PSMB1 Negatively Regulates the Innate Antiviral Immunity by Facilitating Degradation of IKK-ε

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Viruses
2.3. Dual-Luciferase Reporter Assay
2.4. Quantitative Polymerase Chain Reaction (qPCR) Analysis
2.5. Cytokine ELISA Measurements and Type I IFN Bioassays
2.6. Immunoblot Analysis and Immunoprecipitation (IP)
2.7. Statistical Analysis
3. Results
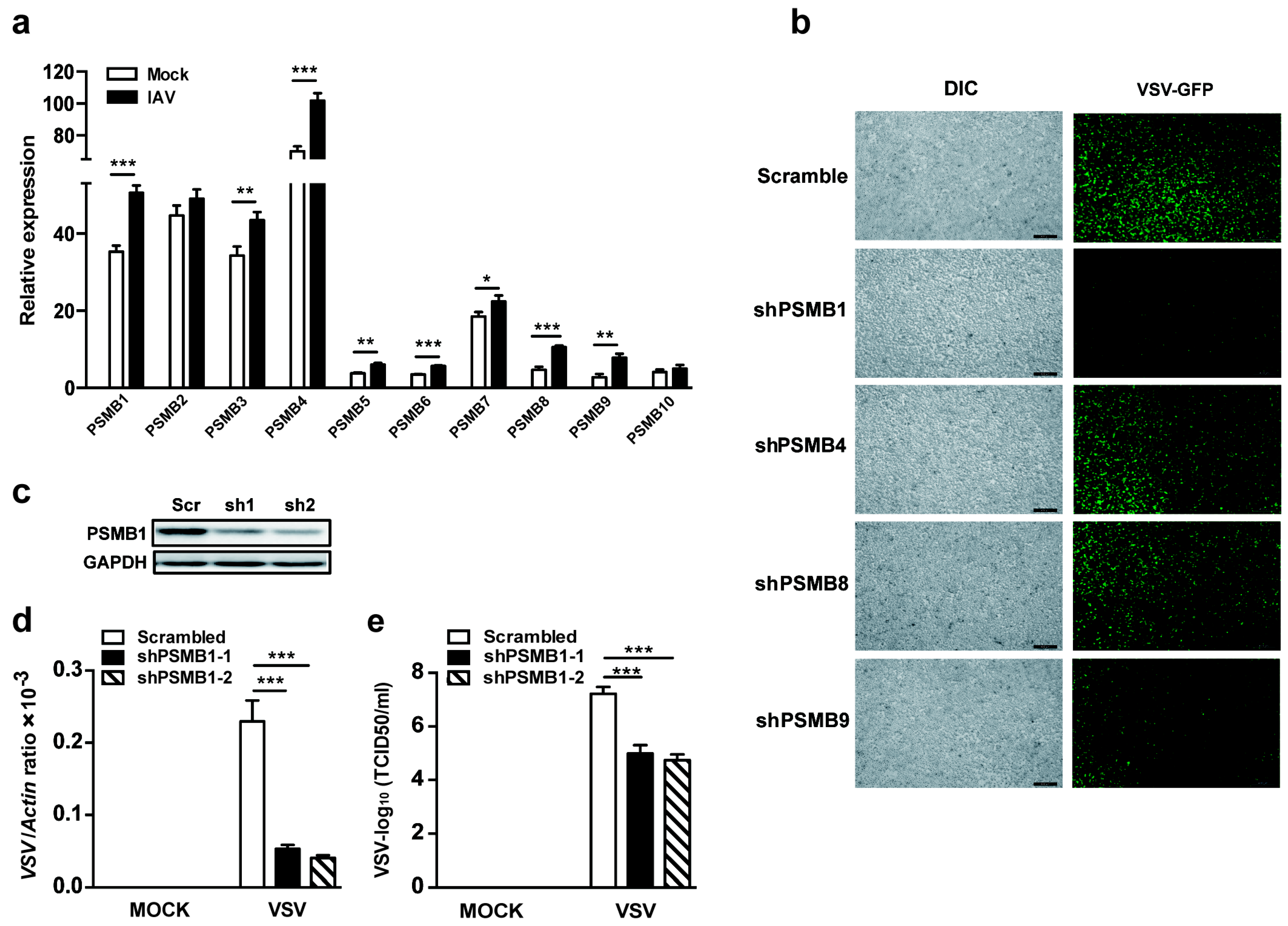
3.1. PSMB1 Is Involved in Cellular Antiviral Responses
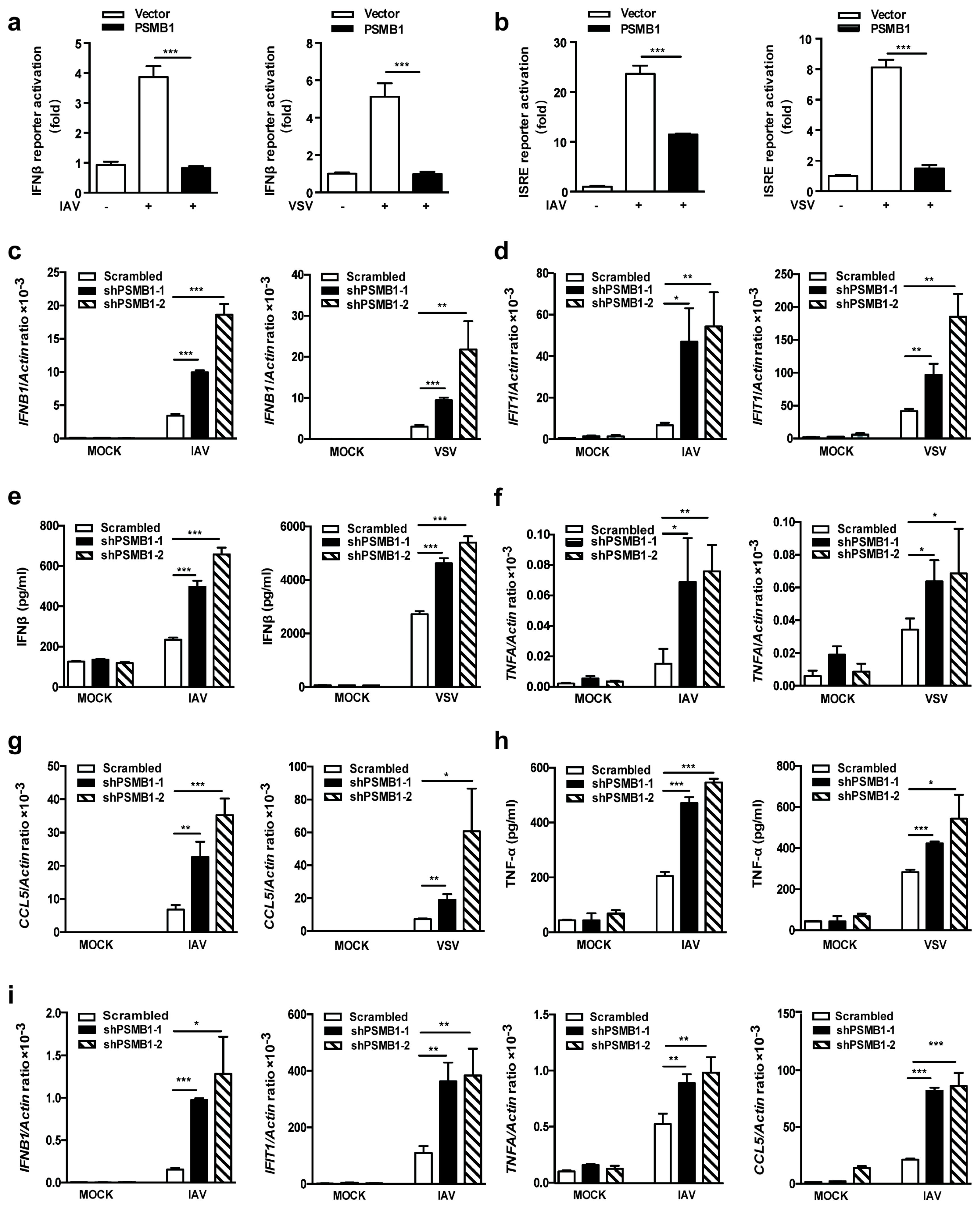
3.2. PSMB1 Negatively Regulates RNA Virus-Induced Innate Immune Responses
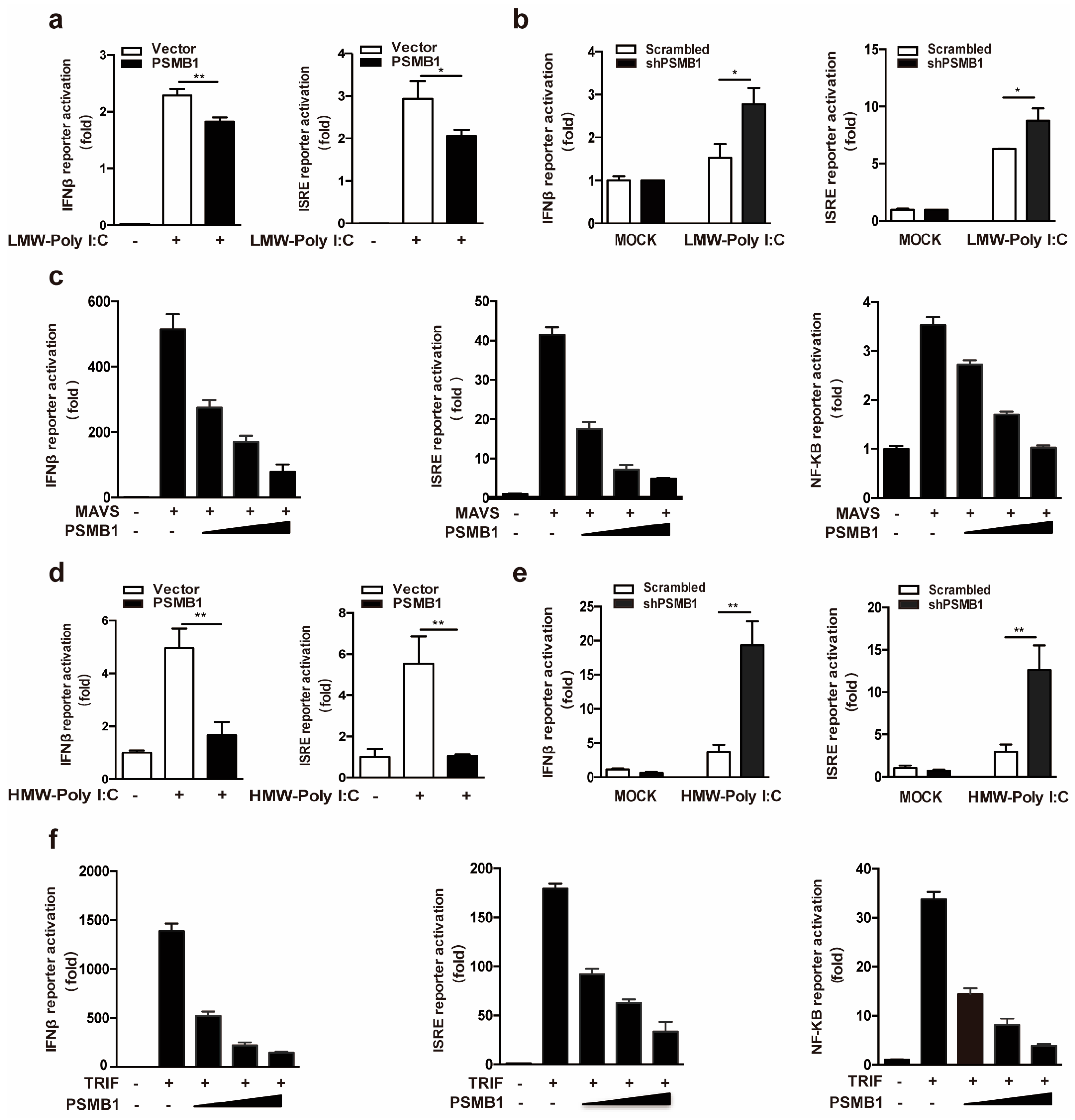
3.3. PSMB1 Inhibits RLR and TLR3 Signaling
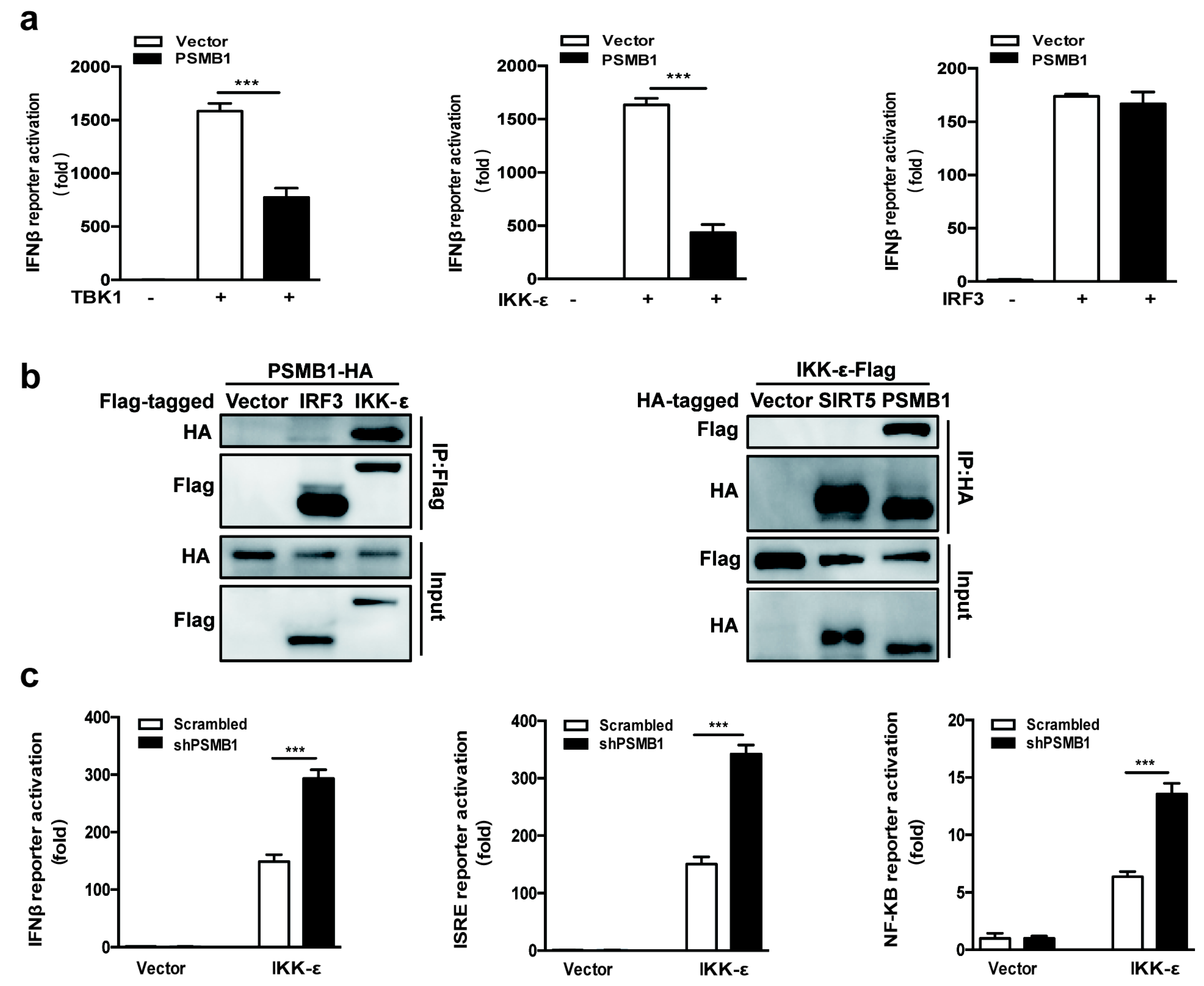
3.4. IKK-ε Is the Target of PSMB1
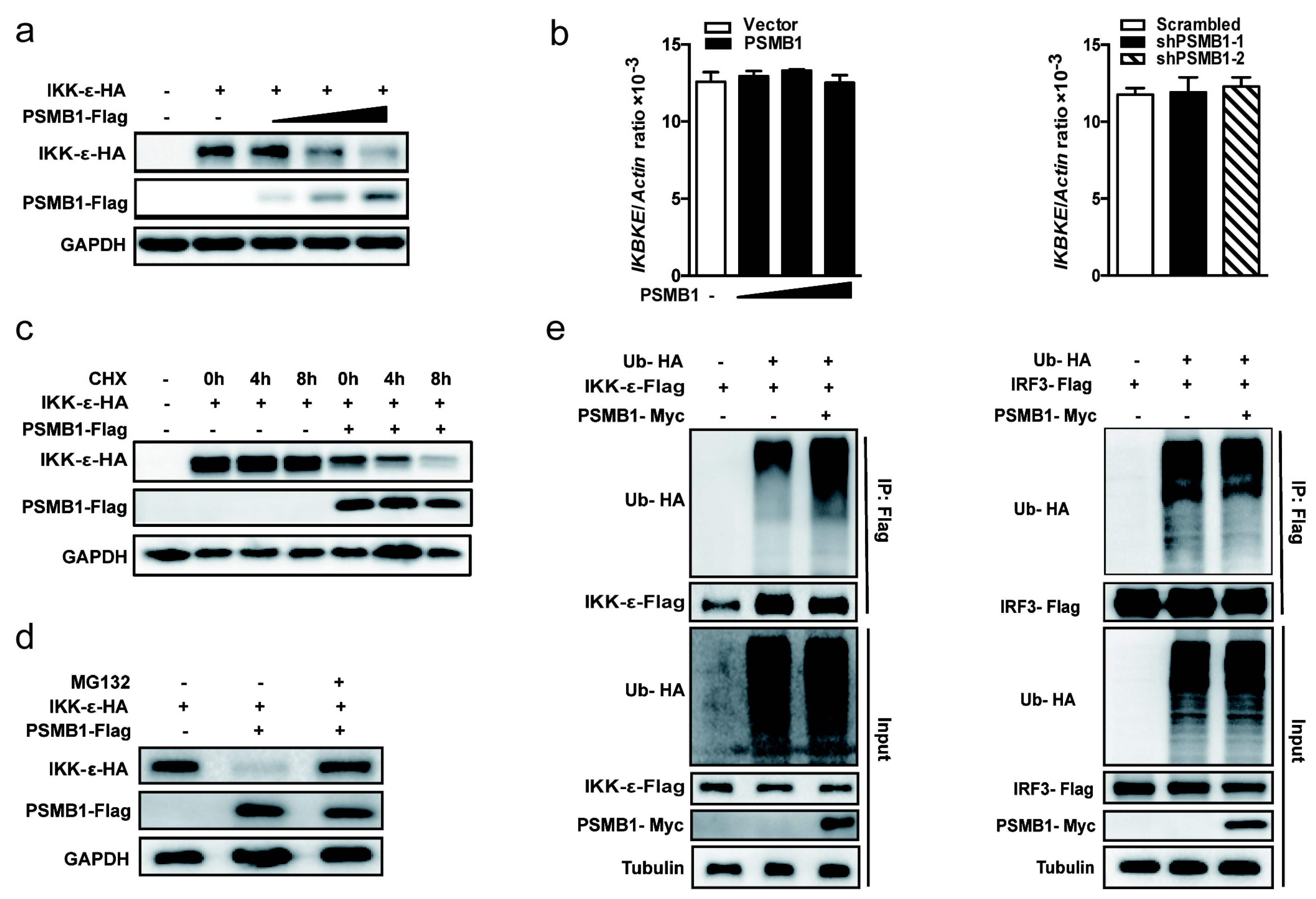
3.5. PSMB1 Promotes IKK-ε Degradation
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern Recognition Receptors and the Innate Immune Response to Viral Infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loo, Y.M.; Gale, M. Immune Signaling by RIG-I-like Receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Tschopp, J. Toll-like receptors and RNA helicases: Two parallel ways to trigger antiviral responses. Mol. Cell 2006, 22, 561–569. [Google Scholar] [CrossRef]
