Three YXXL Sequences of a Bovine Leukemia Virus Transmembrane Protein are Independently Required for Fusion Activity by Controlling Expression on the Cell Membrane

Abstract
:1. Introduction
2. Materials and Methods
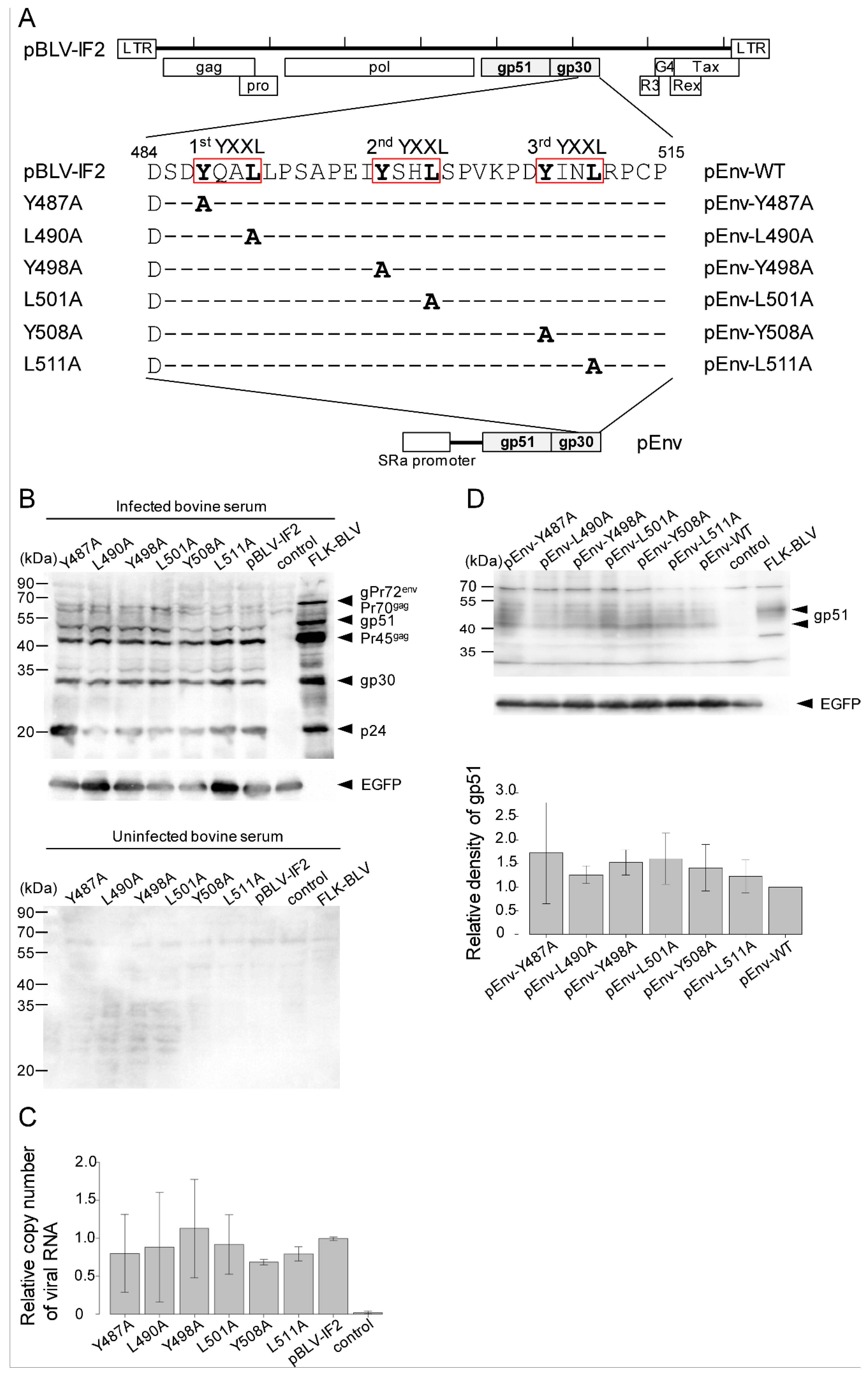
2.1. Plasmids and Construction
2.2. Cell Culture and Transfections
2.3. The Concentration of Virus Particles
2.4. Western Blotting Analysis
2.5. RT-qPCR
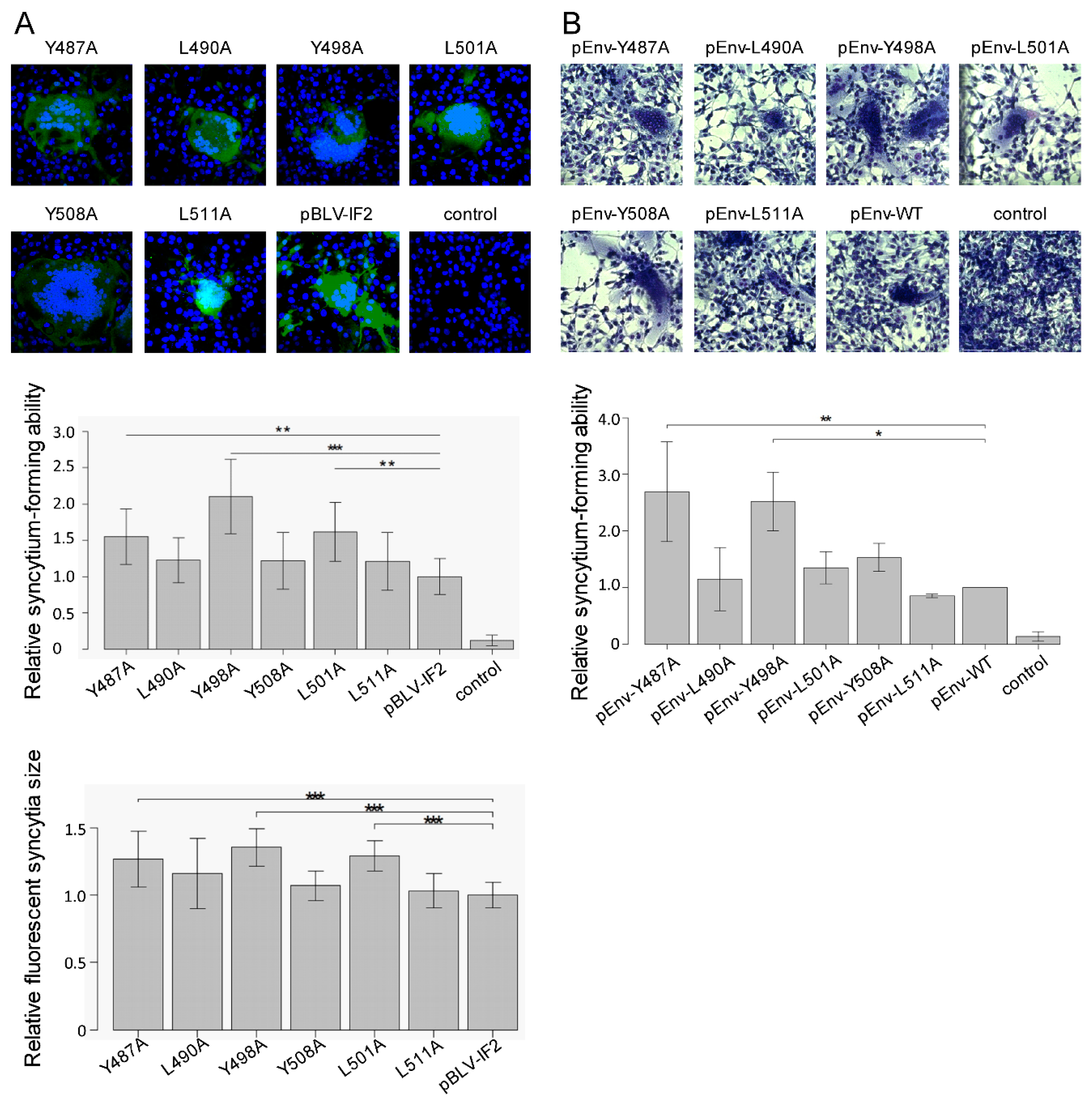
2.6. Syncytia Formation Assay
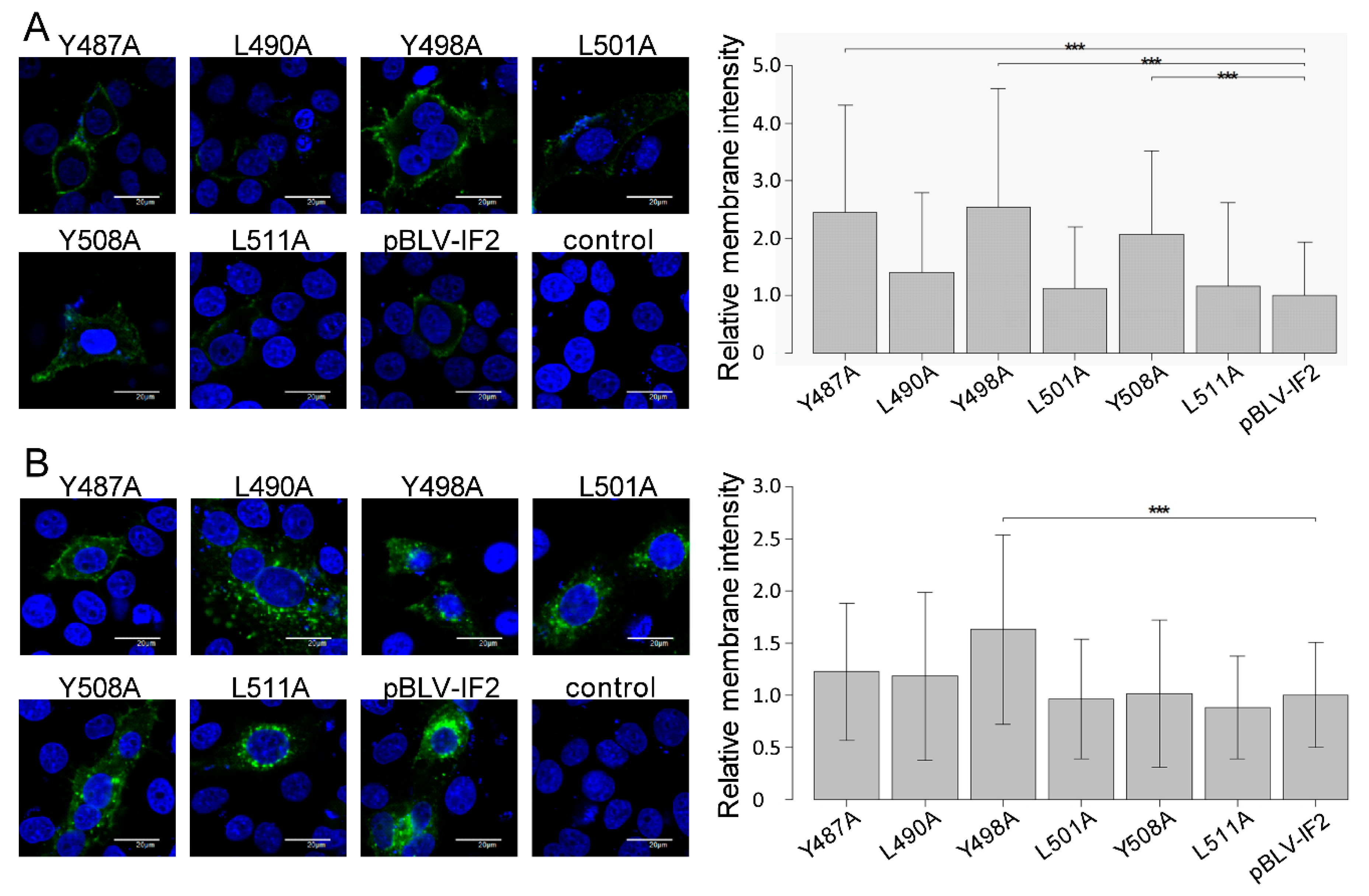
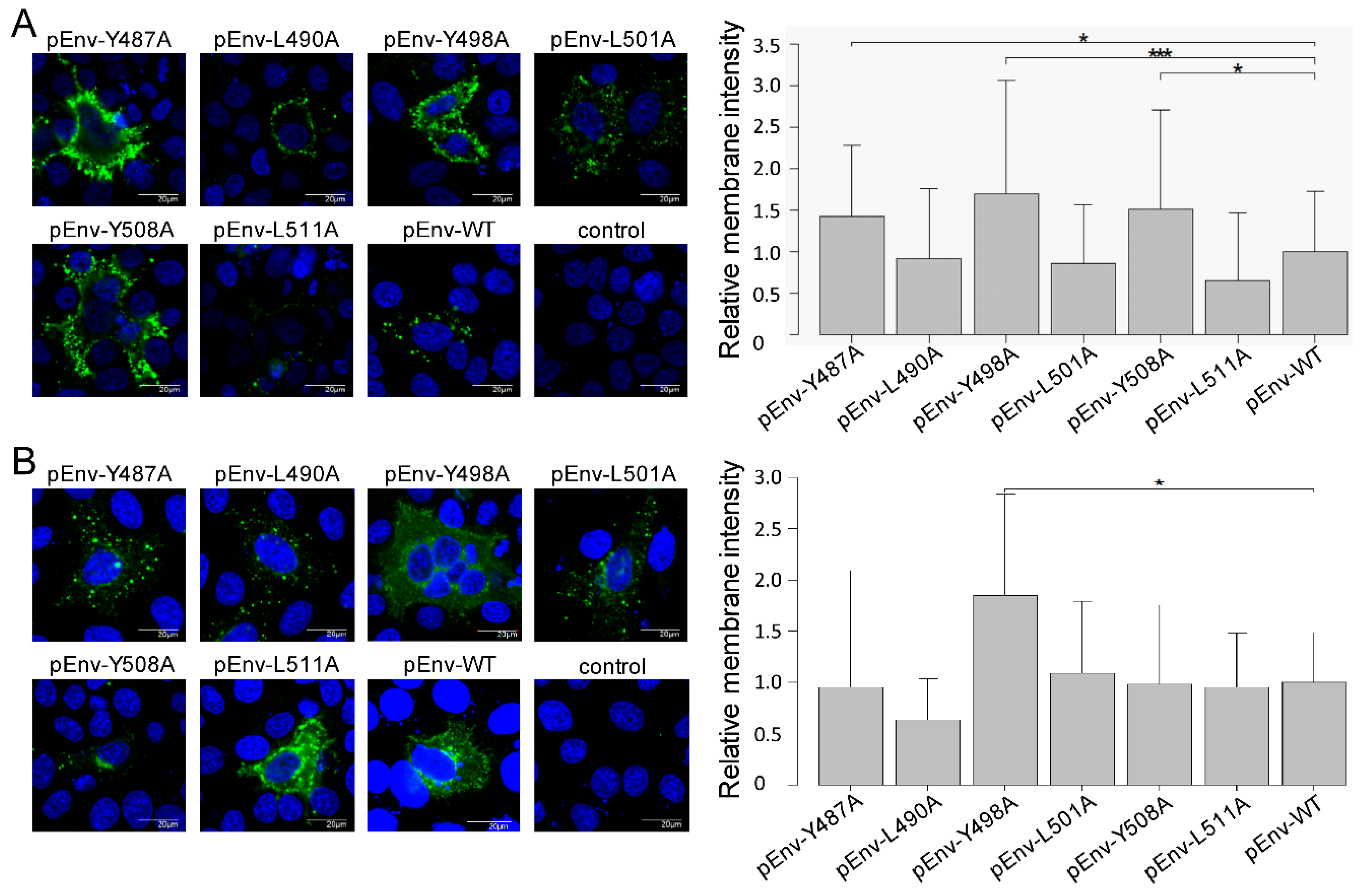
2.7. Fluorescence Microscopy
2.8. Quantification of Intensity of Env Protein on the Cell Membrane
2.9. Quantification of Colocalization
2.10. Statistical Analysis
3. Results
3.1. Mutations in YXXL Sequences in pBLV-IF2 and Env Expression Plasmids Do not Affect the Expression of Viral Protein and the Release of Virus
3.2. Enhanced Syncytium-Forming Ability by All Tyrosine Mutant Forms of the Infectious Molecular Clone pBLV-IF2 and Env Expression Plasmid pEnv
3.3. Enhanced Cell Surface Expression of gp51 by All Tyrosine Mutant Forms of the Infectious Molecular Clone pBLV-IF2 and Env Expression Plasmid pEnv
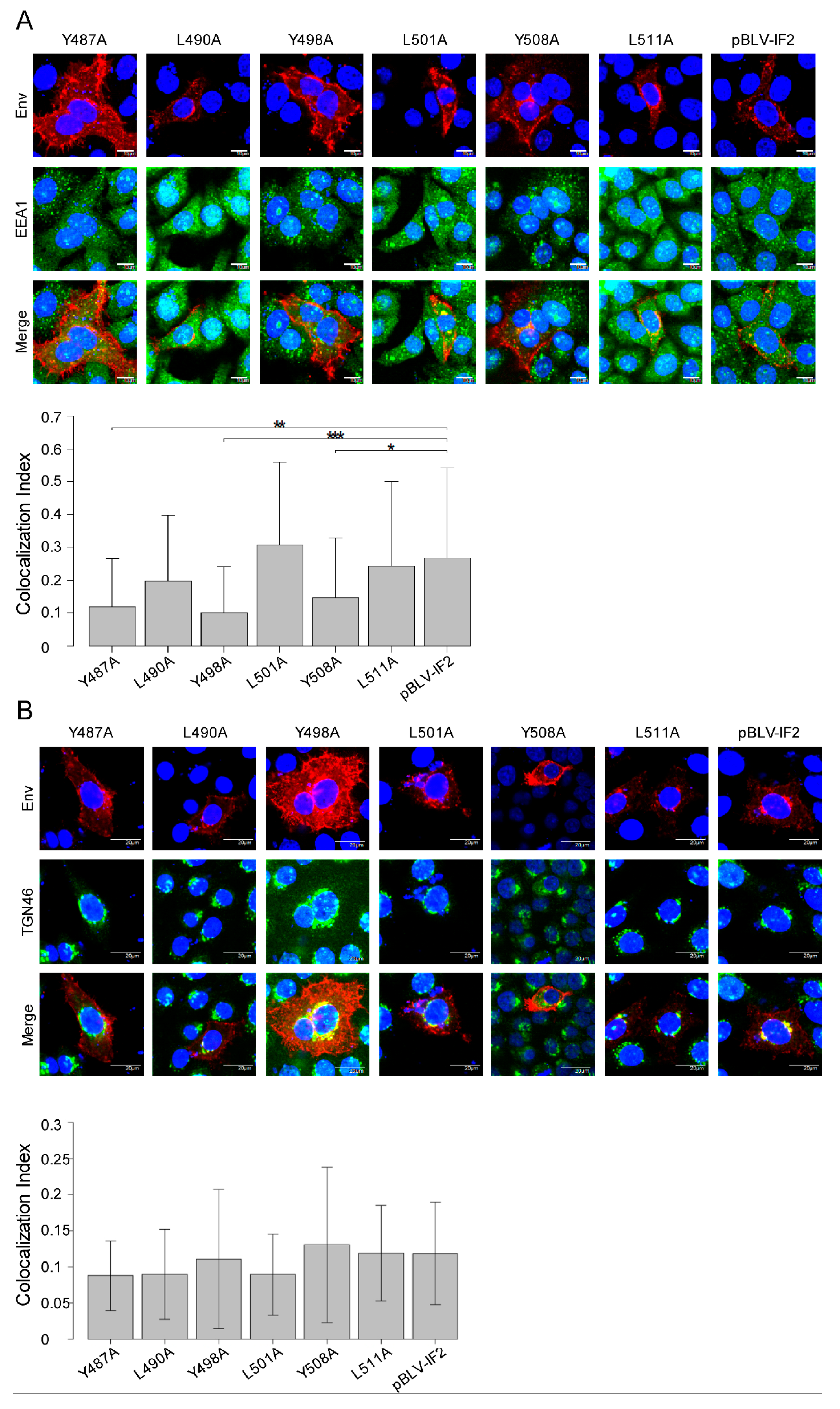
3.4. Localization of gp51 Decreases at the Early Endosome Despite no Effects at the Trans-Golgi Network by Tyrosine Mutant Forms of the Infectious Molecular Clone pBLV-IF2
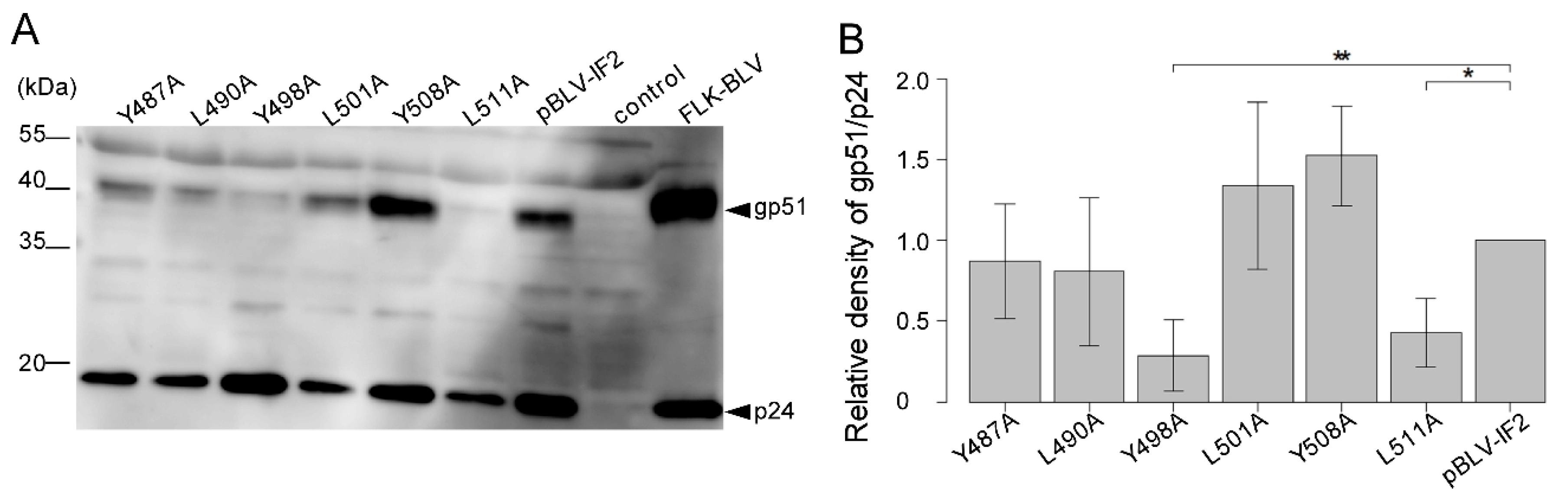
3.5. Y498A and L511A Mutants Interrupt the Incorporation of gp51 into Virions
3.6. Effect of Mutations in the YXXL Sequences on Syncytium-Forming Ability, gp51 Localization, and gp51 Incorporation into Virions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reth, M. Antigen receptor tail clue. Nature 1989, 338, 383–384. