Characterization of Host and Bacterial Contributions to Lung Barrier Dysfunction Following Co-infection with 2009 Pandemic Influenza and Methicillin Resistant Staphylococcus aureus

 , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus, Bacteria, and Cell Conditions
2.2. Viral and Bacterial Infection of Alveolar Epithelial Cells
2.3. Quantification of Bacterial and Viral Replication Kinetics
2.4. Kinome Peptide Array Analysis
2.5. Pathway Overrepresentation and Gene Ontology Analysis
2.6. RNA Extraction, cDNA Synthesis, and Quantitative qPCR
2.7. Barrier Integrity Determination
3. Results
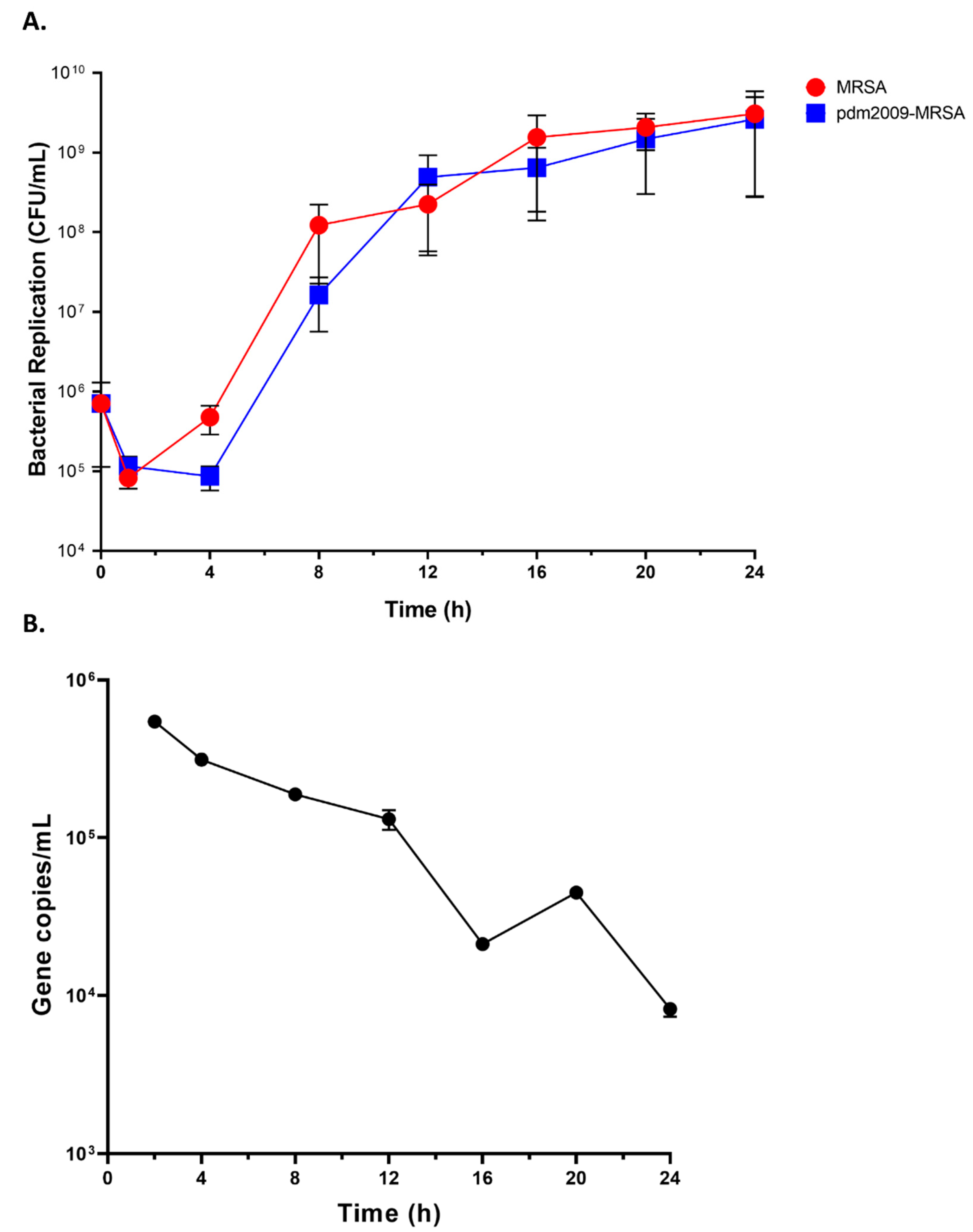
3.1. MRSA Replication Kinetics Are Similar during Bacterial Infection-Alone and pdm2009-MRSA Infection
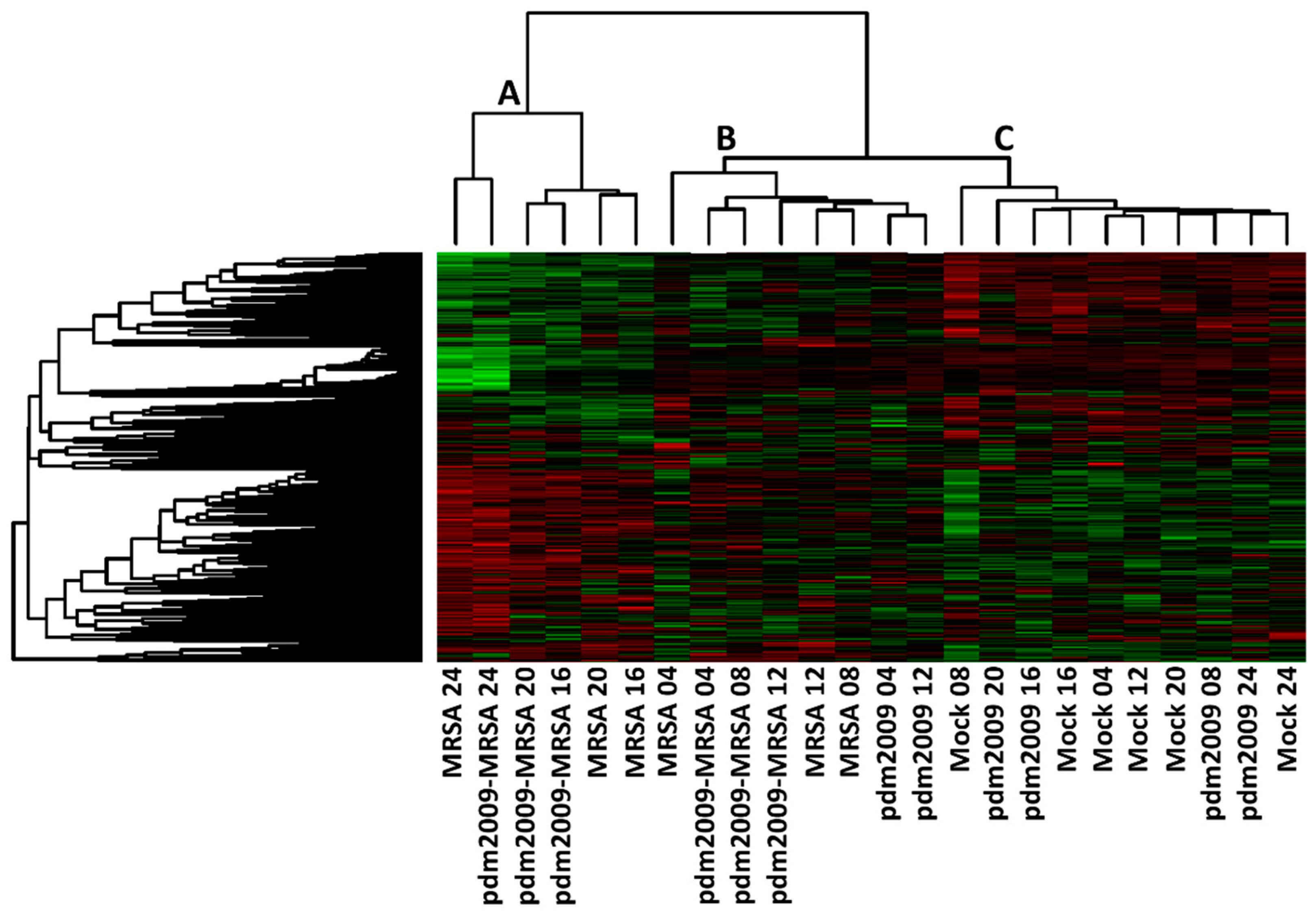
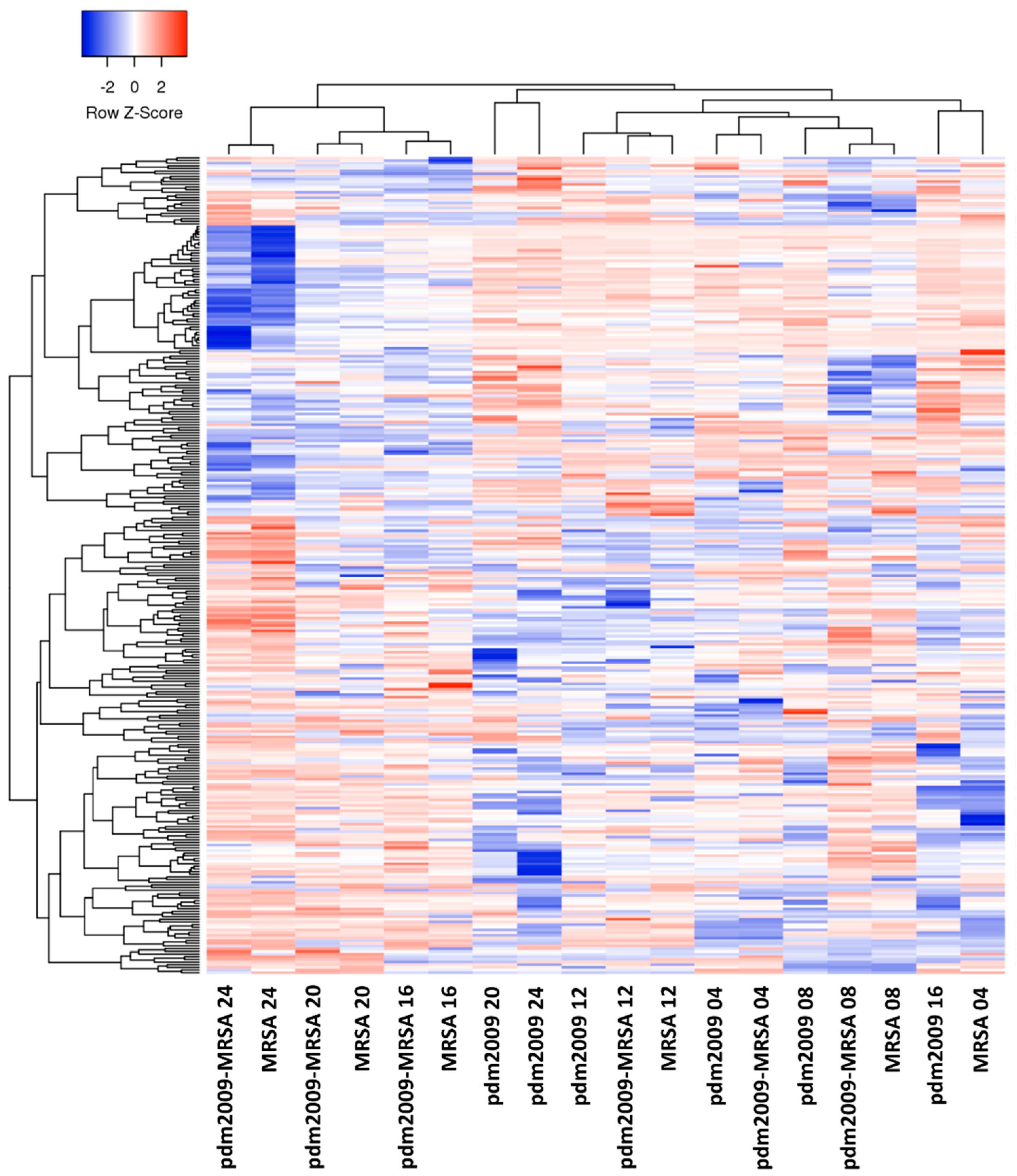