- Verhelst, K.; Verstrepen, L.; Carpentier, I.; Beyaert, R. I kappa B kinase epsilon (IKK epsilon): A therapeutic target in inflammation and cancer. Biochem. Pharm. 2013, 85, 873–880. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKK epsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef]
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 2006, 17, 1807–1819. [Google Scholar] [CrossRef]
- Verbrugge, S.E.; Scheper, R.J.; Lems, W.F.; Gruijl, T.D.; Jansen, G. Proteasome inhibitors as experimental therapeutics of autoimmune diseases. Arthritis Res. 2015, 17, 17. [Google Scholar] [CrossRef] [PubMed]
- Paul, S. Dysfunction of the ubiquitin-proteasome system in multiple disease conditions: Therapeutic approaches. Bioessays 2008, 30, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Dantuma, N.P.; Bott, L.C. The ubiquitin-proteasome system in neurodegenerative diseases: Precipitating factor, yet part of the solution. Front. Mol. Neurosci. 2014, 7, 70. [Google Scholar] [CrossRef]
- Sorokin, A.V.; Kim, E.R.; Ovchinnikov, L.P. Proteasome system of protein degradation and processing. Biochem. (Mosc.) 2009, 74, 1411–1442. [Google Scholar] [CrossRef]
- Li, P.C.; Jin, Y.F.; Qi, F.; Wu, F.Y.; Luo, S.S.; Cheng, Y.J. SIRT6 Acts as a Negative Regulator in Dengue Virus-Induced Inflammatory Response by Targeting the DNA Binding Domain of NF-kappa B p65. Front. Cell. Infect. Microbiol. 2018, 8, 113. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Jin, Y.F.; Zeng, N.X.; Ruan, Q.W.; Qian, F. SOD2 Facilitates the Antiviral Innate Immune Response by Scavenging Reactive Oxygen Species. Viral Immunol. 2017, 30, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Oslund, K.L.; Baumgarth, N. Influenza-induced innate immunity: Regulators of viral replication, respiratory tract pathology & adaptive immunity. Future Virol. 2011, 6, 951–962. [Google Scholar] [PubMed]
- Gack, M.U. Mechanisms of RIG-I-Like Receptor Activation and Manipulation by Viral Pathogens. J. Virol. 2014, 88, 5213–5216. [Google Scholar] [CrossRef] [Green Version]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Yasutomo, K. Dysregulation of immunoproteasomes in autoinflammatory syndromes. Int. Immunol. 2018. [Google Scholar] [CrossRef]
- Vankaer, L.; Ashtonrickardt, P.G.; Eichelberger, M.; Gaczynska, M.; Nagashima, K.; Rock, K.L.; Goldberg, A.L.; Doherty, P.C.; Tonegawa, S. Altered Peptidase and Viral-Specific T-Cell Response in Lmp2 Mutant Mice. Immunity 1994, 1, 533–541. [Google Scholar] [CrossRef]