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Veazey, R.; Williams, K.; Li, M.; Guo, J.; Neipel, F.; Fleckenstein, B.; Lackner, A.; Desrosiers, R.C.; Jung, J.U. Deregulation of cell growth by the K1 gene of Kaposi’s sarcoma-associated herpesvirus. Nat. Med. 1998, 4, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Katz, E.; Lareef, M.H.; Rassa, J.C.; Grande, S.M.; King, L.B.; Russo, J.; Ross, S.R.; Monroe, J.G. MMTV Env encodes an ITAM responsible for transformation of mammary epithelial cells in three-dimensional culture. J. Exp. Med. 2005, 201, 431–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanier, L.L. Viral immunoreceptor tyrosine-based activation motif (ITAM)-mediated signaling in cell transformation and cancer. Trends Cell Biol. 2006, 16, 388–390. [Google Scholar] [CrossRef]
- Aida, Y.; Murakami, H.; Takahashi, M.; Takeshima, S.N. Mechanisms of pathogenesis induced by bovine leukemia virus as a model for human T-cell leukemia virus. Front. Microbiol. 2013, 4, 328. [Google Scholar] [CrossRef] [Green Version]
- Webster, R.G.; Granoff, A. Encyclopedia of Virology: A-Fur; Academic Press: Cambridge, MA, USA, 1994. [Google Scholar]
- Sagata, N.; Yasunaga, T.; Ohishi, K.; Tsuzuku-Kawamura, J.; Onuma, M.; Ikawa, Y. Comparison of the entire genomes of bovine leukemia virus and human T-cell leukemia virus and characterization of their unidentified open reading frames. EMBO J. 1984, 3, 3231–3237. [Google Scholar] [CrossRef]
- Johnston, E.R.; Radke, K. The SU and TM Envelope protein subunits of bovine leukemia virus are linked by disulfide bonds, both in cells and in virions. J. Virol. 2000, 74, 2930–2935. [Google Scholar] [CrossRef] [Green Version]
- Hunter, E. Viral Entry and Receptors. In Retroviruses; Coffin, J.M., Hughes, S.H., Varmus, H.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997. [Google Scholar]
- Bai, L.; Sato, H.; Kubo, Y.; Wada, S.; Aida, Y. CAT1/SLC7A1 acts as a cellular receptor for bovine leukemia virus infection. FASEB Lett. 2019, 33, 14516–14527. [Google Scholar] [CrossRef] [Green Version]
- Voneche, V.; Callebaut, I.; Kettmann, R.; Brasseur, R.; Burny, A.; Portetelle, D. The 19-27 amino acid segment of gp51 adopts an amphiphilic structure and plays a key role in the fusion events induced by bovine leukemia virus. J. Biol. Chem. 1992, 267, 15193–15197. [Google Scholar]