3.2. Temporal Analysis of Host Kinome Responses During pdm2009-MRSA Infection
3.3. Bacterial Invasion- and Attachment-Related Virulence Factor Expression Patterns Are Modulated Early during pdm2009-MRSA Infection
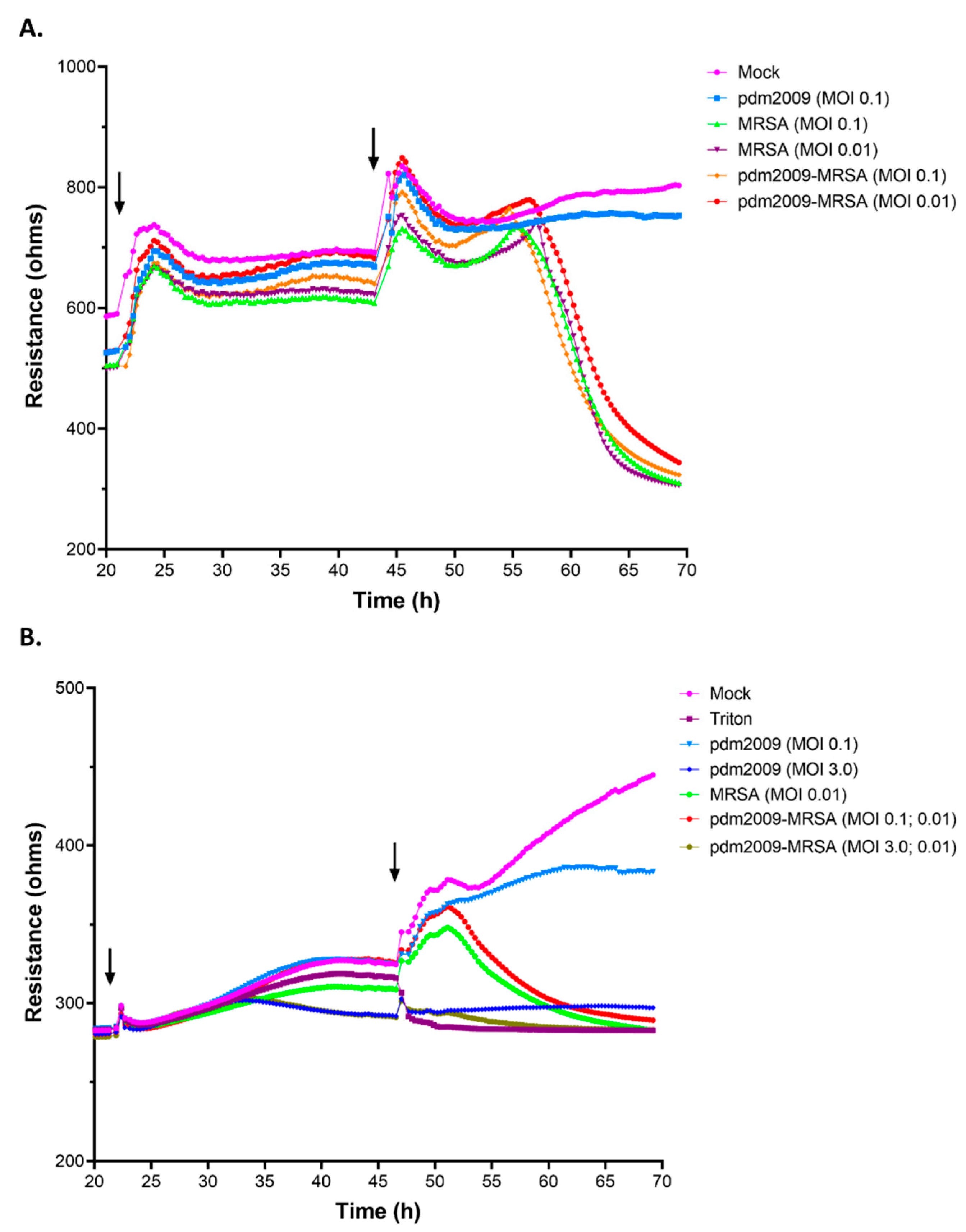
3.4. MRSA-Alone and pdm2009-MRSA Infection Result in Alveolar Epithelial Cell Barrier Dysfunction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis Primers 2018, 4, 3. [Google Scholar] [CrossRef]
- Stephenson, I. Influenza: Molecular virology. Expert Rev. Vaccines 2010, 9, 719–720. [Google Scholar] [CrossRef]
- Taubenberger, J.K.; Morens, D.M. Pandemic influenza—Including a risk assessment of h5n1. Rev. Sci. Tech. 2009, 28, 187–202. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention (CDC). Influenza. Available online: https://www.cdc.gov/flu/index.htm (accessed on 1 November 2018).