- Muchamuel, T.; Basler, M.; Aujay, M.A.; Suzuki, E.; Kalim, K.W.; Lauer, C.; Sylvain, C.; Ring, E.R.; Shields, J.; Jiang, J.; et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med. 2009, 15, 781–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, H.; Oh, S.W.; Fujita, T. RIG-I-Like Receptors and Type I Interferonopathies. J. Interf. Cytok. Res. 2017, 37, 207–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arimoto, K.I.; Miyauchi, S.; Stoner, S.A.; Fan, J.B.; Zhang, D.E. Negative regulation of type I IFN signaling. J. Leukoc. Biol. 2018, 103, 1099–1116. [Google Scholar] [CrossRef]
- Allen, I.C.; Moore, C.B.; Schneider, M.; Lei, Y.; Davis, B.K.; Scull, M.A.; Gris, D.; Roney, K.E.; Zimmermann, A.G.; Bowzard, J.B.; et al. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-kappaB signaling pathways. Immunity 2011, 34, 854–865. [Google Scholar] [CrossRef]
- Pan, Y.; Li, R.; Meng, J.L.; Mao, H.T.; Zhang, Y.; Zhang, J. Smurf2 Negatively Modulates RIG-I-Dependent Antiviral Response by Targeting VISA/MAVS for Ubiquitination and Degradation. J. Immunol. 2014, 192, 4758–4764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, D.; Wang, Z.N.; Huang, A.F.; Zhao, Y.; Qin, F.X. A Novel Function of F-Box Protein FBXO17 in Negative Regulation of Type I IFN Signaling by Recruiting PP2A for IFN Regulatory Factor 3 Deactivation. J. Immunol. 2017, 198, 808–819. [Google Scholar] [CrossRef]
