Treatment of Metastatic Disease through Natural Killer Cell Modulation by Infected Cell Vaccines


{kind=link}
Abstract
:1. Introduction
2. Checkpoint Inhibitors and the Emergence of Cancer Immunotherapies
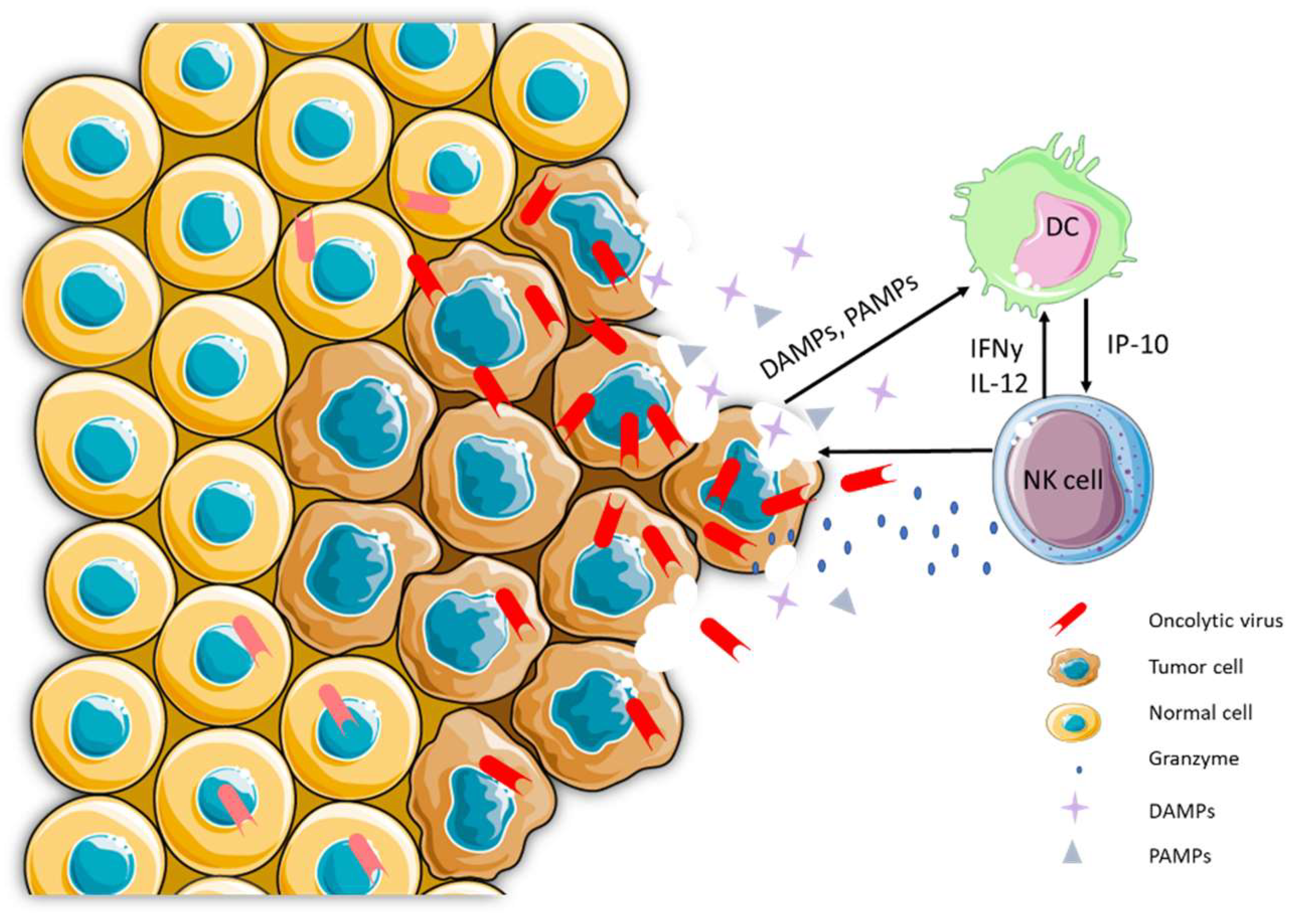
3. Oncolytic Viruses Can Make Cold Tumors Hot
4. OV Infected Tumor Cells Reveal Neo-Epitopes through Immunogenic Cell Death (ICD), Leading to Anti-Tumor Immune Responses
5. OV-Based Infected Cell Vaccines (ICVs) Exploit the Properties of ICD Propagated through Viral Replication
6. Evidence for NK Cell Activation with OVs and ICVs
7. The Importance of NK Cell Monitoring in OV and ICV Therapies
8. Clinical Development Potential of OVs and ICVs
9. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Gottschalk, A.; Sharma, S.; Ford, J.; Durieux, M.E.; Tiouririne, M. Review article: The role of the perioperative period in recurrence after cancer surgery. Anesth. Analg. 2010, 110, 1636–1643. [Google Scholar] [CrossRef] [PubMed]
- Coffey, J.C.; Wang, J.H.; Smith, M.J.; Bouchier-Hayes, D.; Cotter, T.G.; Redmond, H.P. Excisional surgery for cancer cure: Therapy at a cost. Lancet Oncol. 2003, 4, 760–768. [Google Scholar] [CrossRef]
- Loi, S.; Michiels, S.; Salgado, R.; Sirtaine, N.; Jose, V.; Fumagalli, D.; Kellokumpu-Lehtinen, P.L.; Bono, P.; Kataja, V.; Desmedt, C.; et al. Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: Results from the finher trial. Ann. Oncol. 2014, 25, 1544–1550. [Google Scholar] [CrossRef]
- Adams, S.; Gray, R.J.; Demaria, S.; Goldstein, L.; Perez, E.A.; Shulman, L.N.; Martino, S.; Wang, M.; Jones, V.E.; Saphner, T.J.; et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase iii randomized adjuvant breast cancer trials: Ecog 2197 and ecog 1199. J. Clin. Oncol. 2014, 32, 2959–2966. [Google Scholar] [CrossRef] [PubMed]
- Oble, D.A.; Loewe, R.; Yu, P.; Mihm, M.C., Jr. Focus on tils: Prognostic significance of tumor infiltrating lymphocytes in human melanoma. Cancer Immun. 2009, 9, 3. [Google Scholar]
- Ohtani, H. Focus on tils: Prognostic significance of tumor infiltrating lymphocytes in human colorectal cancer. Cancer Immun. 2007, 7, 4. [Google Scholar]
- Liakou, C.I.; Narayanan, S.; Ng Tang, D.; Logothetis, C.J.; Sharma, P. Focus on tils: Prognostic significance of tumor infiltrating lymphocytes in human bladder cancer. Cancer Immun. 2007, 7, 10. [Google Scholar] [PubMed]
- Trujillo, J.A.; Sweis, R.F.; Bao, R.; Luke, J.J. T cell-inflamed versus non-t cell-inflamed tumors: A conceptual framework for cancer immunotherapy drug development and combination therapy selection. Cancer Immunol. Res. 2018, 6, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- Kitano, S.; Nakayama, T.; Yamashita, M. Biomarkers for immune checkpoint inhibitors in melanoma. Front. Oncol. 2018, 8, 270. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Cicchelero, L.; de Rooster, H.; Sanders, N.N. Various ways to improve whole cancer cell vaccines. Expert Rev. Vaccines 2014, 13, 721–735. [Google Scholar] [CrossRef]