- Beaufils, P.; Choquet, D.; Mamoun, R.Z.; Malissen, B. The (YXXL/I) 2 signalling motif found in the cytoplasmic segments of the bovine leukaemia virus Envelope protein and Epstein-Barr virus latent membrane protein 2A can elicit early and late lymphocyte activation events. EMBO J. 1993, 12, 5105–5112. [Google Scholar] [CrossRef]
- Murakami, H.; Kuroiwa, T.; Suzuki, K.; Miura, Y.; Sentsui, H. Analysis of Syk expression in bovine lymphoma and persistent lymphocytosis induced by bovine leukemia virus. J. Vet. Med. Sci. 2011, 73, 41–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, V.T.; Stone, D.M.; Pritchard, S.M.; Cantor, G.H. Bovine leukemia virus gp30 transmembrane (TM) protein is not tyrosine phosphorylated: Examining potential interactions with host tyrosine-mediated signaling. Virus Res. 2002, 90, 155–169. [Google Scholar] [CrossRef]
- Inabe, K.; Nishizawa, M.; Tajima, S.; Ikuta, K.; Aida, Y. The YXXL sequences of a transmembrane protein of bovine leukemia virus are required for viral entry and incorporation of viral Envelope protein into virions. J. Virol. 1999, 73, 1293–1301. [Google Scholar]
- Ohno, H.; Stewart, J.; Fournier, M.C.; Bosshart, H.; Rhee, I.; Miyatake, S.; Saito, T.; Gallusser, A.; Kirchhausen, T.; Bonifacino, J.S. Interaction of tyrosine-based sorting signals with clathrin-associated proteins. Science 1995, 269, 1872–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mettlen, M.; Chen, P.H.; Srinivasan, S.; Danuser, G.; Schmid, S.L. Regulation of Clathrin-Mediated Endocytosis. Annu. Rev. Biochem. 2018, 87, 871–896. [Google Scholar] [CrossRef]
- Boge, M.; Wyss, S.; Bonifacino, J.S.; Thali, M. A membrane-proximal tyrosine-based signal mediates internalization of the HIV-1 Envelope glycoprotein via interaction with the AP-2 clathrin adaptor. J. Biol. Chem. 1998, 273, 15773–15778. [Google Scholar] [CrossRef] [Green Version]
- Yuste, E.; Reeves, J.D.; Doms, R.W.; Desrosiers, R.C. Modulation of Env content in virions of simian immunodeficiency virus: Correlation with cell surface expression and virion infectivity. J. Virol. 2004, 78, 6775–6785. [Google Scholar] [CrossRef] [Green Version]
