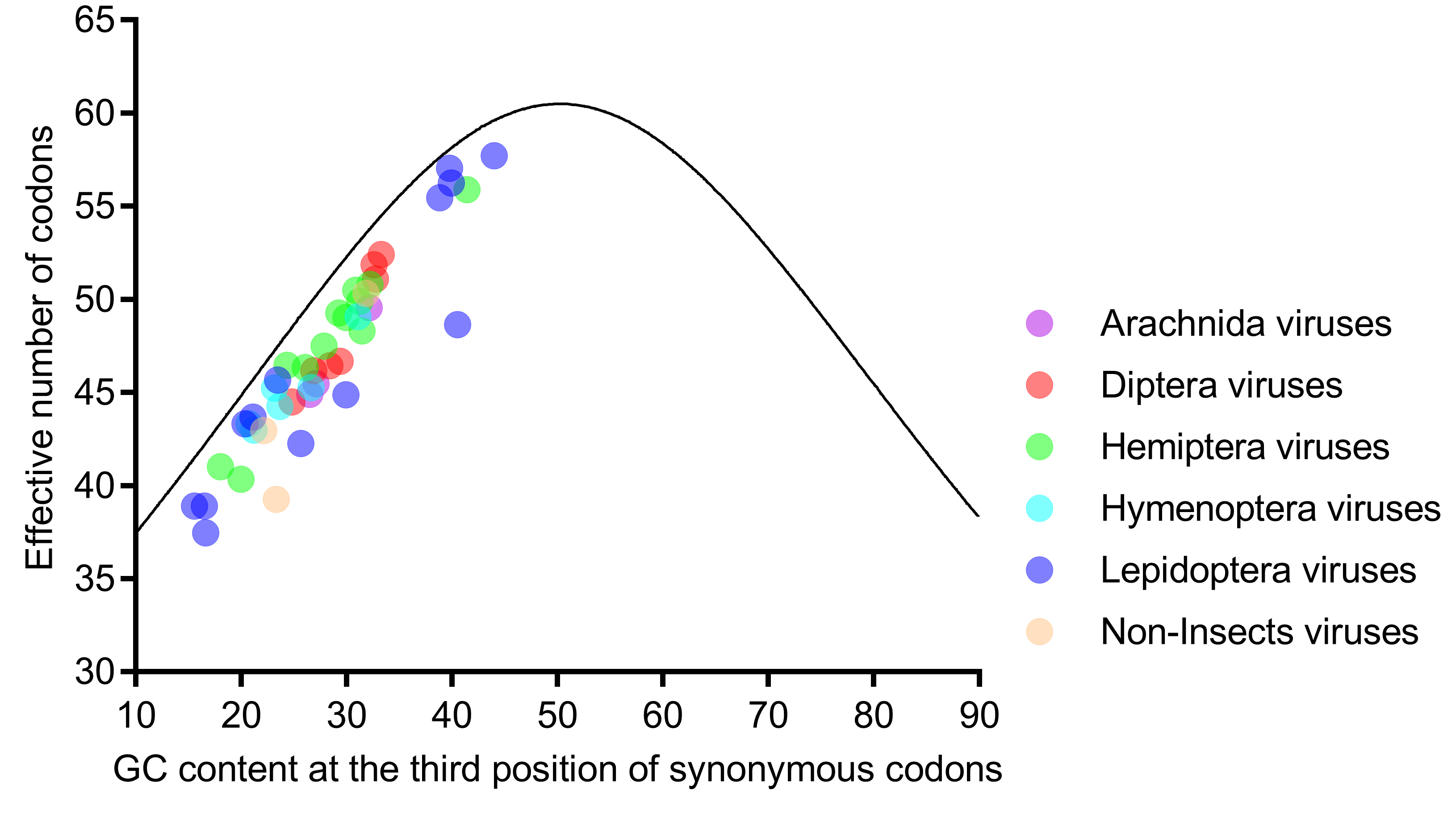
3.4. ENc Value and ENc-Plot Analysis
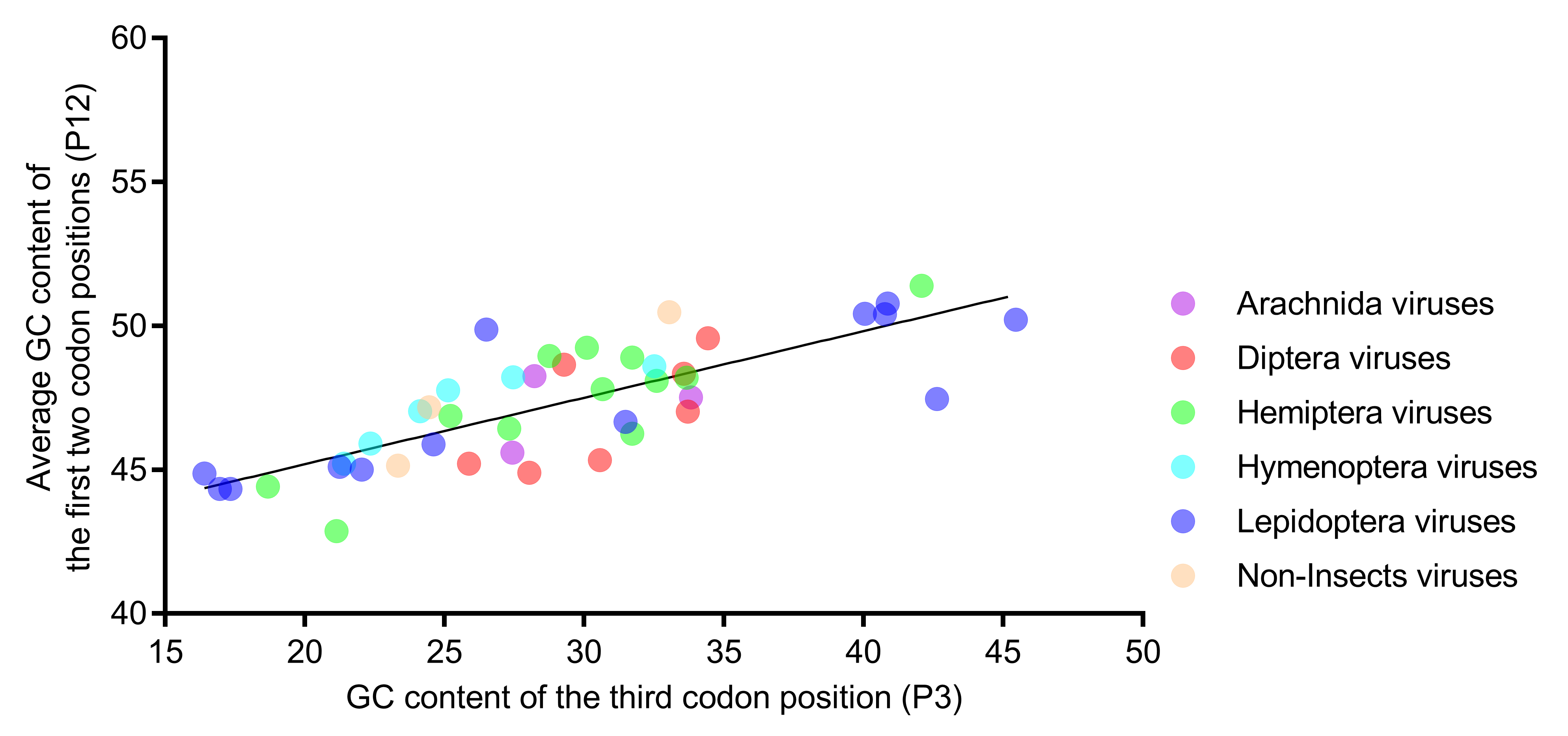
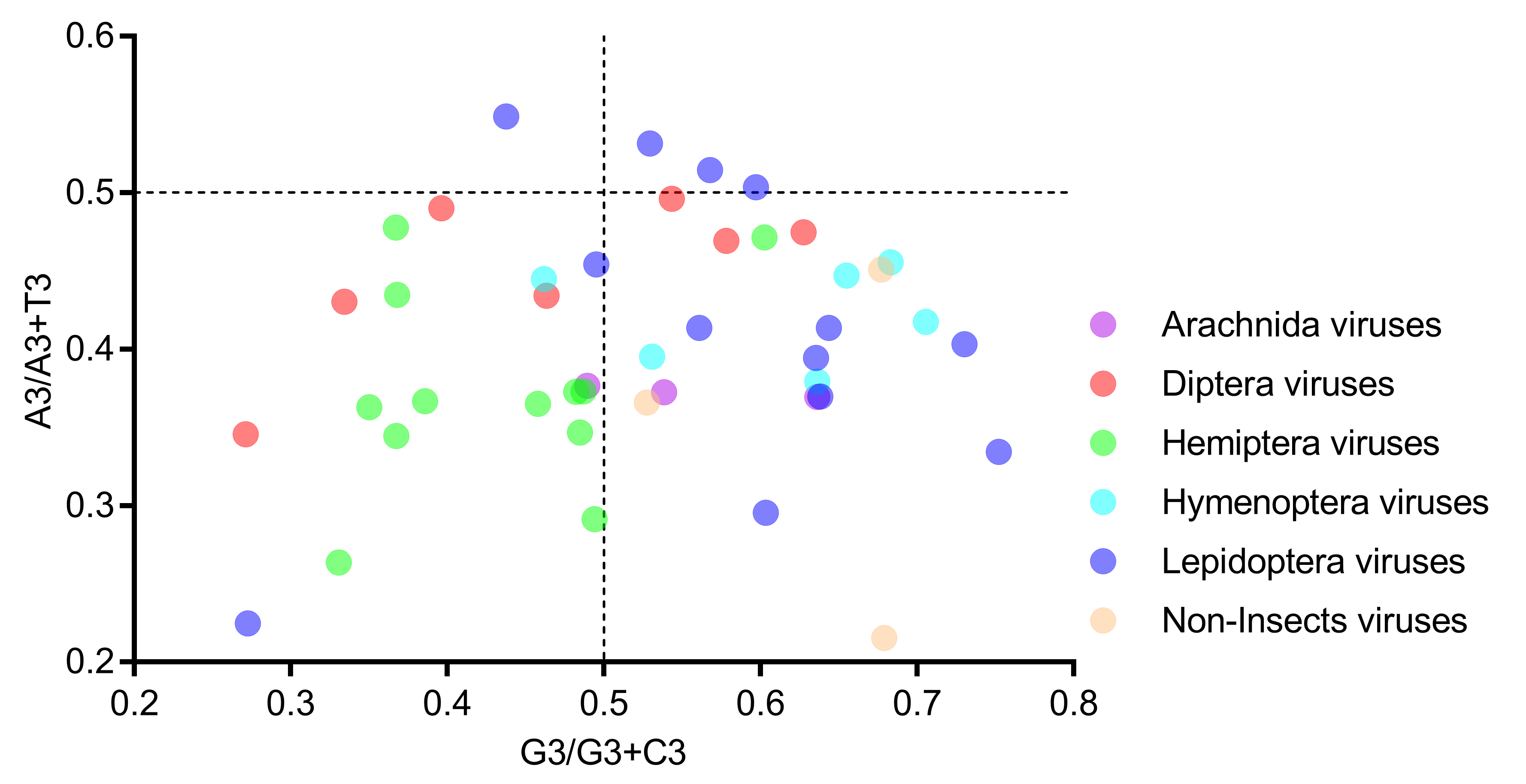
3.5. Relative Neutrality Plot and PR2 Bias Plot
3.6. Correspondence Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Plotkin, J.B.; Kudla, G. Synonymous but not the same: The causes and consequences of codon bias. Nat. Rev. Genet. 2011, 12, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Chaney, J.L.; Clark, P.L. Roles for Synonymous Codon Usage in Protein Biogenesis. Annu Rev. Biophys. 2015, 44, 143–166. [Google Scholar] [CrossRef] [PubMed]
- Supek, F. The Code of Silence: Widespread Associations Between Synonymous Codon Biases and Gene Function. J. Mol. Evol. 2016, 82, 65–73. [Google Scholar] [CrossRef]
- Im, E.-H.; Choi, S.S. Synonymous Codon Usage Controls Various Molecular Aspects. Genomics Inform. 2017, 15, 123–127. [Google Scholar] [CrossRef]
- Mittal, P.; Brindle, J.; Stephen, J.; Plotkin, J.B.; Kudla, G. Codon usage influences fitness through RNA toxicity. Proc. Natl. Acad. Sci. USA 2018, 115, 8639. [Google Scholar] [CrossRef]
- Shackelton, L.A.; Parrish, C.R.; Holmes, E.C. Evolutionary basis of codon usage and nucleotide composition bias in vertebrate DNA viruses. J. Mol. Evol. 2006, 62, 551–563. [Google Scholar] [CrossRef]
- Yao, H.; Chen, M.; Tang, Z. Analysis of Synonymous Codon Usage Bias in Flaviviridae Virus. Biomed. Res. Int. 2019, 2019, 12. [Google Scholar] [CrossRef]
- Shi, S.-L.; Jiang, Y.-R.; Liu, Y.-Q.; Xia, R.-X.; Qin, L. Selective pressure dominates the synonymous codon usage in parvoviridae. Virus Genes 2013, 46, 10–19. [Google Scholar] [CrossRef]
- Valles, S.M.; Chen, Y.; Firth, A.E.; Guérin, D.M.A.; Hashimoto, Y.; Herrero, S.; de Miranda, J.R.; Ryabov, E.; Consortium, I.R. ICTV Virus Taxonomy Profile: Iflaviridae. J. Gen. Virol. 2017, 98, 527–528. [Google Scholar] [CrossRef]
- Wilfert, L.; Long, G.; Leggett, H.C.; Schmid-Hempel, P.; Butlin, R.; Martin, S.J.; Boots, M. Deformed wing virus is a recent global epidemic in honeybees driven by Varroa mites. Science 2016, 351, 594–597. [Google Scholar] [CrossRef]
