The Impact of HIV-1 Genetic Diversity on CRISPR-Cas9 Antiviral Activity and Viral Escape

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Cell Culture, Transfection and Transduction
2.3. Virus Stocks and Infection
2.4. Sequencing Analysis and Virus Rescue Assay
3. Results
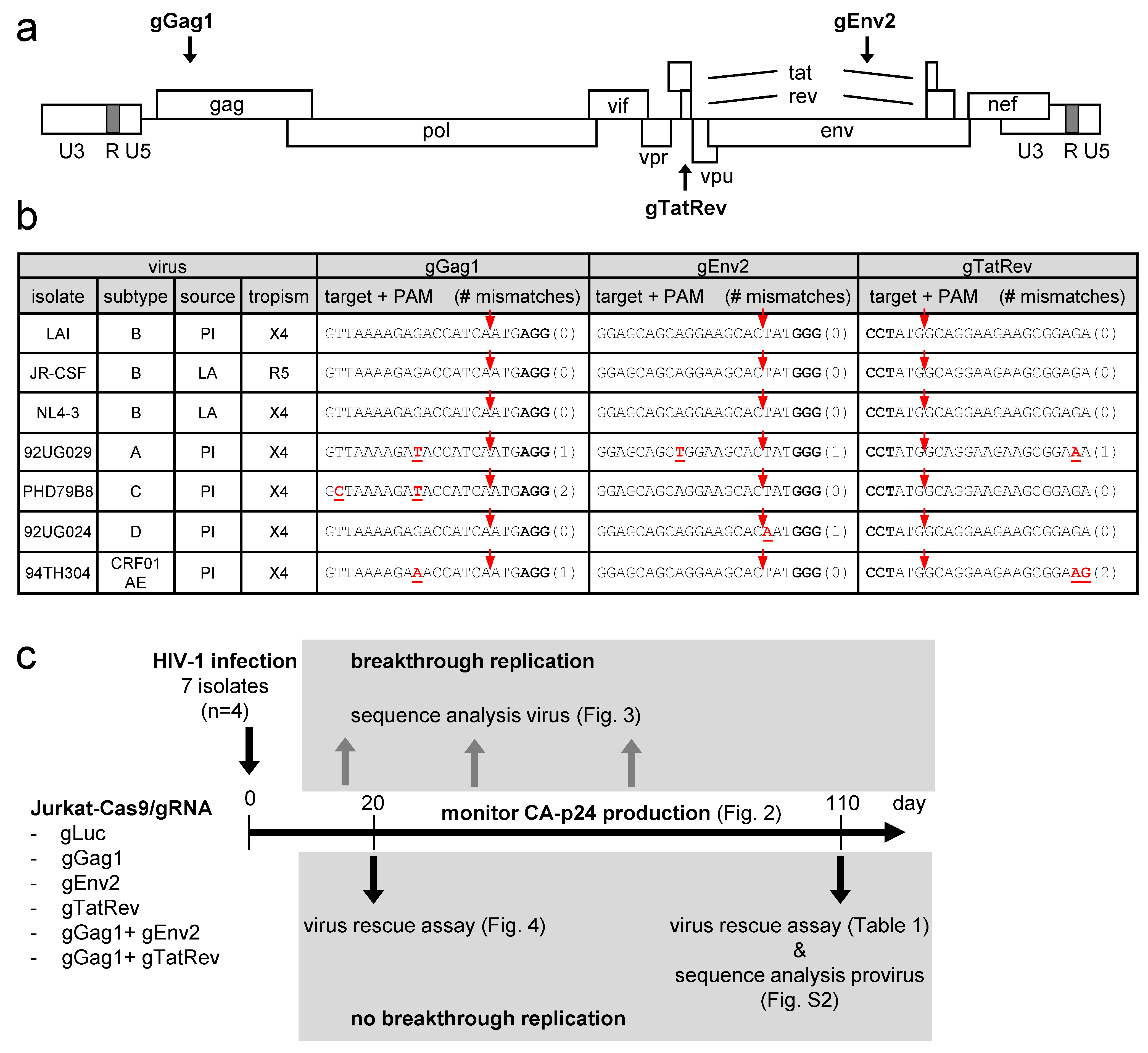
3.1. Experimental Design for Evaluating the Breadth of the CRISPR-Cas9 Dual-gRNA Therapy against Diverse HIV-1 Isolates
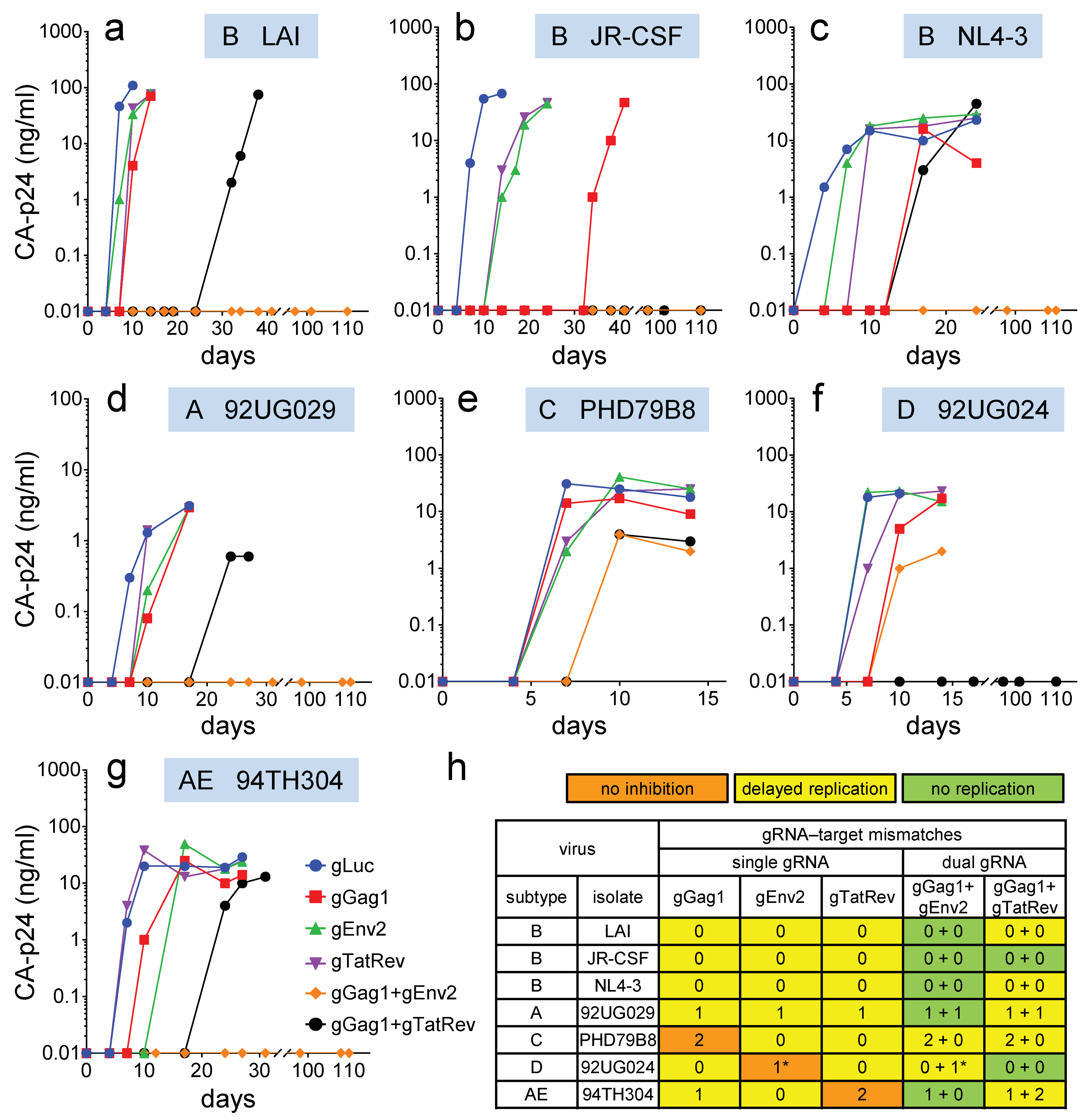
3.2. Impact of Sequence Variation in the gRNA Target Region on the Efficacy of Single gRNAs
3.3. Combination of Effective gRNAs Can Durably Block Virus Replication
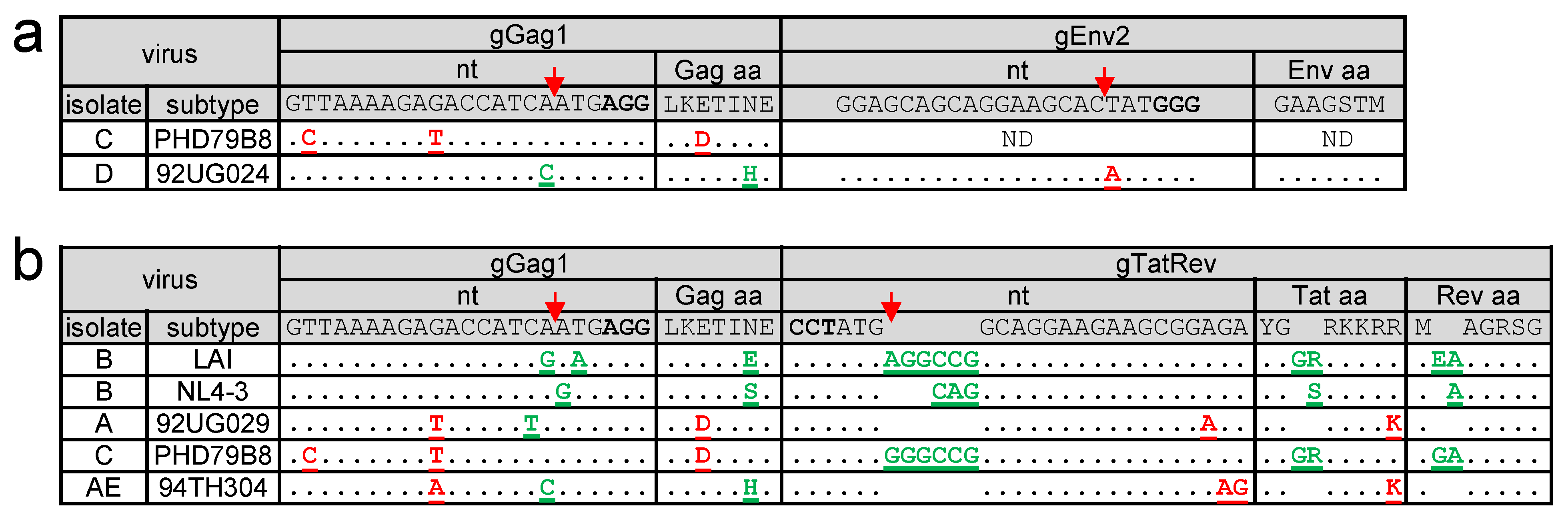
3.4. Virus Escape in Dual-gRNA Protected Cells
3.5. Complete Virus Inactivation in Dual-gRNA Protected Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Davey, R.T., Jr.; Bhat, N.; Yoder, C.; Chun, T.W.; Metcalf, J.A.; Dewar, R.; Natarajan, V.; Lempicki, R.A.; Adelsberger, J.W.; Miller, K.D.; et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc. Natl. Acad. Sci. USA 1999, 96, 15109–15114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitney, J.B.; Hill, A.L.; Sanisetty, S.; Penaloza-MacMaster, P.; Liu, J.; Shetty, M.; Parenteau, L.; Cabral, C.; Shields, J.; Blackmore, S.; et al. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature 2014, 512, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Darcis, G.; Van Driessche, B.; Van Lint, C. HIV latency: Should we shock or lock? Trends Immunol. 2017, 38, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Colby, D.J.; Trautmann, L.; Pinyakorn, S.; Leyre, L.; Pagliuzza, A.; Kroon, E.; Rolland, M.; Takata, H.; Buranapraditkun, S.; Intasan, J.; et al. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in Fiebig I acute HIV infection. Nat. Med. 2018, 24, 923–926. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.K.; Yukl, S.A. Tissue reservoirs of HIV. Curr. Opin. HIV AIDS 2016, 11, 362–370. [Google Scholar] [CrossRef]