- Delamarre, L.; Pique, C.; Rosenberg, A.R.; Blot, V.; Grange, M.P.; Le Blanc, I.; Dokhelar, M.C. The Y-S-L-I tyrosine-based motif in the cytoplasmic domain of the human T-cell leukemia virus type 1 Envelope is essential for cell-to-cell transmission. J. Virol. 1999, 73, 9659–9663. [Google Scholar]
- Lambele, M.; Labrosse, B.; Roch, E.; Moreau, A.; Verrier, B.; Barin, F.; Roingeard, P.; Mammano, F.; Brand, D. Impact of natural polymorphism within the gp41 cytoplasmic tail of human immunodeficiency virus type 1 on the intracellular distribution of Envelope glycoproteins and viral assembly. J. Virol. 2007, 81, 125–140. [Google Scholar] [CrossRef] [Green Version]
- Novakovic, S.; Sawai, E.T.; Radke, K. Dileucine and YXXL motifs in the cytoplasmic tail of the bovine leukemia virus transmembrane Envelope protein affect protein expression on the cell surface. J. Virol. 2004, 78, 8301–8311. [Google Scholar] [CrossRef] [Green Version]
- De Gassart, A.; Trentin, B.; Martin, M.; Hocquellet, A.; Bette-Bobillo, P.; Mamoun, R.; Vidal, M. Exosomal sorting of the cytoplasmic domain of bovine leukemia virus TM Env protein. Cell Biol. Int. 2009, 33, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Willems, L.; Gatot, J.S.; Mammerickx, M.; Portetelle, D.; Burny, A.; Kerkhofs, P.; Kettmann, R. The YXXL signalling motifs of the bovine leukemia virus transmembrane protein are required for in vivo infection and maintenance of high viral loads. J. Virol. 1995, 69, 4137–4141. [Google Scholar] [PubMed]
- Inabe, K.; Ikuta, K.; Aida, Y. Transmission and propagation in cell culture of virus produced by cells transfected with an infectious molecular clone of bovine leukemia virus. Virology 1998, 245, 53–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cormack, B.P.; Valdivia, R.H.; Falkow, S. FACS-optimized mutants of the green fluorescent protein (GFP). Gene 1996, 173, 33–38. [Google Scholar] [CrossRef]