- Geng, P.; Li, W.; de Miranda, J.R.; Qian, Z.; An, L.; Terenius, O. Studies on the transmission and tissue distribution of Antheraea pernyi iflavirus in the Chinese oak silkmoth Antheraea pernyi. Virology 2017, 502, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Vootla, S.K.; Lu, X.M.; Kari, N.; Gadwala, M.; Lu, Q. Rapid detection of infectious flacherie virus of the silkworm, Bombyx mori, using RT-PCR and nested PCR. J. Insect Sci. 2013, 13, 120. [Google Scholar] [CrossRef] [PubMed]
- Carballo, A.; Murillo, R.; Jakubowska, A.; Herrero, S.; Williams, T.; Caballero, P. Co-infection with iflaviruses influences the insecticidal properties of Spodoptera exigua multiple nucleopolyhedrovirus occlusion bodies: Implications for the production and biosecurity of baculovirus insecticides. PLoS ONE 2017, 12, e0177301. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Tripp, J.; Bonning, B.C.; Miller, W.A. Challenges associated with research on RNA viruses of insects. Curr. Opin. Insect. Sci. 2015, 8, 62–68. [Google Scholar] [CrossRef]
- Yuan, H.; Xu, P.; Yang, X.; Graham, R.I.; Wilson, K.; Wu, K. Characterization of a novel member of genus Iflavirus in Helicoverpa armigera. J. Invertebr. Pathol. 2017, 144, 65–73. [Google Scholar] [CrossRef]
- Suzuki, T.; Takeshima, Y.; Mikamoto, T.; Saeki, J.D.; Kato, T.; Park, E.Y.; Kawagishi, H.; Dohra, H. Genome Sequence of a Novel Iflavirus from mRNA Sequencing of the Pupa of Bombyx mori Inoculated with Cordyceps militaris. Genome Announc 2015, 3, e01039-15. [Google Scholar] [CrossRef]
- Silva, L.A.; Ardisson-Araujo, D.M.; Tinoco, R.S.; Fernandes, O.A.; Melo, F.L.; Ribeiro, B.M. Complete genome sequence and structural characterization of a novel iflavirus isolated from Opsiphanes invirae (Lepidoptera: Nymphalidae). J. Invertebr. Pathol. 2015, 130, 136–140. [Google Scholar] [CrossRef]
- Martin, S.J.; Brettell, L.E. Deformed Wing Virus in Honeybees and Other Insects. Annu. Rev. Virol. 2019, 6, 49–69. [Google Scholar] [CrossRef]