- Keenan, B.P.; Jaffee, E.M. Whole cell vaccines--past progress and future strategies. Semin. Oncol. 2012, 39, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Vaha-Koskela, M.J.; Heikkila, J.E.; Hinkkanen, A.E. Oncolytic viruses in cancer therapy. Cancer Lett. 2007, 254, 178–216. [Google Scholar] [CrossRef] [PubMed]
- Parato, K.A.; Lichty, B.D.; Bell, J.C. Diplomatic immunity: Turning a foe into an ally. Curr. Opin. Mol. Ther. 2009, 11, 13–21. [Google Scholar]
- Rehman, H.; Silk, A.W.; Kane, M.P.; Kaufman, H.L. Into the clinic: Talimogene laherparepvec (t-vec), a first-in-class intratumoral oncolytic viral therapy. J. Immunother. Cancer 2016, 4, 53. [Google Scholar] [CrossRef]
- Stojdl, D.F.; Lichty, B.D.; tenOever, B.R.; Paterson, J.M.; Power, A.T.; Knowles, S.; Marius, R.; Reynard, J.; Poliquin, L.; Atkins, H.; et al. Vsv strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 2003, 4, 263–275. [Google Scholar] [CrossRef]
- Bridle, B.W.; Stephenson, K.B.; Boudreau, J.E.; Koshy, S.; Kazdhan, N.; Pullenayegum, E.; Brunelliere, J.; Bramson, J.L.; Lichty, B.D.; Wan, Y. Potentiating cancer immunotherapy using an oncolytic virus. Mol. Ther. 2010, 18, 1430–1439. [Google Scholar] [CrossRef]
- Diaz, R.M.; Galivo, F.; Kottke, T.; Wongthida, P.; Qiao, J.; Thompson, J.; Valdes, M.; Barber, G.; Vile, R.G. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 2007, 67, 2840–2848. [Google Scholar] [CrossRef]
- Brun, J.; McManus, D.; Lefebvre, C.; Hu, K.; Falls, T.; Atkins, H.; Bell, J.C.; McCart, J.A.; Mahoney, D.; Stojdl, D.F. Identification of genetically modified maraba virus as an oncolytic rhabdovirus. Mol. Ther. 2010, 18, 1440–1449. [Google Scholar] [CrossRef]
- Arens, R. Rational design of vaccines: Learning from immune evasion mechanisms of persistent viruses and tumors. Adv. Immunol. 2012, 114, 217–243. [Google Scholar]
- Kim, J.W.; Gulley, J.L. Poxviral vectors for cancer immunotherapy. Expert Opin. Biol. Ther. 2012, 12, 463–478. [Google Scholar] [CrossRef] [Green Version]
- Pitt, J.M.; Kroemer, G.; Zitvogel, L. Immunogenic and non-immunogenic cell death in the tumor microenvironment. Adv. Exp. Med. Biol. 2017, 1036, 65–79. [Google Scholar] [PubMed]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Chaput, N.; De Botton, S.; Obeid, M.; Apetoh, L.; Ghiringhelli, F.; Panaretakis, T.; Flament, C.; Zitvogel, L.; Kroemer, G. Molecular determinants of immunogenic cell death: Surface exposure of calreticulin makes the difference. J. Mol. Med. (Berl) 2007, 85, 1069–1076. [Google Scholar] [CrossRef]