- Amaya, M.; Keck, F.; Bailey, C.; Narayanan, A. The role of the IKK complex in viral infections. Pathog. Dis. 2014, 72, 32–44. [Google Scholar] [CrossRef] [Green Version]
- Parvatiyar, K.; Barber, G.N.; Harhaj, E.W. TAX1BP1 and A20 Inhibit Antiviral Signaling by Targeting TBK1-IKKi Kinases. J. Biol. Chem. 2010, 285, 14999–15009. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.Y.; Wu, X.F.; Lee, A.J.; Jin, W.; Chang, M.; Wright, A.; Imaizumi, T.; Sun, S.C. Regulation of I kappa B kinase-related kinases and antiviral responses by tumor suppressor CYLD. J. Biol. Chem. 2008, 283, 18621–18626. [Google Scholar] [CrossRef]
- Maelfait, J.; Beyaert, R. Emerging Role of Ubiquitination in Antiviral RIG-I Signaling. Microbiol. Mol. Biol. Rev. 2012, 76, 33–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.X.; Song, T.; Wei, W.N.; Ni, C.F.; Zheng, Z.R.; Xu, Q.B.; Ma, H.F.; Li, L.; Zhang, Y.H.; He, X.; et al. Negative Regulation of MAVS-Mediated Innate Immune Response by PSMA7. J. Immunol. 2009, 183, 4241–4248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arimoto, K.I.; Takahashi, H.; Hishiki, T.; Konishi, H.; Fujita, T.; Shimotohno, K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc. Natl. Acad. Sci. USA 2007, 104, 7500–7505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castanier, C.; Zemirli, N.; Portier, A.; Garcin, D.; Bidere, N.; Vazquez, A.; Arnoult, D. MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. 