- Tian, L.; Shen, X.; Murphy, R.W.; Shen, Y. The adaptation of codon usage of +ssRNA viruses to their hosts. Infect. Genet. Evol. 2018, 63, 175–179. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Karlin, S.; Burge, C. Dinucleotide relative abundance extremes: A genomic signature. Trends Genet. 1995, 11, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Tuohy, T.M.F.; Mosurski, K.R. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986, 14, 5125–5143. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.L.; Jiang, Y.R.; Yang, R.S.; Wang, Y.; Qin, L. Codon usage in Alphabaculovirus and Betabaculovirus hosted by the same insect species is weak, selection dominated and exhibits no more similar patterns than expected. Infect. Genet. Evol 2016, 44, 412–417. [Google Scholar] [CrossRef]
- Zhou, J.H.; Zhang, J.; Sun, D.J.; Ma, Q.; Chen, H.T.; Ma, L.N.; Ding, Y.Z.; Liu, Y.S. The distribution of synonymous codon choice in the translation initiation region of dengue virus. PLoS ONE 2013, 8, e77239. [Google Scholar] [CrossRef]
- Wright, F. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Roychoudhury, S.; Pan, A.; Mukherjee, D. Genus specific evolution of codon usage and nucleotide compositional traits of poxviruses. Virus Genes 2011, 42, 189–199. [Google Scholar] [CrossRef]
- Chen, Y.; Shi, Y.; Deng, H.; Gu, T.; Xu, J.; Ou, J.; Jiang, Z.; Jiao, Y.; Zou, T.; Wang, C. Characterization of the porcine epidemic diarrhea virus codon usage bias. Infect. Genet. Evol. 2014, 28, 95–100. [Google Scholar] [CrossRef]
- Sueoka, N. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. USA 1988, 85, 2653. [Google Scholar] [CrossRef]
- Sueoka, N. Intrastrand parity rules of DNA base composition and usage biases of synonymous codons. J. Mol. Evol. 1995, 40, 318–325. [Google Scholar] [CrossRef]
- Sueoka, N. Translation-coupled violation of Parity Rule 2 in human genes is not the cause of heterogeneity of the DNA G+C content of third codon position. Gene 1999, 238, 53–58. [Google Scholar] [CrossRef]