- Darcis, G.; Bouchat, S.; Kula, A.; Van Driessche, B.; Delacourt, N.; Vanhulle, C.; Avettand-Fenoel, V.; De Wit, S.; Rohr, O.; Rouzioux, C.; et al. Reactivation capacity by latency-reversing agents ex vivo correlates with the size of the HIV-1 reservoir. AIDS 2017, 31, 181–189. [Google Scholar] [CrossRef]
- Chen, H.C.; Martinez, J.P.; Zorita, E.; Meyerhans, A.; Filion, G.J. Position effects influence HIV latency reversal. Nat. Struct. Mol. Biol. 2017, 24, 47–54. [Google Scholar] [CrossRef]
- Chun, T.W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 1997, 387, 183–188. [Google Scholar] [CrossRef]
- Khoury, G.; Darcis, G.; Lee, M.Y.; Bouchat, S.; Van Driessche, B.; Purcell, D.F.J.; Van Lint, C. The molecular biology of HIV latency. Adv. Exp. Med. Biol. 2018, 1075, 187–212. [Google Scholar] [CrossRef]
- Buchholz, F.; Hauber, J. In vitro evolution and analysis of HIV-1 LTR-specific recombinases. Methods 2011, 53, 102–109. [Google Scholar] [CrossRef]
- Karpinski, J.; Hauber, I.; Chemnitz, J.; Schafer, C.; Paszkowski-Rogacz, M.; Chakraborty, D.; Beschorner, N.; Hofmann-Sieber, H.; Lange, U.C.; Grundhoff, A.; et al. Directed evolution of a recombinase that excises the provirus of most HIV-1 primary isolates with high specificity. Nat. Biotechnol. 2016, 34, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, I.; Hauber, I.; Hauber, J.; Buchholz, F. HIV-1 proviral DNA excision using an evolved recombinase. Science 2007, 316, 1912–1915. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, R.; Berges, B.K.; Solis-Leal, A.; Igbinedion, O.; Strong, C.L.; Schiller, M.R. TALEN gene editing takes aim on HIV. Hum. Genet. 2016, 135, 1059–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manjunath, N.; Yi, G.; Dang, Y.; Shankar, P. Newer gene editing technologies toward HIV gene therapy. Viruses 2013, 5, 2748–2766. [Google Scholar] [CrossRef] [PubMed]
