Assessment of Immunogenicity and Neutralisation Efficacy of Viral-Vectored Vaccines Against Chikungunya Virus

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bioinformatics Analysis of CHIKV Genomes and Proteins
2.2. Transgene Design
2.3. Viral-Vectored Vaccine Production
2.4. Animals
2.5. Immunisation of Mice
2.6. Cell Culture
2.7. Small Scale Transfection and Western Blot Analysis
2.8. Electron Microscopy (TEM)
2.9. Ex-Vivo IFNγ ELISpot Assay
2.10. CHIKV E2 Protein Production
2.11. Enzyme-Linked Immunosorbent Assay
2.12. Neutralisation Assay
2.13. Ethics Statement
2.14. Data Availability
3. Results
3.1. Designing of the CHIKV Antigen Cassette
3.2. Characterisation of the CHIKV Antigen-Expression
3.3. CHIKV-Cellular Responses after Vaccination
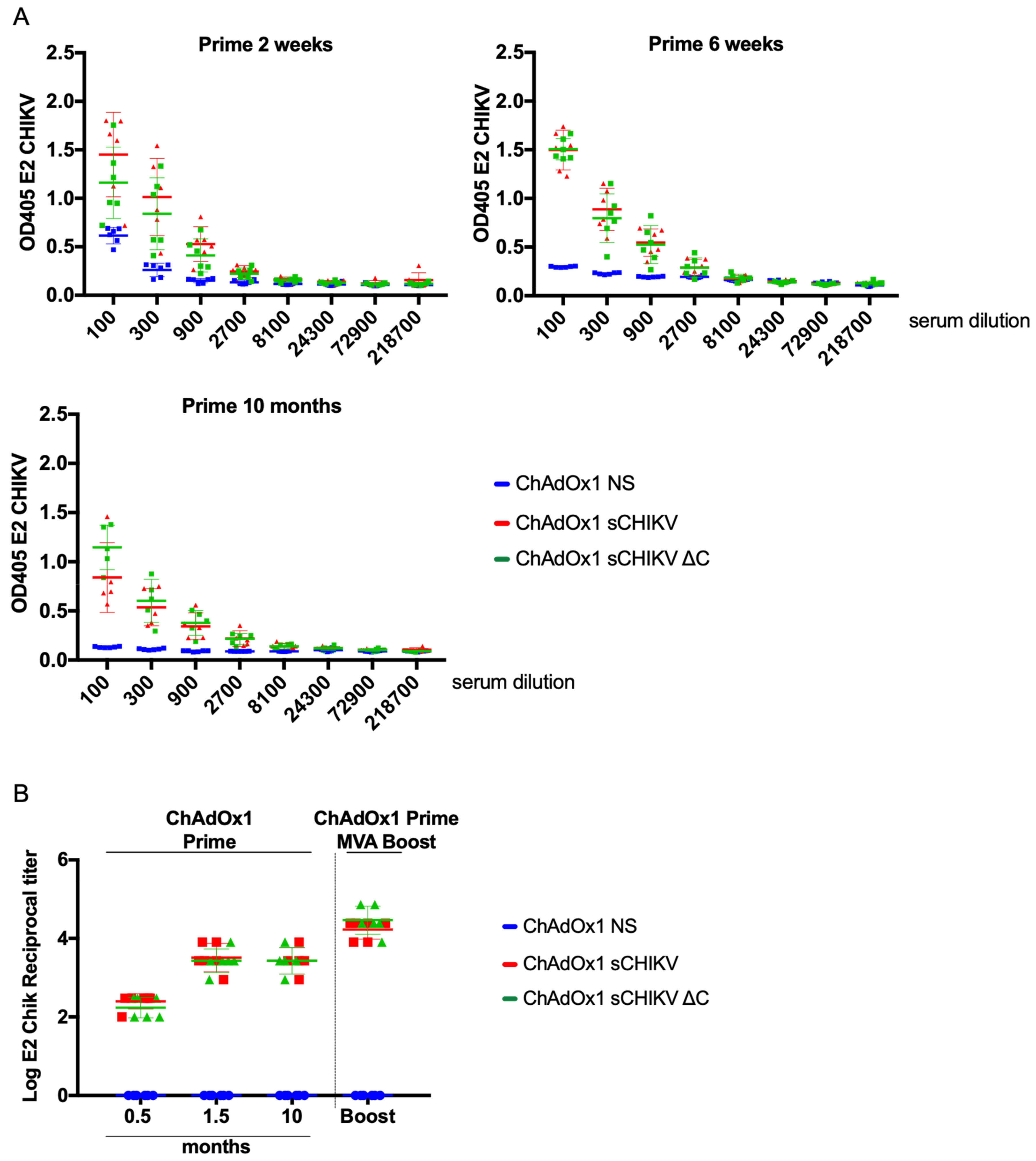
3.4. CHIKV-Humoral Responses after Vaccination
3.5. CHIKV-Neutralising Capacity in Vaccinated-Mice Sera
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weaver, S.C.; Lecuit, M. Chikungunya Virus and the Global Spread of a Mosquito-Borne Disease. N. Engl. J. Med. 2015, 372, 1231–1239. [Google Scholar] [CrossRef]
- Schmaljohn, A.L.; McClain, D. Alphaviruses (Togaviridae) and Flaviviruses (Flaviviridae). In Medical Microbiology, 4th ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Halstead, S.B. Reappearance of chikungunya, formerly called dengue, in the Americas. Emerg. Infect. Dis. 2015, 21, 557–561. [Google Scholar] [CrossRef]
- Vignuzzi, M.; Higgs, S. The Bridges and Blockades to Evolutionary Convergence on the Road to Predicting Chikungunya Virus Evolution. Annu. Rev. Virol. 2017, 4, 181–200. [Google Scholar] [CrossRef]
- Petersen, L.R.; Powers, A.M. Chikungunya: Epidemiology. Research 2016, 5, 82. [Google Scholar] [CrossRef]
- Weaver, S.C.; Forrester, N.L. Chikungunya: Evolutionary history and recent epidemic spread. Antivir. Res. 2015, 120, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Lo Presti, A.; Cella, E.; Angeletti, S.; Ciccozzi, M. Molecular epidemiology, evolution and phylogeny of Chikungunya virus: An updating review. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2016, 41, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Soni, A.; Pandey, K.M.; Ray, P.; Jayaram, B. Genomes to hits in silico—A country path today, a highway tomorrow: A case study of chikungunya. Curr. Pharm. Des. 2013, 19, 4687–4700. [Google Scholar] [CrossRef]
- Solignat, M.; Gay, B.; Higgs, S.; Briant, L.; Devaux, C. Replication cycle of chikungunya: A re-emerging arbovirus. Virology 2009, 393, 183–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.; Kesari, P.; Kumar, P.; Tomar, S. Structure-function insights into chikungunya virus capsid protein: Small molecules targeting capsid hydrophobic pocket. Virology 2018, 515, 223–234. [Google Scholar] [CrossRef]