- Garg, A.D.; Galluzzi, L.; Apetoh, L.; Baert, T.; Birge, R.B.; Bravo-San Pedro, J.M.; Breckpot, K.; Brough, D.; Chaurio, R.; Cirone, M.; et al. Molecular and translational classifications of damps in immunogenic cell death. Front. Immunol. 2015, 6, 588. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Panaretakis, T.; Tufi, R.; Joza, N.; van Endert, P.; Ghiringhelli, F.; Apetoh, L.; Chaput, N.; Flament, C.; et al. Ecto-calreticulin in immunogenic chemotherapy. Immunol. Rev. 2007, 220, 22–34. [Google Scholar] [CrossRef]
- Boisgerault, N.; Tangy, F.; Gregoire, M. New perspectives in cancer virotherapy: Bringing the immune system into play. Immunotherapy 2010, 2, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Sukkurwala, A.Q.; Martins, I.; Shen, S.; Zitvogel, L.; Kroemer, G. Subversion of the chemotherapy-induced anticancer immune response by the ecto-atpase cd39. Oncoimmunology 2012, 1, 393–395. [Google Scholar] [CrossRef]
- Obeid, M.; Panaretakis, T.; Tesniere, A.; Joza, N.; Tufi, R.; Apetoh, L.; Ghiringhelli, F.; Zitvogel, L.; Kroemer, G. Leveraging the immune system during chemotherapy: Moving calreticulin to the cell surface converts apoptotic death from "silent" to immunogenic. Cancer Res. 2007, 67, 7941–7944. [Google Scholar] [CrossRef]
- Guillerme, J.B.; Boisgerault, N.; Roulois, D.; Menager, J.; Combredet, C.; Tangy, F.; Fonteneau, J.F.; Gregoire, M. Measles virus vaccine-infected tumor cells induce tumor antigen cross-presentation by human plasmacytoid dendritic cells. Clin. Cancer Res. 2013, 19, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, J.E.; Bridle, B.W.; Stephenson, K.B.; Jenkins, K.M.; Brunelliere, J.; Bramson, J.L.; Lichty, B.D.; Wan, Y. Recombinant vesicular stomatitis virus transduction of dendritic cells enhances their ability to prime innate and adaptive antitumor immunity. Mol. Ther. 2009, 17, 1465–1472. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, J.E.; Stephenson, K.B.; Wang, F.; Ashkar, A.A.; Mossman, K.L.; Lenz, L.L.; Rosenthal, K.L.; Bramson, J.L.; Lichty, B.D.; Wan, Y. Il-15 and type i interferon are required for activation of tumoricidal nk cells by virus-infected dendritic cells. Cancer Res. 2011, 71, 2497–2506. [Google Scholar] [CrossRef] [PubMed]
- Gastl, G.; Finstad, C.L.; Guarini, A.; Bosl, G.; Gilboa, E.; Bander, N.H.; Gansbacher, B. Retroviral vector-mediated lymphokine gene transfer into human renal cancer cells. Cancer Res. 1992, 52, 6229–6236. [Google Scholar] [PubMed]
- Disis, M.L.; Schiffman, K.; Gooley, T.A.; McNeel, D.G.; Rinn, K.; Knutson, K.L. Delayed-type hypersensitivity response is a predictor of peripheral blood t-cell immunity after her-2/neu peptide immunization. Clin. Cancer Res. 2000, 6, 1347–1350. [Google Scholar]
- Bridle, B.W.; Boudreau, J.E.; Lichty, B.D.; Brunelliere, J.; Stephenson, K.; Koshy, S.; Bramson, J.L.; Wan, Y. Vesicular stomatitis virus as a novel cancer vaccine vector to prime antitumor immunity amenable to rapid boosting with adenovirus. Mol. Ther. 2009, 17, 1814–1821. [Google Scholar] [CrossRef]