- Stone, D.; Kiem, H.P.; Jerome, K.R. Targeted gene disruption to cure HIV. Curr. Opin. HIV AIDS 2013, 8, 217–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, D.; Niyonzima, N.; Jerome, K.R. Genome editing and the next generation of antiviral therapy. Hum. Genet. 2016, 135, 1071–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. CRISPR-Cas based antiviral strategies against HIV-1. Virus Res. 2018, 244, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Darcis, G.; Das, A.T.; Berkhout, B. Tackling HIV persistence: Pharmacological versus CRISPR-based shock strategies. Viruses 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Soppe, J.A.; Lebbink, R.J. Antiviral goes viral: Harnessing CRISPR/Cas9 to combat viruses in humans. Trends Microbiol. 2017, 25, 833–850. [Google Scholar] [CrossRef]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. CRISPR-Cas9 can inhibit HIV-1 replication but NHEJ repair facilitates virus escape. Mol. Ther. 2016, 24, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Wainberg, M.A.; Das, A.T.; Berkhout, B. CRISPR/Cas9: A double-edged sword when used to combat HIV infection. Retrovirology 2016, 13, 37. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Pan, Q.; Gendron, P.; Zhu, W.; Guo, F.; Cen, S.; Wainberg, M.A.; Liang, C. CRISPR/Cas9-derived mutations both inhibit HIV-1 replication and accelerate viral escape. Cell Rep. 2016, 15, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. A Combinatorial CRISPR-Cas9 Attack on HIV-1 DNA Extinguishes All Infectious Provirus in Infected T Cell Cultures. Cell Rep. 2016, 17, 2819–2826. [Google Scholar] [CrossRef] [PubMed]
- Lebbink, R.J.; de Jong, D.C.; Wolters, F.; Kruse, E.M.; van Ham, P.M.; Wiertz, E.J.; Nijhuis, M. A combinational CRISPR/Cas9 gene-editing approach can halt HIV replication and prevent viral escape. Sci. Rep. 2017, 7, 41968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geretti, A.M. HIV-1 subtypes: Epidemiology and significance for HIV management. Curr. Opin. Infect. Dis. 2006, 19, 1–7. [Google Scholar] [CrossRef]