- Sun, S.; Xiang, Y.; Akahata, W.; Holdaway, H.; Pal, P.; Zhang, X.; Diamond, M.S.; Nabel, G.J.; Rossmann, M.G. Structural analyses at pseudo atomic resolution of Chikungunya virus and antibodies show mechanisms of neutralization. eLife 2013, 2, e00435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lescar, J.; Roussel, A.; Wien, M.W.; Navaza, J.; Fuller, S.D.; Wengler, G.; Wengler, G.; Rey, F.A. The Fusion glycoprotein shell of Semliki Forest virus: An icosahedral assembly primed for fusogenic activation at endosomal pH. Cell 2001, 105, 137–148. [Google Scholar] [CrossRef]
- Zhang, W.; Heil, M.; Kuhn, R.J.; Baker, T.S. Heparin binding sites on Ross River virus revealed by electron cryo-microscopy. Virology 2005, 332, 511–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Figueiredo, M.L.G.; Figueiredo, L.T.M. Emerging alphaviruses in the Americas: Chikungunya and Mayaro. Rev. Soc. Bras. Med. Trop. 2014, 47, 677–683. [Google Scholar] [CrossRef] [Green Version]
- Burt, F.J.; Chen, W.; Miner, J.J.; Lenschow, D.J.; Merits, A.; Schnettler, E.; Kohl, A.; Rudd, P.A.; Taylor, A.; Herrero, L.J.; et al. Chikungunya virus: An update on the biology and pathogenesis of this emerging pathogen. Lancet Infect. Dis. 2017, 17, e107–e117. [Google Scholar] [CrossRef]
- Brito, C.A.A. de Alert: Severe cases and deaths associated with Chikungunya in Brazil. Rev. Soc. Bras. Med. Trop. 2017, 50, 585–589. [Google Scholar] [CrossRef]
- Rezza, G. Dengue and chikungunya: Long-distance spread and outbreaks in naïve areas. Pathog. Glob. Health 2014, 108, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Sandoval, A. 51 years in of Chikungunya clinical vaccine development: A historical perspective. Hum. Vaccines Immunother. 2019. [Google Scholar] [CrossRef]
- Erasmus, J.H.; Rossi, S.L.; Weaver, S.C. Development of vaccines for chikungunya fever. J. Infect. Dis. 2016, 214 (Suppl. 5), S488–S496. [Google Scholar] [CrossRef]
- Rezza, G.; Weaver, S.C. Chikungunya as a paradigm forz emerging viral diseases: Evaluating disease impact and hurdles to vaccine development. PLoS Negl. Trop. Dis. 2019, 13, e0006919. [Google Scholar] [CrossRef] [PubMed]
- Milligan, G.N.; Schnierle, B.S.; McAuley, A.J.; Beasley, D.W.C. Defining a correlate of protection for chikungunya virus vaccines. Vaccine 2018. in Press. [Google Scholar] [CrossRef]