- Shillitoe, E.J.; Pellenz, C. Factors that limit the effectiveness of herpes simplex virus type 1 for treatment of oral cancer in mice. Clin. Cancer Res. 2005, 11, 3109–3116. [Google Scholar] [CrossRef]
- Derubertis, B.G.; Stiles, B.M.; Bhargava, A.; Gusani, N.J.; Hezel, M.; D’Angelica, M.; Fong, Y. Cytokine-secreting herpes viral mutants effectively treat tumor in a murine metastatic colorectal liver model by oncolytic and t-cell-dependent mechanisms. Cancer Gene Ther. 2007, 14, 590–597. [Google Scholar] [CrossRef]
- Martinez, J.; Huang, X.; Yang, Y. Direct tlr2 signaling is critical for nk cell activation and function in response to vaccinia viral infection. PLoS Pathog. 2010, 6, e1000811. [Google Scholar] [CrossRef]
- Dokun, A.O.; Kim, S.; Smith, H.R.; Kang, H.S.; Chu, D.T.; Yokoyama, W.M. Specific and nonspecific nk cell activation during virus infection. Nat. Immunol. 2001, 2, 951–956. [Google Scholar] [CrossRef]
- Natuk, R.J.; Welsh, R.M. Accumulation and chemotaxis of natural killer/large granular lymphocytes at sites of virus replication. J. Immunol. 1987, 138, 877–883. [Google Scholar] [PubMed]
- Drake, C.G. Prostate cancer as a model for tumour immunotherapy. Nat. Rev. Immunol. 2010, 10, 580–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gujar, S.A.; Pan, D.A.; Marcato, P.; Garant, K.A.; Lee, P.W. Oncolytic virus-initiated protective immunity against prostate cancer. Mol. Ther. 2011, 19, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.G.; Fraser, N.W. Requirement of an integrated immune response for successful neuroattenuated hsv-1 therapy in an intracranial metastatic melanoma model. Mol. Ther. 2003, 7, 741–747. [Google Scholar] [CrossRef]
- Jarahian, M.; Watzl, C.; Fournier, P.; Arnold, A.; Djandji, D.; Zahedi, S.; Cerwenka, A.; Paschen, A.; Schirrmacher, V.; Momburg, F. Activation of natural killer cells by newcastle disease virus hemagglutinin-neuraminidase. J. Virol. 2009, 83, 8108–8121. [Google Scholar] [CrossRef] [PubMed]
- Rintoul, J.L.; Lemay, C.G.; Tai, L.H.; Stanford, M.M.; Falls, T.J.; de Souza, C.T.; Bridle, B.W.; Daneshmand, M.; Ohashi, P.S.; Wan, Y.; et al. Orfv: A novel oncolytic and immune stimulating parapoxvirus therapeutic. Mol. Ther. 2012, 20, 1148–1157. [Google Scholar] [CrossRef]
- Zhang, J.; Tai, L.H.; Ilkow, C.S.; Alkayyal, A.A.; Ananth, A.A.; de Souza, C.T.; Wang, J.; Sahi, S.; Ly, L.; Lefebvre, C.; et al. Maraba mg1 virus enhances natural killer cell function via conventional dendritic cells to reduce postoperative metastatic disease. Mol. Ther. 2014, 22, 1320–1332. [Google Scholar] [CrossRef] [PubMed]
- Krebs, P.; Barnes, M.J.; Lampe, K.; Whitley, K.; Bahjat, K.S.; Beutler, B.; Janssen, E.; Hoebe, K. Nk-cell-mediated killing of target cells triggers robust antigen-specific t-cell-mediated and humoral responses. Blood 2009, 113, 6593–6602. [Google Scholar] [CrossRef]