- Robertson, D.L.; Anderson, J.P.; Bradac, J.A.; Carr, J.K.; Foley, B.; Funkhouser, R.K.; Gao, F.; Hahn, B.H.; Kalish, M.L.; Kuiken, C.; et al. HIV-1 nomenclature proposal. Science 2000, 288, 55–56. [Google Scholar] [CrossRef]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [Green Version]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Roychoudhury, P.; De Silva Feelixge, H.; Reeves, D.; Mayer, B.T.; Stone, D.; Schiffer, J.T.; Jerome, K.R. Viral diversity is an obligate consideration in CRISPR/Cas9 designs for targeting the HIV reservoir. BMC Biol. 2018, 16, 75. [Google Scholar] [CrossRef] [PubMed]
- Dampier, W.; Sullivan, N.T.; Mell, J.C.; Pirrone, V.; Ehrlich, G.D.; Chung, C.H.; Allen, A.G.; DeSimone, M.; Zhong, W.; Kercher, K.; et al. Broad-spectrum and personalized guide RNAs for CRISPR/Cas9 HIV-1 therapeutics. AIDS Res. Hum. Retroviruses 2018, 34, 950–960. [Google Scholar] [CrossRef] [PubMed]
- Pham, H.; Kearns, N.A.; Maehr, R. Transcriptional Regulation with CRISPR/Cas9 Effectors in Mammalian Cells. Methods Mol. Biol. 2016, 1358, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [Green Version]
- Das, A.T.; Harwig, A.; Berkhout, B. The HIV-1 Tat protein has a versatile role in activating viral transcription. J. Virol. 2011, 85, 9506–9516. [Google Scholar] [CrossRef] [PubMed]
- Darcis, G.; Kula, A.; Bouchat, S.; Fujinaga, K.; Corazza, F.; Ait-Ammar, A.; Delacourt, N.; Melard, A.; Kabeya, K.; Vanhulle, C.; et al. An in-depth comparison of latency-reversing agent combinations in various in vitro and ex vivo HIV-1 latency models identified Bryostatin-1+JQ1 and Ingenol-B+JQ1 to potently reactivate viral rene expression. PLoS Pathogens. 2015, 11, e1005063. [Google Scholar] [CrossRef] [PubMed]