- Hagiwara, K.; Murakami, T.; Xue, G.; Shimizu, Y.; Takeda, E.; Hashimoto, Y.; Honda, K.; Kondoh, Y.; Osada, H.; Tsunetsugu-Yokota, Y.; et al. Identification of a novel Vpr-binding compound that inhibits HIV-1 multiplication in macrophages by chemical array. Biochem. Biophys. Res. Commun. 2010, 403, 40–45. [Google Scholar] [CrossRef]
- Sato, H.; Watanuki, S.; Bai, L.; Borjigin, L.; Ishizaki, H.; Matsumoto, Y.; Hachiya, Y.; Sentsui, H.; Aida, Y. A sensitive luminescence syncytium induction assay (LuSIA) based on a reporter plasmid containing a mutation in the glucocorticoid response element in the long terminal repeat U3 region of bovine leukemia virus. Virol. J. 2019, 16, 66. [Google Scholar] [CrossRef] [Green Version]
- Bai, L.; Yokoyama, K.; Watanuki, S.; Ishizaki, H.; Takeshima, S.N.; Aida, Y. Development of a new recombinant p24 ELISA system for diagnosis of bovine leukemia virus in serum and milk. Arch. Virol. 2019, 164, 201–211. [Google Scholar] [CrossRef]
- Jimba, M.; Takeshima, S.N.; Matoba, K.; Endoh, D.; Aida, Y. BLV-CoCoMo-qPCR: Quantitation of bovine leukemia virus proviral load using the CoCoMo algorithm. Retrovirology 2010, 7, 91. [Google Scholar] [CrossRef] [Green Version]
- Jimba, M.; Takeshima, S.N.; Murakami, H.; Kohara, J.; Kobayashi, N.; Matsuhashi, T.; Ohmori, T.; Nunoya, T.; Aida, Y. BLV-CoCoMo-qPCR: A useful tool for evaluating bovine leukemia virus infection status. BMC Vet. Res. 2012, 8, 167. [Google Scholar] [CrossRef] [Green Version]
- Takeshima, S.N.; Watanuki, S.; Ishizaki, H.; Matoba, K.; Aida, Y. Development of a direct blood-based PCR system to detect BLV provirus using CoCoMo primers. Arch. Virol. 2016, 161, 1539–1546. [Google Scholar] [CrossRef]
- Sato, H.; Watanuki, S.; Murakami, H.; Sato, R.; Ishizaki, H.; Aida, Y. Development of a luminescence syncytium induction assay (LuSIA) for easily detecting and quantitatively measuring bovine leukemia virus infection. Arch. Virol. 2018, 163, 1519–1530. [Google Scholar] [CrossRef] [PubMed]