2012, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.W.; Zhu, C.L.; Wang, T.C.; Jiang, H.; Ren, Y.H.; Zhang, Q.; Wu, K.L.; Liu, F.; Liu, Y.L.; Wu, J.G. GP73 represses host innate immune response to promote virus replication by facilitating MAVS and TRAF6 degradation. PLoS Pathog. 2017, 13, e1006321. [Google Scholar] [CrossRef] [PubMed]
- Nakhaei, P.; Mesplede, T.; Solis, M.; Sun, Q.; Zhao, T.J.; Yang, L.; Chuang, T.H.; Ware, C.F.; Lin, R.T.; Hiscott, J. The E3 Ubiquitin Ligase Triad3A Negatively Regulates the RIG-I/MAVS Signaling Pathway by Targeting TRAF3 for Degradation. PLoS Pathog. 2009, 5, e1000650. [Google Scholar] [CrossRef]
- Cho, S.; Choi, Y.J.; Kim, J.; Jeong, S.; Kim, J.; Kim, S.; Ryu, S. Binding and regulation of HIF-1a by a subunit of the proteasome complex PSMA7. FEBS Lett. 2001, 498, 62–66. [Google Scholar] [CrossRef]
- Keutgens, A.; Zhang, X.; Shostak, K.; Robert, I.; Olivier, S.; Vanderplasschen, A.; Chapelle, J.; Viatour, P.; Merville, M.P.; Bex, F.; et al. BCL-3 Degradation Involves Its Polyubiquitination through a FBW7-independent Pathway and Its Binding to the Proteasome Subunit PSMB1. J. Biol. Chem. 2010, 285, 25831–25840. [Google Scholar] [CrossRef] [Green Version]