- Ter Brake, O.; Konstantinova, P.; Ceylan, M.; Berkhout, B. Silencing of HIV-1 with RNA interference: A multiple shRNA approach. Mol. Ther. 2006, 14, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Carrillo, E.; Berkhout, B. The impact of HIV-1 genetic diversity on the efficacy of a combinatorial RNAi-based gene therapy. Gene Ther. 2015, 22, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Pollakis, G.; Abebe, A.; Kliphuis, A.; Chalaby, M.I.; Bakker, M.; Mengistu, Y.; Brouwer, M.; Goudsmit, J.; Schuitemaker, H.; Paxton, W.A. Phenotypic and genotypic comparisons of CCR5- and CXCR4-tropic human immunodeficiency virus type 1 biological clones isolated from subtype C-infected individuals. J. Virol. 2004, 78, 2841–2852. [Google Scholar] [CrossRef]
- Herrera-Carrillo, E.; Paxton, W.A.; Berkhout, B. The search for a T cell line for testing novel antiviral strategies against HIV-1 isolates of diverse receptor tropism and subtype origin. J. Virol. Methods 2014, 203, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Jeeninga, R.E.; Jan, B.; van den Berg, H.; Berkhout, B. Construction of doxycyline-dependent mini-HIV-1 variants for the development of a virotherapy against leukemias. Retrovirology 2006, 3, 64. [Google Scholar] [CrossRef] [PubMed]
- Dampier, W.; Sullivan, N.T.; Chung, C.H.; Mell, J.C.; Nonnemacher, M.R.; Wigdahl, B. Designing broad-spectrum anti-HIV-1 gRNAs to target patient-derived variants. Sci. Rep. 2017, 7, 14413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Duan, D.; Chen, S.J. CRISPR-Cas9 cleavage efficiency correlates strongly with target-sgRNA folding stability: From physical mechanism to off-target assessment. Sci. Rep. 2017, 7, 143. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.E.; Taussig, D.; Steinfeld, I.; Phadnis, S.M.; Lunstad, B.D.; Singh, M.; Vuong, X.; Okochi, K.D.; McCaffrey, R.; Olesiak, M.; et al. Improving CRISPR-Cas specificity with chemical modifications in single-guide RNAs. Nucleic Acids Res. 2018, 46, 792–803. [Google Scholar] [CrossRef]
- Chuai, G.H.; Wang, Q.L.; Liu, Q. In silico meets in vivo: Towards computational CRISPR-based sgRNA design. Trends Biotechnol. 2017, 35, 12–21. [Google Scholar] [CrossRef]