- Villalta, J.I.; Galli, S.; Iacaruso, M.F.; Antico Arciuch, V.G.; Poderoso, J.J.; Jares-Erijman, E.A.; Pietrasanta, L.I. New algorithm to determine true colocalization in combination with image restoration and time-lapse confocal microscopy to MAP kinases in mitochondria. PLoS ONE 2011, 6, e19031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lifson, J.D.; Reyes, G.R.; McGrath, M.S.; Stein, B.S.; Engleman, E.G. AIDS retrovirus induced cytopathology: Giant cell formation and involvement of CD4 antigen. Science 1986, 232, 1123–1127. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Fischman, D.A.; Steck, T.L. Selective solubilization of proteins and phospholipids from red blood cell membranes by nonionic detergents. J. Supramol. Struct. 1973, 1, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ma, T.; Lau, P.K.; Wang, J.; Zhao, T.; Du, S.; Loy, M.M.T.; Guo, Y. Visualization of Protein Sorting at the Trans-Golgi Network and Endosomes Through Super-Resolution Imaging. Front. Cell Dev. Biol. 2019, 7, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, F.T.; Callaghan, J.M.; Steele-Mortimer, O.; Stenmark, H.; Parton, R.G.; Campbell, P.L.; McCluskey, J.; Yeo, J.P.; Tock, E.P.; Toh, B.H. EEA1, an early endosome-associated protein. EEA1 is a conserved alpha-helical peripheral membrane protein flanked by cysteine “fingers” and contains a calmodulin-binding IQ motif. J. Biol. Chem. 1995, 270, 13503–13511. [Google Scholar] [CrossRef] [Green Version]
- Ganley, I.G.; Espinosa, E.; Pfeffer, S.R. A syntaxin 10-SNARE complex distinguishes two distinct transport routes from endosomes to the trans-Golgi in human cells. J. Cell Biol. 2008, 180, 159–172. [Google Scholar] [CrossRef] [Green Version]
- Bruck, C.; Portetelle, D.; Burny, A.; Zavada, J. Topographical analysis by monoclonal antibodies of BLV-gp51 epitopes involved in viral functions. Virology 1982, 122, 353–362. [Google Scholar] [CrossRef]
- Gatot, J.S.; Callebaut, I.; Mornon, J.P.; Portetelle, D.; Burny, A.; Kerkhofs, P.; Kettmann, R.; Willems, L. Conservative mutations in the immunosuppressive region of the bovine leukemia virus transmembrane protein affect fusion but not infectivity in vivo. J. Biol. Chem. 1998, 273, 12870–12880. [Google Scholar] [CrossRef] [Green Version]