- Suzuki, H.; Brown, C.J.; Forney, L.J.; Top, E.M. Comparison of correspondence analysis methods for synonymous codon usage in bacteria. DNA Res. 2008, 15, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Wang, Y.; Liu, Z.; Cheng, H.; Xue, Y. HemI: A Toolkit for Illustrating Heatmaps. PLoS ONE 2014, 9, e111988. [Google Scholar] [CrossRef] [PubMed]
- Hanson, G.; Coller, J. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell. Biol. 2017, 19, 20. [Google Scholar] [CrossRef] [PubMed]
- Biswas, K.K.; Palchoudhury, S.; Chakraborty, P.; Bhattacharyya, U.K.; Ghosh, D.K.; Debnath, P.; Ramadugu, C.; Keremane, M.L.; Khetarpal, R.K.; Lee, R.F. Codon Usage Bias Analysis of Citrus tristeza virus: Higher Codon Adaptation to Citrus reticulata Host. Viruses 2019, 11, 331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karniychuk, U.U. Analysis of the synonymous codon usage bias in recently emerged enterovirus D68 strains. Virus Res. 2016, 223, 73–79. [Google Scholar] [CrossRef]
- Kumar, N.; Kulkarni, D.D.; Lee, B.; Kaushik, R.; Bhatia, S.; Sood, R.; Pateriya, A.K.; Bhat, S.; Singh, V.P. Evolution of Codon Usage Bias in Henipaviruses Is Governed by Natural Selection and Is Host-Specific. Viruses 2018, 10, 604. [Google Scholar] [CrossRef] [Green Version]
- Khandia, R.; Singhal, S.; Kumar, U.; Ansari, A.; Tiwari, R.; Dhama, K.; Das, J.; Munjal, A.; Singh, R.K. Analysis of Nipah Virus Codon Usage and Adaptation to Hosts. Front. Microbiol 2019, 10, 886. [Google Scholar] [CrossRef] [Green Version]
- Cristina, J.; Moreno, P.; Moratorio, G.; Musto, H. Genome-wide analysis of codon usage bias in Ebolavirus. Virus Res. 2015, 196, 87–93. [Google Scholar] [CrossRef]
- Cristina, J.; Fajardo, A.; Sonora, M.; Moratorio, G.; Musto, H. A detailed comparative analysis of codon usage bias in Zika virus. Virus Res. 2016, 223, 147–152. [Google Scholar] [CrossRef]