- Pal, P.; Dowd, K.A.; Brien, J.D.; Edeling, M.A.; Gorlatov, S.; Johnson, S.; Lee, I.; Akahata, W.; Nabel, G.J.; Richter, M.K.S.; et al. Development of a Highly Protective Combination Monoclonal Antibody Therapy against Chikungunya Virus. PLoS Pathog. 2013, 9, e1003312. [Google Scholar] [CrossRef] [PubMed]
- Kam, Y.W.; Simarmata, D.; Chow, A.; Her, Z.; Teng, T.S.; Ong, E.K.S.; Rénia, L.; Leo, Y.S.; Ng, L.F.P. Early appearance of neutralizing immunoglobulin G3 antibodies is associated with chikungunya virus clearance and long-term clinical protection. J. Infect. Dis. 2012, 205, 1147–1154. [Google Scholar] [CrossRef]
- Yoon, I.K.; Alera, M.T.; Lago, C.B.; Tac-An, I.A.; Villa, D.; Fernandez, S.; Thaisomboonsuk, B.; Klungthong, C.; Levy, J.W.; Velasco, J.M.; et al. High Rate of Subclinical Chikungunya Virus Infection and Association of Neutralizing Antibody with Protection in a Prospective Cohort in The Philippines. PLoS Negl. Trop. Dis. 2015, 9, e0003764. [Google Scholar] [CrossRef]
- Coughlan, L.; Sridhar, S.; Payne, R.; Edmans, M.; Milicic, A.; Venkatraman, N.; Lugonja, B.; Clifton, L.; Qi, C.; Folegatti, P.M.; et al. Heterologous Two-Dose Vaccination with Simian Adenovirus and Poxvirus Vectors Elicits Long-Lasting Cellular Immunity to Influenza Virus A in Healthy Adults. EBioMedicine 2018, 29, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.J.; Sebastian, S.; Spencer, A.J.; Gilbert, S.C. Simian adenoviruses as vaccine vectors. Future Virol. 2016, 11, 649–659. [Google Scholar] [CrossRef]
- Stylianou, E.; Griffiths, K.L.; Poyntz, H.C.; Harrington-Kandt, R.; Dicks, M.D.; Stockdale, L.; Betts, G.; McShane, H. Improvement of BCG protective efficacy with a novel chimpanzee adenovirus and a modified vaccinia Ankara virus both expressing Ag85A. Vaccine 2015, 33, 6800–6808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antrobus, R.D.; Coughlan, L.; Berthoud, T.K.; Dicks, M.D.; Hill, A.V.; Lambe, T.; Gilbert, S.C. Clinical Assessment of a Novel Recombinant Simian Adenovirus ChAdOx1 as a Vectored Vaccine Expressing Conserved Influenza A Antigens. Mol. Ther. 2014, 22, 668–674. [Google Scholar] [CrossRef] [PubMed]
- López-Camacho, C.; Abbink, P.; Larocca, R.A.; Dejnirattisai, W.; Boyd, M.; Badamchi-Zadeh, A.; Wallace, Z.R.; Doig, J.; Velazquez, R.S.; Neto, R.D.L.; et al. Rational Zika vaccine design via the modulation of antigen membrane anchors in chimpanzee adenoviral vectors. Nat. Commun. 2018, 9, 2441. [Google Scholar] [CrossRef]
- Chojnacki, S.; Cowley, A.; Lee, J.; Foix, A.; Lopez, R. Programmatic access to bioinformatics tools from EMBL-EBI update: 2017. Nucleic Acids Res. 2017, 45, W550–W553. [Google Scholar] [CrossRef] [Green Version]