- Ghebrehewet, S.; MacPherson, P.; Ho, A. Influenza. BMJ 2016, 355, i6258. [Google Scholar] [CrossRef]
- Moghadami, M. A narrative review of influenza: A seasonal and pandemic disease. Iran. J. Med. Sci 2017, 42, 2–13. [Google Scholar]
- McCullers, J.A. The co-pathogenesis of influenza viruses with bacteria in the lung. Nat. Rev. Microbiol. 2014, 12, 252–262. [Google Scholar] [CrossRef]
- Taubenberger, J.K.; Morens, D.M. 1918 influenza: The mother of all pandemics. Emerg Infect. Dis 2006, 12, 15–22. [Google Scholar] [CrossRef]
- Morens, D.M.; Taubenberger, J.K.; Fauci, A.S. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: Implications for pandemic influenza preparedness. J. Infect. Dis. 2008, 198, 962–970. [Google Scholar] [CrossRef]
- Rudd, J.M.; Ashar, H.K.; Chow, V.T.; Teluguakula, N. Lethal synergism between influenza and streptococcus pneumoniae. J. Infect. Pulm. Dis. 2016, 2. [Google Scholar] [CrossRef]
- Rynda-Apple, A.; Robinson, K.M.; Alcorn, J.F. Influenza and bacterial superinfection: Illuminating the immunologic mechanisms of disease. Infect. Immun. 2015, 83, 3764–3770. [Google Scholar] [CrossRef]
- Louria, D.B.; Blumenfeld, H.L.; Ellis, J.T.; Kilbourne, E.D.; Rogers, D.E. Studies on influenza in the pandemic of 1957-1958. Ii. Pulmonary complications of influenza. J. Clin. Investig. 1959, 38, 213–265. [Google Scholar] [CrossRef]
- Mark, D.D. The pathology of 1957 (asian) influenza. Am. Rev. Tuberc. 1959, 79, 440–449. [Google Scholar]
- Oseasohn, R.; Adelson, L.; Kaji, M. Clinicopathologic study of thirty-three fatal cases of asian influenza. N. Engl. J. Med. 1959, 260, 509–518. [Google Scholar] [CrossRef]
- Oswald, N.C.; Shooter, R.A.; Curwen, M.P. Pneumonia complicating asian influenza. Br. Med. J. 1958, 2, 1305–1311. [Google Scholar] [CrossRef]
- Robertson, L.; Caley, J.P.; Moore, J. Importance of staphylococcus aureus in pneumonia in the 1957 epidemic of influenza a. Lancet 1958, 2, 233–236. [Google Scholar] [CrossRef]
- Chertow, D.S.; Memoli, M.J. Bacterial coinfection in influenza: A grand rounds review. JAMA 2013, 309, 275–282. [Google Scholar] [CrossRef]
- Louie, J.; Jean, C.; Chen, T.H.; Park, S.; Ueki, R.; Harper, T.; Chmara, E.; Myers, J.; Stoppacher, R.; Catanese, C.; et al. Bacterial coinfections in lung tissue specimens from fatal cases of 2009 pandemic influenza a (h1n1)—United states, may-august 2009. Mmwr Morb Mortal Wkly. Rep. 2009, 58, 1071–1074. [Google Scholar]
- Gill, J.R.; Sheng, Z.M.; Ely, S.F.; Guinee, D.G.; Beasley, M.B.; Suh, J.; Deshpande, C.; Mollura, D.J.; Morens, D.M.; Bray, M.; et al. Pulmonary pathologic findings of fatal 2009 pandemic influenza a/h1n1 viral infections. Arch. Pathol. Lab. Med. 2010, 134, 235–243. [Google Scholar]
- Mauad, T.; Hajjar, L.A.; Callegari, G.D.; da Silva, L.F.; Schout, D.; Galas, F.R.; Alves, V.A.; Malheiros, D.M.; Auler, J.O., Jr.; Ferreira, A.F.; et al. Lung pathology in fatal novel human influenza a (h1n1) infection. Am. J. Respir. Crit. Care Med. 2010, 181, 72–79. [Google Scholar] [CrossRef]
- Gillet, Y.; Vanhems, P.; Lina, G.; Bes, M.; Vandenesch, F.; Floret, D.; Etienne, J. Factors predicting mortality in necrotizing community-acquired pneumonia caused by Staphylococcus aureus containing panton-valentine leukocidin. Clin. Infect. Dis. 2007, 45, 315–321. [Google Scholar] [CrossRef]
- Randolph, A.G.; Vaughn, F.; Sullivan, R.; Rubinson, L.; Thompson, B.T.; Yoon, G.; Smoot, E.; Rice, T.W.; Loftis, L.L.; Helfaer, M.; et al. Critically ill children during the 2009-2010 influenza pandemic in the united states. Pediatrics 2011, 128, e1450–e1458. [Google Scholar] [CrossRef]
- Rice, T.W.; Rubinson, L.; Uyeki, T.M.; Vaughn, F.L.; John, B.B.; Miller, R.R., 3rd; Higgs, E.; Randolph, A.G.; Smoot, B.E.; Thompson, B.T.; et al. Critical illness from 2009 pandemic influenza a virus and bacterial coinfection in the united states. Crit. Care Med. 2012, 40, 1487–1498. [Google Scholar] [CrossRef]