- Kliman, R.M.; Bernal, C.A. Unusual usage of AGG and TTG codons in humans and their viruses. Gene 2005, 352, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Palidwor, G.A.; Perkins, T.J.; Xia, X. A General Model of Codon Bias Due to GC Mutational Bias. PLoS ONE 2010, 5, e13431. [Google Scholar] [CrossRef] [PubMed]
- Svensson, E.I.L.; Berger, D. The Role of Mutation Bias in Adaptive Evolution. Trends Ecol. Evol. 2019, 34, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, L.; He, W.; Zhang, X.; Wen, B.; Wang, C.; Xu, Q.; Li, G.; Zhou, J.; Veit, M.; et al. Genetic Evolution and Molecular Selection of the HE Gene of Influenza C Virus. Viruses 2019, 11, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasrullah, I.; Butt, A.M.; Tahir, S.; Idrees, M.; Tong, Y. Genomic analysis of codon usage shows influence of mutation pressure, natural selection, and host features on Marburg virus evolution. BMC Evol. Biol. 2015, 15, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butt, A.M.; Nasrullah, I.; Tong, Y. Genome-wide analysis of codon usage and influencing factors in chikungunya viruses. PLoS ONE 2014, 9, e90905. [Google Scholar] [CrossRef] [Green Version]
- Giallonardo, F.D.; Schlub, T.E.; Shi, M.; Holmes, E.C. Dinucleotide Composition in Animal RNA Viruses Is Shaped More by Virus Family than by Host Species. J. Virol. 2017, 91, e02381-16. [Google Scholar] [CrossRef] [Green Version]
- Sexton, N.R.; Ebel, G.D. Effects of Arbovirus Multi-Host Life Cycles on Dinucleotide and Codon Usage Patterns. Viruses 2019, 11, 643. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Chen, Y.; Bonning, B.C. RNA virus discovery in insects. Curr. Opin. Insect Sci. 2015, 8, 54–61. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Preferred Codons (aa) | Arachnida | Diptera | Hemiptera | Hymenoptera | Lepidoptera | Non-Insects |
|---|---|---|---|---|---|---|
| GCT(Ala) | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| AGA(Arg) | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| CGT(Arg) | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| AAT(Asn) | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| GAT(Asp) | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| TGT(Cys) | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| CAA(Gln) | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| GAA(Glu) | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| GGA(Gly) | ✗ | ✓ | ✗ | ✗ | ✗ | ✗ |