- Schneider, J.; Gilbert, S.C.; Blanchard, T.J.; Hanke, T.; Robson, K.J.; Hannan, C.M.; Becker, M.; Sinden, R.; Smith, G.L.; Hill, A.V.S. Enhanced immunogenicity for CD8+ T cell induction and complete protective efficacy of malaria DNA vaccination by boosting with modified vaccinia virus Ankara. Nat. Med. 1998, 4, 397–402. [Google Scholar] [CrossRef]
- Aricescu, A.R.; Lu, W.; Jones, E.Y. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Gläsker, S.; Lulla, A.; Lulla, V.; Couderc, T.; Drexler, J.F.; Liljeström, P.; Lecuit, M.; Drosten, C.; Merits, A.; Kümmerer, B.M. Virus replicon particle based Chikungunya virus neutralization assay using Gaussia luciferase as readout. Virol. J. 2013, 10. [Google Scholar] [CrossRef] [PubMed]
- Akahata, W.; Yang, Z.-Y.; Andersen, H.; Sun, S.; Holdaway, H.A.; Kong, W.-P.; Lewis, M.G.; Higgs, S.; Rossmann, M.G.; Rao, S.; et al. A virus-like particle vaccine for epidemic Chikungunya virus protects nonhuman primates against infection. Nat. Med. 2010, 16, 334–348. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Suhrbier, A.; Penn-Nicholson, A.; Woraratanadharm, J.; Gardner, J.; Luo, M.; Le, T.T.; Anraku, I.; Sakalian, M.; Einfeld, D.; et al. A complex adenovirus vaccine against chikungunya virus provides complete protection against viraemia and arthritis. Vaccine 2011, 29, 2803–2809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraswat, S.; Athmaram, T.N.; Parida, M.; Agarwal, A.; Saha, A.; Dash, P.K. Expression and Characterization of Yeast Derived Chikungunya Virus Like Particles (CHIK-VLPs) and Its Evaluation as a Potential Vaccine Candidate. PLoS Negl. Trop. Dis. 2016, 10, e0004782. [Google Scholar] [CrossRef]
- Dorange, F.; Piver, E.; Bru, T.; Collin, C.; Roingeard, P.; Pagès, J.-C. Vesicular stomatitis virus glycoprotein: A transducing coat for SFV-based RNA vectors. J. Gene Med. 2004, 6, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Tuekprakhon, A.; Puiprom, O.; Sasaki, T.; Michiels, J.; Bartholomeeusen, K.; Nakayama, E.E.; Meno, M.K.; Phadungsombat, J.; Huits, R.; Ariën, K.K.; et al. Broad-spectrum monoclonal antibodies against chikungunya virus structural proteins: Promising candidates for antibody-based rapid diagnostic test development. PLoS ONE 2018, 13, e0208851. [Google Scholar] [CrossRef]
- Reyes-Sandoval, A.; Berthoud, T.; Alder, N.; Siani, L.; Gilbert, S.C.; Nicosia, A.; Colloca, S.; Cortese, R.; Hill, A.V.S. Prime-boost immunization with adenoviral and modified vaccinia virus Ankara vectors enhances the durability and polyfunctionality of protective malaria CD8+ T-cell responses. Infect. Immun. 2010, 78, 145–153. [Google Scholar] [CrossRef]