- Tong, S.Y.C.; Davis, J.S.; Eichenberger, E.; Holland, T.L., Jr.; Vance, G. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef]
- Bellinghausen, C.; Rohde, G.G.U.; Savelkoul, P.H.M.; Wouters, E.F.M.; Stassen, F.R.M. Viral-bacterial interactions in the respiratory tract. J. Gen. Virol. 2016, 97, 3089–3102. [Google Scholar] [CrossRef]
- Okamoto, S.; Kawabata, S.; Nakagawa, I.; Okuno, Y.; Goto, T.; Sano, K.; Hamada, S. Influenza a virus-infected hosts boost an invasive type of streptococcus pyogenes infection in mice. J. Virol. 2003, 77, 4104–4112. [Google Scholar] [CrossRef]
- Herold, S.; Becker, C.; Ridge, K.M.; Budinger, G.R.S. Influenza virus-induced lung injury: Pathogenesis and implications for treatment. Eur. Respir. J. 2015, 45, 1463–1478. [Google Scholar] [CrossRef]
- Short, K.R.; Kasper, J.; van der Aa, S.; Andeweg, A.C.; Zaaraoui-Boutahar, F.; Goeijenbier, M.; Richard, M.; Herold, S.; Becker, C.; Scott, D.P.; et al. Influenza virus damages the alveolar barrier by disrupting epithelial cell tight junctions. Eur. Respir. J. 2016, 47, 954–966. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, A.K.; Vipat, V.C.; Mukherjee, S.; Singh, R.; Pawar, S.D.; Mishra, A.C. Host gene expression profiling in influenza a virus-infected lung epithelial (a549) cells: A comparative analysis between highly pathogenic and modified h5n1 viruses. Virol. J. 2010, 7, 219. [Google Scholar] [CrossRef]
- Dapat, C.; Saito, R.; Suzuki, H.; Horigome, T. Quantitative phosphoproteomic analysis of host responses in human lung epithelial (a549) cells during influenza virus infection. Virus Res. 2014, 179, 53–63. [Google Scholar] [CrossRef]
- Lin, X.; Wang, R.; Zou, W.; Sun, X.; Liu, X.; Zhao, L.; Wang, S.; Jin, M. The influenza virus h5n1 infection can induce ros production for viral replication and host cell death in a549 cells modulated by human cu/zn superoxide dismutase (sod1) overexpression. Viruses 2016, 8, 13. [Google Scholar] [CrossRef]
- Te Velthuis, A.J.W.; Long, J.C.; Bauer, D.L.V.; Fan, R.L.Y.; Yen, H.L.; Sharps, J.; Siegers, J.Y.; Killip, M.J.; French, H.; Oliva-Martin, M.J.; et al. Mini viral rnas act as innate immune agonists during influenza virus infection. Nat. Microbiol. 2018, 3, 1234–1242. [Google Scholar] [CrossRef]
- Zhou, B.; Li, J.; Liang, X.; Yang, Z.; Jiang, Z. Transcriptome profiling of influenza a virus-infected lung epithelial (a549) cells with lariciresinol-4-beta-d-glucopyranoside treatment. PLoS ONE 2017, 12, e0173058. [Google Scholar]
- Qi, L.; Kash, J.C.; Dugan, V.G.; Wang, R.; Jin, G.; Cunningham, R.E.; Taubenberger, J.K. Taubenberger. Role of sialic acid binding specificity of the 1918 influenza virus hemagglutinin protein in virulence and pathogenesis for mice. J. Virol. 2009, 83, 3754–3761. [Google Scholar] [CrossRef]
- Kindrachuk, J.; Ork, B.; Hart, B.J.; Mazur, S.; Holbrook, M.R.; Frieman, M.B.; Traynor, D.; Johnson, R.F.; Dyall, J.; Kuhn, J.H.; et al. Antiviral potential of erk/mapk and pi3k/akt/mtor signaling modulation for middle east respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob Agents Chemother. 2015, 59, 1088–1099. [Google Scholar] [CrossRef]
- Kindrachuk, J.; Wahl-Jensen, V.; Safronetz, D.; Trost, B.; Hoenen, T.; Arsenault, R.; Feldmann, F.; Traynor, D.; Postnikova, E.; Kusalik, A.; et al. Ebola virus modulates transforming growth factor beta signaling and cellular markers of mesenchyme-like transition in hepatocytes. J. Virol. 2014, 88, 9877–9892. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.; Kindrachuk, J.; Maattanen, P.; Napper, S.; Kusalik, A. Piika 2: An expanded, web-based platform for analysis of kinome microarray data. PLoS ONE 2013, 8, e80837. [Google Scholar] [CrossRef] [PubMed]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef] [PubMed]