- Tycko, J.; Myer, V.E.; Hsu, P.D. Methods for optimizing CRISPR-Cas9 genome editing specificity. Mol. Cell 2016, 63, 355–370. [Google Scholar] [CrossRef]
- Schwartz, C.; Bouchat, S.; Marban, C.; Gautier, V.; Van Lint, C.; Rohr, O.; Le Douce, V. On the way to find a cure: Purging latent HIV-1 reservoirs. Biochem. Pharmacol. 2017, 146, 10–22. [Google Scholar] [CrossRef]
- Darcis, G.; Van Driessche, B.; Bouchat, S.; Kirchhoff, F.; Van Lint, C. Molecular control of HIV and SIV latency. Curr Top. Microbiol. Immunol. 2018, 417, 1–22. [Google Scholar] [CrossRef]
- Darcis, G.; Coombs, R.W.; Van Lint, C. Exploring the anatomical HIV reservoirs: Role of the testicular tissue. AIDS 2016, 30, 2891–2893. [Google Scholar] [CrossRef]
- Campbell, L.A.; Coke, L.M.; Richie, C.T.; Fortuno, L.V.; Park, A.Y.; Harvey, B.K. Gesicle-mediated delivery of CRISPR/Cas9 ribonucleoprotein complex for inactivating the HIV provirus. Mol. Ther. 2019, 27, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [PubMed]
- Bella, R.; Kaminski, R.; Mancuso, P.; Young, W.B.; Chen, C.; Sariyer, R.; Fischer, T.; Amini, S.; Ferrante, P.; Jacobson, J.M.; et al. Removal of HIV DNA by CRISPR from patient blood engrafts in humanized mice. Mol. Ther. Nucleic Acids 2018, 12, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, F.; Dang, L.; Liang, C.; Wang, C.; He, B.; Liu, J.; Li, D.; Wu, X.; Xu, X.; et al. In vivo delivery systems for therapeutic genome editing. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, C.; Zhang, T.; Qu, X.; Zhang, Y.; Putatunda, R.; Xiao, X.; Li, F.; Xiao, W.; Zhao, H.; Dai, S.; et al. In vivo excision of HIV-1 provirus by saCas9 and multiplex single-guide RNAs in animal models. Mol. Ther. 2017, 25, 1168–1186. [Google Scholar] [CrossRef]
- Gao, Z.; Herrera-Carrillo, E.; Berkhout, B. A single H1 promoter can drive both guide RNA and endonuclease expression in the CRISPR-Cas9 system. Mol. Ther. Nucleic Acids 2018, 14, 32–40. [Google Scholar] [CrossRef]