- Johnston, E.R.; Albritton, L.M.; Radke, K. Envelope proteins containing single amino acid substitutions support a structural model of the receptor-binding domain of bovine leukemia virus surface protein. J. Virol. 2002, 76, 10861–10872. [Google Scholar] [CrossRef] [Green Version]
- Spies, C.P.; Compans, R.W. Effects of cytoplasmic domain length on cell surface expression and syncytium-forming capacity of the simian immunodeficiency virus Envelope glycoprotein. Virology 1994, 203, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Sauter, M.M.; Pelchen-Matthews, A.; Bron, R.; Marsh, M.; LaBranche, C.C.; Vance, P.J.; Romano, J.; Haggarty, B.S.; Hart, T.K.; Lee, W.M.; et al. An internalization signal in the simian immunodeficiency virus transmembrane protein cytoplasmic domain modulates expression of Envelope glycoproteins on the cell surface. J. Cell Biol. 1996, 132, 795–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyss, S.; Berlioz-Torrent, C.; Boge, M.; Blot, G.; Honing, S.; Benarous, R.; Thali, M. The highly conserved C-terminal dileucine motif in the cytosolic domain of the human immunodeficiency virus type 1 Envelope glycoprotein is critical for its association with the AP-1 clathrin adaptor [correction of adapter]. J. Virol. 2001, 75, 2982–2992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, M.; Chu, H.; Chen, X.; Choi, J.; Wen, X.; Hammonds, J.; Ding, L.; Hunter, E.; Spearman, P. A tyrosine-based motif in the HIV-1 Envelope glycoprotein tail mediates cell-type- and Rab11-FIP1C-dependent incorporation into virions. Proc. Natl. Acad. Sci. USA 2015, 112, 7575–7580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirschman, J.; Qi, M.; Ding, L.; Hammonds, J.; Dienger-Stambaugh, K.; Wang, J.J.; Lapierre, L.A.; Goldenring, J.R.; Spearman, P. HIV-1 Envelope Glycoprotein Trafficking through the Endosomal Recycling Compartment Is Required for Particle Incorporation. J. Virol. 2018, 92, e01893-17. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid | YXXL Sequence | Mutation | Syncytium-Forming Ability 1 | Localization | Incorporation in Virions 5 | ||