- Lemay, C.G.; Rintoul, J.L.; Kus, A.; Paterson, J.M.; Garcia, V.; Falls, T.J.; Ferreira, L.; Bridle, B.W.; Conrad, D.P.; Tang, V.A.; et al. Harnessing oncolytic virus-mediated antitumor immunity in an infected cell vaccine. Mol. Ther. 2012, 20, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Alkayyal, A.A.; Tai, L.H.; Kennedy, M.A.; de Souza, C.T.; Zhang, J.; Lefebvre, C.; Sahi, S.; Ananth, A.A.; Mahmoud, A.B.; Makrigiannis, A.P.; et al. Nk-cell recruitment is necessary for eradication of peritoneal carcinomatosis with an il12-expressing maraba virus cellular vaccine. Cancer Immunol. Res. 2017, 5, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, R.J.; Errington, F.; Steele, L.P.; Ilett, E.J.; Morgan, R.S.; Harrington, K.J.; Pandha, H.S.; Selby, P.J.; Vile, R.G.; Melcher, A.A. Reciprocal human dendritic cell-natural killer cell interactions induce antitumor activity following tumor cell infection by oncolytic reovirus. J. Immunol. 2009, 183, 4312–4321. [Google Scholar] [CrossRef]
- Ockert, D.; Schirrmacher, V.; Beck, N.; Stoelben, E.; Ahlert, T.; Flechtenmacher, J.; Hagmuller, E.; Buchcik, R.; Nagel, M.; Saeger, H.D. Newcastle disease virus-infected intact autologous tumor cell vaccine for adjuvant active specific immunotherapy of resected colorectal carcinoma. Clin. Cancer Res. 1996, 2, 21–28. [Google Scholar] [PubMed]
- Liebrich, W.; Schlag, P.; Manasterski, M.; Lehner, B.; Stohr, M.; Moller, P.; Schirrmacher, V. In vitro and clinical characterisation of a newcastle disease virus-modified autologous tumour cell vaccine for treatment of colorectal cancer patients. Eur. J. Cancer 1991, 27, 703–710. [Google Scholar] [CrossRef]
- Schirrmacher, V. Clinical trials of antitumor vaccination with an autologous tumor cell vaccine modified by virus infection: Improvement of patient survival based on improved antitumor immune memory. Cancer Immunol. Immunother. 2005, 54, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Conrad, D.P.; Tsang, J.; Maclean, M.; Diallo, J.S.; Le Boeuf, F.; Lemay, C.G.; Falls, T.J.; Parato, K.A.; Bell, J.C.; Atkins, H.L. Leukemia cell-rhabdovirus vaccine: Personalized immunotherapy for acute lymphoblastic leukemia. Clin. Cancer Res. 2013, 19, 3832–3843. [Google Scholar] [CrossRef] [PubMed]
- Joffre, O.P.; Segura, E.; Savina, A.; Amigorena, S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012, 12, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Tai, L.H.; de Souza, C.T.; Belanger, S.; Ly, L.; Alkayyal, A.A.; Zhang, J.; Rintoul, J.L.; Ananth, A.A.; Lam, T.; Breitbach, C.J.; et al. Preventing postoperative metastatic disease by inhibiting surgery-induced dysfunction in natural killer cells. Cancer Res. 2013, 73, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Degli-Esposti, M.A.; Smyth, M.J. Close encounters of different kinds: Dendritic cells and nk cells take centre stage. Nat. Rev. Immunol. 2005, 5, 112–124. [Google Scholar] [CrossRef]