- Alharbi, N.K.; Padron-Regalado, E.; Thompson, C.P.; Kupke, A.; Wells, D.; Sloan, M.A.; Grehan, K.; Temperton, N.; Lambe, T.; Warimwe, G.; et al. ChAdOx1 and MVA based vaccine candidates against MERS-CoV elicit neutralising antibodies and cellular immune responses in mice. Vaccine 2017, 35, 3780–3788. [Google Scholar] [CrossRef]
- Liljeström, P.; Garoff, H. Internally located cleavable signal sequences direct the formation of Semliki Forest virus membrane proteins from a polyprotein precursor. J. Virol. 1991, 65, 147–154. [Google Scholar]
- Chattopadhyay, A.; Wang, E.; Seymour, R.; Weaver, S.C.; Rose, J.K. A Chimeric Vesiculo/Alphavirus Is an Effective Alphavirus Vaccine. J. Virol. 2012, 87, 395–402. [Google Scholar] [CrossRef] [Green Version]
- Muthumani, K.; Lankaraman, K.M.; Laddy, D.J.; Sundaram, S.G.; Chung, C.W.; Sako, E.; Wu, L.; Khan, A.; Sardesai, N.; Kim, J.J.; et al. Immunogenicity of novel consensus-based DNA vaccines against Chikungunya virus. Vaccine 2008, 26, 5128–5134. [Google Scholar] [CrossRef] [PubMed]
- García-Arriaza, J.; Cepeda, V.; Hallengärd, D.; Sorzano, C.Ó.S.; Kümmerer, B.M.; Liljeström, P.; Esteban, M. A novel poxvirus-based vaccine, MVA-CHIKV, is highly immunogenic and protects mice against chikungunya infection. J. Virol. 2014, 88, 3527–3547. [Google Scholar] [CrossRef]
- Teo, T.-H.; Lum, F.-M.; Claser, C.; Lulla, V.; Lulla, A.; Merits, A.; Rénia, L.; Ng, L.F.P. A pathogenic role for CD4+ T cells during Chikungunya virus infection in mice. J. Immunol. Baltim. Md. 1950 2013, 190, 259–269. [Google Scholar] [CrossRef]
- Chu, H.; Das, S.C.; Fuchs, J.F.; Suresh, M.; Weaver, S.C.; Stinchcomb, D.T.; Partidos, C.D.; Osorio, J.E. Deciphering the protective role of adaptive immunity to CHIKV/IRES a novel candidate vaccine against Chikungunya in the A129 mouse model. Vaccine 2013, 31, 3353–3360. [Google Scholar] [CrossRef]
- Hoarau, J.J.; Gay, F.; Pellé, O.; Samri, A.; Jaffar-Bandjee, M.C.; Gasque, P.; Autran, B. Identical strength of the T cell responses against E2, nsP1 and capsid CHIKV proteins in recovered and chronic patients after the epidemics of 2005–2006 in La Reunion Island. PLoS ONE 2013, 8, e84695. [Google Scholar] [CrossRef] [PubMed]
- Roques, P.; Ljungberg, K.; Kümmerer, B.M.; Gosse, L.; Dereuddre-Bosquet, N.; Tchitchek, N.; Hallengärd, D.; García-Arriaza, J.; Meinke, A.; Esteban, M.; et al. Attenuated and vectored vaccines protect nonhuman primates against Chikungunya virus. JCI Insight 2017, 2, e83527. [Google Scholar] [CrossRef] [PubMed]