- Varga, G.; Mikala, G.; Kiss, K.P.; Kosoczki, E.; Szabo, E.; Meggysi, N.; Balassa, K.; Kovy, P.; Tegeze, B.; Szombath, G.; et al. Proteasome Subunit Beta Type 1 P11A Polymorphism Is a New Prognostic Marker in Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2017, 17, 734–742. [Google Scholar] [CrossRef] [Green Version]
- Murata, S.; Yashiroda, H.; Tanaka, K. Molecular mechanisms of proteasome assembly. Nat. Rev. Mol. Cell Biol. 2009, 10, 104–115. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, F.; Niu, Z.; Zhou, B.; Li, P.; Qian, F. PSMB1 Negatively Regulates the Innate Antiviral Immunity by Facilitating Degradation of IKK-ε. Viruses 2019, 11, 99. https://doi.org/10.3390/v11020099
Wu F, Niu Z, Zhou B, Li P, Qian F. PSMB1 Negatively Regulates the Innate Antiviral Immunity by Facilitating Degradation of IKK-ε. Viruses. 2019; 11(2):99. https://doi.org/10.3390/v11020099
Chicago/Turabian StyleWu, Fangyi, Zhenmin Niu, Bin Zhou, Pengcheng Li, and Feng Qian. 2019. "PSMB1 Negatively Regulates the Innate Antiviral Immunity by Facilitating Degradation of IKK-ε" Viruses 11, no. 2: 99. https://doi.org/10.3390/v11020099
APA StyleWu, F., Niu, Z., Zhou, B., Li, P., & Qian, F. (2019). PSMB1 Negatively Regulates the Innate Antiviral Immunity by Facilitating Degradation of IKK-ε. Viruses, 11(2), 99. https://doi.org/10.3390/v11020099