- Bayat, H.; Modarressi, M.H.; Rahimpour, A. The conspicuity of CRISPR-Cpf1 system as a significant breakthrough in genome editing. Curr. Microbiol. 2018, 75, 107–115. [Google Scholar] [CrossRef]
- Gao, Z.; Herrera-Carrillo, E.; Berkhout, B. Improvement of the CRISPR-Cpf1 system with ribozyme-processed crRNA. RNA Biol. 2018, 15, 1458–1467. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Virus | Cultures with Replication-Competent Virus 1 | ||||
|---|---|---|---|---|---|
| gGag1 + gEnv2 | gGag1 + gTatRev | ||||
| Subtype | Isolate | day 20 | day 110 | day 20 | day 110 |
| B | LAI | 1/4 | 0/4 | 4/4 | NT |
| B | JR-CSF | 0/4 | 0/4 | 1/4 | 0/4 |
| B | NL4-3 | 1/4 | 0/4 | 4/4 | NT |
| A | 92UG029 | 1/4 | 0/4 | 4/4 | NT |
| C | PHD79B8 | NT | NT | NT | NT |
| D | 92UG024 | NT | NT | 0/4 | 0/4 |
| AE | 94TH304 | 0/4 | 0/4 | 4/4 | NT |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Darcis, G.; Binda, C.S.; Klaver, B.; Herrera-Carrillo, E.; Berkhout, B.; Das, A.T. The Impact of HIV-1 Genetic Diversity on CRISPR-Cas9 Antiviral Activity and Viral Escape. Viruses 2019, 11, 255. https://doi.org/10.3390/v11030255
Darcis G, Binda CS, Klaver B, Herrera-Carrillo E, Berkhout B, Das AT. The Impact of HIV-1 Genetic Diversity on CRISPR-Cas9 Antiviral Activity and Viral Escape. Viruses. 2019; 11(3):255. https://doi.org/10.3390/v11030255
Chicago/Turabian StyleDarcis, Gilles, Caroline S. Binda, Bep Klaver, Elena Herrera-Carrillo, Ben Berkhout, and Atze T. Das. 2019. "The Impact of HIV-1 Genetic Diversity on CRISPR-Cas9 Antiviral Activity and Viral Escape" Viruses 11, no. 3: 255. https://doi.org/10.3390/v11030255
APA StyleDarcis, G., Binda, C. S., Klaver, B., Herrera-Carrillo, E., Berkhout, B., & Das, A. T. (2019). The Impact of HIV-1 Genetic Diversity on CRISPR-Cas9 Antiviral Activity and Viral Escape. Viruses, 11(3), 255. https://doi.org/10.3390/v11030255