- Lynn, D.J.; Winsor, G.L.; Chan, C.; Richard, N.; Laird, M.R.; Barsky, A.; Gardy, J.L.; Roche, F.M.; Chan, T.H.; Shah, N.; et al. Innatedb: Facilitating systems-level analyses of the mammalian innate immune response. Mol. Syst. Biol. 2008, 4, 218. [Google Scholar] [CrossRef]
- Li, Y.; Arsenault, R.J.; Trost, B.; Slind, J.; Griebel, P.J.; Napper, S.; Kusalik, A. A systematic approach for analysis of peptide array kinome data. Sci. Signal. 2012, 5, pl2. [Google Scholar] [CrossRef]
- Atshan, S.S.; Shamsudin, M.N.; Karunanidhi, A.; van Belkum, A.; Lung, L.T.; Sekawi, Z.; Nathan, J.J.; Ling, K.H.; Seng, J.S.; Ali, A.M.; et al. Quantitative pcr analysis of genes expressed during biofilm development of methicillin resistant staphylococcus aureus (mrsa). Infect. Genet. Evol. 2013, 18, 106–112. [Google Scholar] [CrossRef]
- Miyake, T.; Soda, K.; Itoh, Y.; Sakoda, Y.; Ishigaki, H.; Nagata, T.; Ishida, H.; Nakayama, M.; Ozaki, H.; Tsuchiya, H.; et al. Amelioration of pneumonia with streptococcus pneumoniae infection by inoculation with a vaccine against highly pathogenic avian influenza virus in a non-human primate mixed infection model. J. Med. Primatol. 2010, 39, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; McCullers, J.A. Secondary bacterial infections in influenza virus infection pathogenesis. Curr. Top. Microbiol. Immunol. 2014, 385, 327–356. [Google Scholar] [PubMed]
- Chertow, D.S.; Kindrachuk, J.; Sheng, Z.M.; Pujanauski, L.M.; Cooper, K.; Nogee, D.; Claire, M.S.; Solomon, J.; Perry, D.; Sayre, P.; et al. Influenza a and methicillin-resistant staphylococcus aureus co-infection in rhesus macaques—A model of severe pneumonia. Antivir. Res. 2016, 129, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Kindrachuk, J.; Falcinelli, S.; Wada, J.; Kuhn, J.H.; Hensley, L.E.; Jahrling, P.B. Systems kinomics for characterizing host responses to high-consequence pathogens at the nih/niaid integrated research facility-frederick. Pathog. Dis. 2014, 71, 190–198. [Google Scholar] [CrossRef]
- Arsenault, R.; Griebel, P.; Napper, S. Peptide arrays for kinome analysis: New opportunities and remaining challenges. Proteomics 2011, 11, 4595–4609. [Google Scholar] [CrossRef] [PubMed]
- Stolwijk, J.A.; Matrougui, K.; Renken, C.W.; Trebak, M. Impedance analysis of gpcr-mediated changes in endothelial barrier function: Overview and fundamental considerations for stable and reproducible measurements. Pflügers Arch. 2014, 467, 2193–2218. [Google Scholar] [CrossRef] [PubMed]
- Powers, M.E.; Bubeck Wardenburg, J. Igniting the fire: Staphylococcus aureus virulence factors in the pathogenesis of sepsis. PLoS Pathog. 2014, 10, e1003871. [Google Scholar] [CrossRef] [PubMed]
- Gouaux, E.; Hobaugh, M.; Song, L. A-hemolysin, y-hemolysin, and leukocidin from staphylococcus aureus: Distant in sequence but similar in structure. Protein Sci. 1997, 6, 2631–2635. [Google Scholar] [CrossRef] [PubMed]
- Wardenburg, J.B.; Bae, T.; Otto, M.; DeLeo, F.R.; Schneewind, O. Poring over pores: A-hemolysin and panton-valentine leukocidin in staphylococcus aureus pneumonia. Nat. Med. 2007, 13, 1405–1406. [Google Scholar] [CrossRef]
- Murai, M.; Moriyama, H.; Hata, E.; Takeuchi, F.; Amemura-Maekawa, J. Variation and association of firbonectin-binding protein genes fnba and fnbb in staphylococcus aureus japanese isolates. Microbiol. Immunol. 2016, 60, 312–325. [Google Scholar] [CrossRef]
- Shinji, H.; Yosizawa, Y.; Tajima, A.; Sugimoto, S.; Seki, K.; Mizunoe, Y. Role of fibronectin-binding proteins a and b in in vitro cellular infections and in vivo septic infections by Staphylococcus aureus. Infect. Immun. 2011, 2215–2223. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Cheng, A.G.; Kim, H.-Y.; Missiakas, D.M.; Schneewind, O. Nontoxigenic protein a vaccine for methicillin-resistant staphylococcus aureus infections in mice. J. Exp. Med. 2010, 207, 1863–1870. [Google Scholar] [CrossRef]