|---|---|---|---|---|---|---|---|
| Whole 2 | Early Endosome 3 | Trans-Golgi Network 4 | |||||
| pBLV-IF2 | 1st YXXL | Y487A | +++ | M | − | + | + |
| L490A | + | C | + | + | + | ||
| 2nd YXXL | Y498A | ++++ | M | − | + | − | |
| L501A | + | C | + | + | + | ||
| 3rd YXXL | Y508A | +++ | M | − | + | + | |
| L511A | + | C | + | + | − | ||
| WT | + | C | + | + | + | ||
| pEnv | 1st YXXL | Y487A | ++ | M | |||
| L490A | + | C | |||||
| 2nd YXXL | Y498A | ++ | M | ||||
| L501A | + | C | |||||
| 3rd YXXL | Y508A | + | M | ||||
| L511A | + | C | |||||
| WT | + | C | |||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsuura, R.; Inabe, K.; Otsuki, H.; Kurokawa, K.; Dohmae, N.; Aida, Y. Three YXXL Sequences of a Bovine Leukemia Virus Transmembrane Protein are Independently Required for Fusion Activity by Controlling Expression on the Cell Membrane. Viruses 2019, 11, 1140. https://doi.org/10.3390/v11121140
Matsuura R, Inabe K, Otsuki H, Kurokawa K, Dohmae N, Aida Y. Three YXXL Sequences of a Bovine Leukemia Virus Transmembrane Protein are Independently Required for Fusion Activity by Controlling Expression on the Cell Membrane. Viruses. 2019; 11(12):1140. https://doi.org/10.3390/v11121140
Chicago/Turabian StyleMatsuura, Ryosuke, Kazunori Inabe, Hiroyuki Otsuki, Kazuo Kurokawa, Naoshi Dohmae, and Yoko Aida. 2019. "Three YXXL Sequences of a Bovine Leukemia Virus Transmembrane Protein are Independently Required for Fusion Activity by Controlling Expression on the Cell Membrane" Viruses 11, no. 12: 1140. https://doi.org/10.3390/v11121140
APA StyleMatsuura, R., Inabe, K., Otsuki, H., Kurokawa, K., Dohmae, N., & Aida, Y. (2019). Three YXXL Sequences of a Bovine Leukemia Virus Transmembrane Protein are Independently Required for Fusion Activity by Controlling Expression on the Cell Membrane. Viruses, 11(12), 1140. https://doi.org/10.3390/v11121140