- Hallengärd, D.; Lum, F.-M.; Kümmerer, B.M.; Lulla, A.; Lulla, V.; García-Arriaza, J.; Fazakerley, J.K.; Roques, P.; Le Grand, R.; Merits, A.; et al. Prime-boost immunization strategies against Chikungunya virus. J. Virol. 2014, 88, 13333–13343. [Google Scholar] [CrossRef] [PubMed]
- Panning, M.; Grywna, K.; Van Esbroeck, M.; Emmerich, P.; Drosten, C. Chikungunya fever in travelers returning to Europe from the Indian Ocean Region, 2006. Emerg. Infect. Dis. 2008, 14, 416. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.J.; Dowd, K.A.; Mendoza, F.H.; Saunders, J.G.; Sitar, S.; Plummer, S.H.; Yamshchikov, G.; Sarwar, U.N.; Hu, Z.; Enama, M.E.; et al. Safety and tolerability of chikungunya virus-like particle vaccine in healthy adults: A phase 1 dose-escalation trial. Lancet 2014, 384, 2046–2052. [Google Scholar] [CrossRef]
- Ramsauer, K.; Schwameis, M.; Firbas, C.; Müllner, M.; Putnak, R.J.; Thomas, S.J.; Desprès, P.; Tauber, E.; Jilma, B.; Tangy, F. Immunogenicity, safety, and tolerability of a recombinant measles-virus-based chikungunya vaccine: A randomised, double-blind, placebo-controlled, active-comparator, first-in-man trial. Lancet Infect. Dis. 2015, 15, 519–527. [Google Scholar] [CrossRef]
- Xiang, Z.; Li, Y.; Cun, A.; Yang, W.; Ellenberg, S.; Switzer, W.M.; Kalish, M.L.; Ertl, H.C.J. Chimpanzee adenovirus antibodies in humans, sub-Saharan Africa. Emerg. Infect. Dis. 2006, 12, 1596–1599. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Camacho, C.; Kim, Y.C.; Blight, J.; Lazaro Moreli, M.; Montoya-Diaz, E.; T Huiskonen, J.; Mareike Kümmerer, B.; Reyes-Sandoval, A. Assessment of Immunogenicity and Neutralisation Efficacy of Viral-Vectored Vaccines Against Chikungunya Virus. Viruses 2019, 11, 322. https://doi.org/10.3390/v11040322
López-Camacho C, Kim YC, Blight J, Lazaro Moreli M, Montoya-Diaz E, T Huiskonen J, Mareike Kümmerer B, Reyes-Sandoval A. Assessment of Immunogenicity and Neutralisation Efficacy of Viral-Vectored Vaccines Against Chikungunya Virus. Viruses. 2019; 11(4):322. https://doi.org/10.3390/v11040322
Chicago/Turabian StyleLópez-Camacho, César, Young Chan Kim, Joshua Blight, Marcos Lazaro Moreli, Eduardo Montoya-Diaz, Juha T Huiskonen, Beate Mareike Kümmerer, and Arturo Reyes-Sandoval. 2019. "Assessment of Immunogenicity and Neutralisation Efficacy of Viral-Vectored Vaccines Against Chikungunya Virus" Viruses 11, no. 4: 322. https://doi.org/10.3390/v11040322
APA StyleLópez-Camacho, C., Kim, Y. C., Blight, J., Lazaro Moreli, M., Montoya-Diaz, E., T Huiskonen, J., Mareike Kümmerer, B., & Reyes-Sandoval, A. (2019). Assessment of Immunogenicity and Neutralisation Efficacy of Viral-Vectored Vaccines Against Chikungunya Virus. Viruses, 11(4), 322. https://doi.org/10.3390/v11040322