| GGT(Gly) | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ |
| CAT(His) | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| ATT(Ile) | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| CTT(Leu) | ✗ | ✓ | ✗ | ✗ | ✗ | ✗ |
| TTA(Leu) | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ |
| TTG(Leu) | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| AAA(Lys) | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ |
| TTT(Phe) | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| CCT(Pro) | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| AGT(Ser) | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ |
| TCA(Ser) | ✗ | ✓ | ✗ | ✗ | ✗ | ✗ |
| TCT(Ser) | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| ACT(Thr) | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| UAT(Tyr) | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| GTT(Val) | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, S.-L.; Xia, R.-X. Codon Usage in the Iflaviridae Family Is Not Diverse Though the Family Members Are Isolated from Diverse Host Taxa. Viruses 2019, 11, 1087. https://doi.org/10.3390/v11121087
Shi S-L, Xia R-X. Codon Usage in the Iflaviridae Family Is Not Diverse Though the Family Members Are Isolated from Diverse Host Taxa. Viruses. 2019; 11(12):1087. https://doi.org/10.3390/v11121087
Chicago/Turabian StyleShi, Sheng-Lin, and Run-Xi Xia. 2019. "Codon Usage in the Iflaviridae Family Is Not Diverse Though the Family Members Are Isolated from Diverse Host Taxa" Viruses 11, no. 12: 1087. https://doi.org/10.3390/v11121087
APA StyleShi, S. -L., & Xia, R. -X. (2019). Codon Usage in the Iflaviridae Family Is Not Diverse Though the Family Members Are Isolated from Diverse Host Taxa. Viruses, 11(12), 1087. https://doi.org/10.3390/v11121087