- Basanisi, M.G.; La Bella, G.; Nobili, G.; Franconieri, I.; La Salandra, G. Genotyping of methicillin-resistant Staphylococcus aureus (mrsa) isolated from milk and dairy products in south italy. Food Microbiol. 2017, 62, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Ocal, D.N.; Dolapci, I.; Karahan, Z.C.; Tekeli, A. Investigation of biofilm formation properties of staphylococcus isolates. Mikrobiyoloji Bul. 2017, 51, 10–19. [Google Scholar] [CrossRef]
- Downer, R.; Roche, F.; Park, P.W.; Mecham, R.P.; Foster, T.J. The elastin-binding protein of Staphylococcus aureus (ebps) is expressed at the cell surface as an integral membrane protein and not as a cell wall-associated protein. J. Biol. Chem. 2001, 277, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Ni Eidhin, D.; Perkins, S.; Francois, P.; Vaudaux, P.; Hook, M.; Foster, T.J. Clumping factor b (clfb), a new surface-located fibrinogen-binding adhesin of staphylococcus aureus. Mol. Microbiol. 1998, 30, 245–257. [Google Scholar] [CrossRef]
- Kumar, A.; Tassopoulos, A.M.; Li, Q.; Yu, F.S. Staphylococcus aureus protein a induced inflammatory response in human corneal epithelial cells. Biochem. Biophys. Res. Commun. 2007, 354, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.A.; Stewart, J.J.; Crouch, H.K.; Florez, C.E.; Hospenthal, D.R. Methicillin-resistant staphylococcus aureus (mrsa) nares colonization at hospital admission and its effect on subsequent mrsa infection. Clin. Infect. Dis. 2004, 39, 776–782. [Google Scholar] [CrossRef]
- Defres, S.; Marwick, C.; Nathwani, D. Mrsa as a cause of lung infection including airway infection, community-acquired pneumonia and hospital-acquired pneumonia. Eur. Respir. J. 2009, 34, 1470–1476. [Google Scholar] [CrossRef]
- Sarkar, M.; Niranjan, N.; Banyal, P. Mechanisms of hypoxemia. Lung India 2017, 34, 47–60. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Phosphosite | Phospho-Western Blot (Fold Change) | Kinome Analysis (Fold Change) |
|---|---|---|---|
| PDGFRb | Y751 | 1.81 | 1.61 |
| Fyn | Y420 | 11.12 | 1.54 |
| STAT5b | Y699 | 9.06 | 2.25 |
| Lyn | Y397 | 21.30 | 2.11 |
| Lck | Y394 | 14.42 | 2.03 |
| CREB | S133 | 2.00 | 2.54 |
| β-catenin | Y654 | 8.97 | 2.39 |
| EGFR | Y1086 | 2.25 | 2.24 |
| Akt | S473 | 9.47 | 3.39 |
| p38a | T180/Y182 | 2.65 | 2.13 |
| ERK1/2 | T202/Y204; T185/Y187 | 29.17 | 2.62 |
| GSK3a/b | S21/S9 | 1.75 | 1.73 |
| HSP60 | S70 | 2.02 | 1.83 |
| STAT3 | S727 | 1.36 | 1.87 |
| Pyk2 | Y402 | 1.76 | 2.53 |
| PLCg1 | Y783 | 1.67 | 1.31 |
| c-Jun | S63 | 1.87 | 2.49 |
| p53 | S392 | 3.04 | 2.58 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nickol, M.E.; Ciric, J.; Falcinelli, S.D.; Chertow, D.S.; Kindrachuk, J. Characterization of Host and Bacterial Contributions to Lung Barrier Dysfunction Following Co-infection with 2009 Pandemic Influenza and Methicillin Resistant Staphylococcus aureus. Viruses 2019, 11, 116. https://doi.org/10.3390/v11020116
Nickol ME, Ciric J, Falcinelli SD, Chertow DS, Kindrachuk J. Characterization of Host and Bacterial Contributions to Lung Barrier Dysfunction Following Co-infection with 2009 Pandemic Influenza and Methicillin Resistant Staphylococcus aureus. Viruses. 2019; 11(2):116. https://doi.org/10.3390/v11020116
Chicago/Turabian StyleNickol, Michaela E., Justine Ciric, Shane D. Falcinelli, Daniel S. Chertow, and Jason Kindrachuk. 2019. "Characterization of Host and Bacterial Contributions to Lung Barrier Dysfunction Following Co-infection with 2009 Pandemic Influenza and Methicillin Resistant Staphylococcus aureus" Viruses 11, no. 2: 116. https://doi.org/10.3390/v11020116
APA StyleNickol, M. E., Ciric, J., Falcinelli, S. D., Chertow, D. S., & Kindrachuk, J. (2019). Characterization of Host and Bacterial Contributions to Lung Barrier Dysfunction Following Co-infection with 2009 Pandemic Influenza and Methicillin Resistant Staphylococcus aureus. Viruses, 11(2), 116. https://doi.org/10.3390/v11020116