- Albertsson, P.A.; Basse, P.H.; Hokland, M.; Goldfarb, R.H.; Nagelkerke, J.F.; Nannmark, U.; Kuppen, P.J. Nk cells and the tumour microenvironment: Implications for nk-cell function and anti-tumour activity. Trends Immunol. 2003, 24, 603–609. [Google Scholar] [CrossRef]
- Malmberg, K.J.; Bryceson, Y.T.; Carlsten, M.; Andersson, S.; Bjorklund, A.; Bjorkstrom, N.K.; Baumann, B.C.; Fauriat, C.; Alici, E.; Dilber, M.S.; et al. Nk cell-mediated targeting of human cancer and possibilities for new means of immunotherapy. Cancer Immunol. Immunother. 2008, 57, 1541–1552. [Google Scholar] [CrossRef]
- Woo, C.Y.; Clay, T.M.; Lyerly, H.K.; Morse, M.A.; Osada, T. Role of natural killer cell function in dendritic cell-based vaccines. Expert Rev. Vaccines 2006, 5, 55–65. [Google Scholar] [CrossRef]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Lowdell, M.W.; Craston, R.; Samuel, D.; Wood, M.E.; O’Neill, E.; Saha, V.; Prentice, H.G. Evidence that continued remission in patients treated for acute leukaemia is dependent upon autologous natural killer cells. Br. J. Haematol. 2002, 117, 821–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, M.; Lanier, L.L.; Sigal, L.J. A role for nkg2d in nk cell-mediated resistance to poxvirus disease. PLoS Pathog. 2008, 4, e30. [Google Scholar] [CrossRef] [PubMed]
- Hamerman, J.A.; Ogasawara, K.; Lanier, L.L. Nk cells in innate immunity. Curr. Opin. Immunol. 2005, 17, 29–35. [Google Scholar] [CrossRef]
- Big Pharma Could Turn to Viruses to Boost Cancer Immunotherapies. Available online: https://pharmaphorum.com/news/big-pharma-turn-viruses-boost-cancer-immunotherapies/ (accessed on 10 February 2019).
- Global Cancer Vaccine Market & Clinical Trial Insight 2025 Report Highlight. Available online: https://www.researchandmarkets.com/reports/4620934/global-cancer-vaccine-market-and-clinical-trial (accessed on 5 March 2019).
- 2017 Immuno-Oncology Medicines in Development. Available online: http://phrma-docs.phrma.org/files/dmfile/MID_Immuno-Oncology-2017_Drug-List1.pdf (accessed on 21 March 2019).

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niavarani, S.R.; Lawson, C.; Tai, L.-H. Treatment of Metastatic Disease through Natural Killer Cell Modulation by Infected Cell Vaccines. Viruses 2019, 11, 434. https://doi.org/10.3390/v11050434
Niavarani SR, Lawson C, Tai L-H. Treatment of Metastatic Disease through Natural Killer Cell Modulation by Infected Cell Vaccines. Viruses. 2019; 11(5):434. https://doi.org/10.3390/v11050434
Chicago/Turabian StyleNiavarani, Seyedeh Raheleh, Christine Lawson, and Lee-Hwa Tai. 2019. "Treatment of Metastatic Disease through Natural Killer Cell Modulation by Infected Cell Vaccines" Viruses 11, no. 5: 434. https://doi.org/10.3390/v11050434
APA StyleNiavarani, S. R., Lawson, C., & Tai, L. -H. (2019). Treatment of Metastatic Disease through Natural Killer Cell Modulation by Infected Cell Vaccines. Viruses, 11(5), 434. https://doi.org/10.3390/v11050434