The Interplay between Host Innate Immunity and Hepatitis E Virus

{kind=link}
Abstract
:1. Introduction
2. Innate Immune Response to HEV Infection
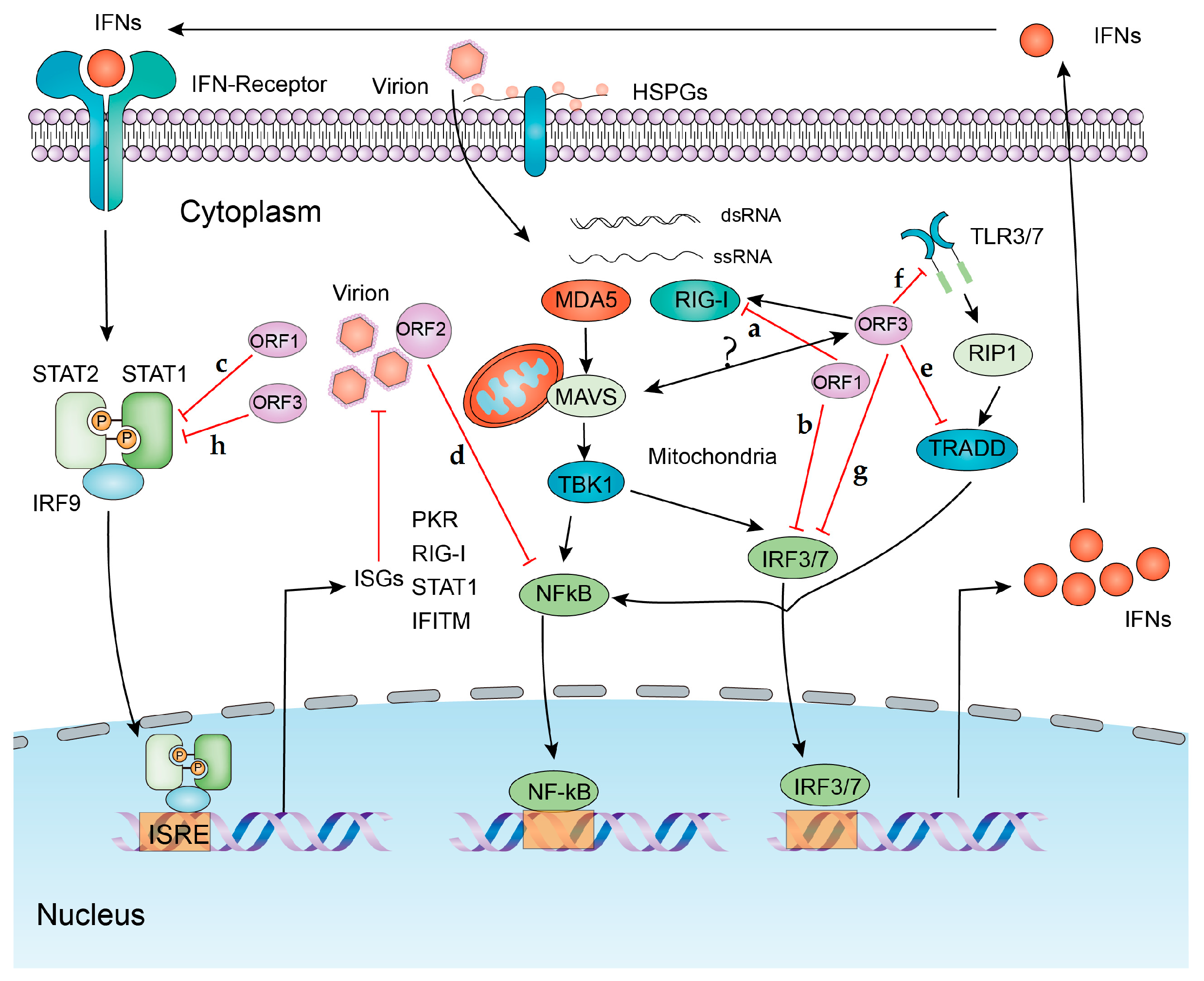
2.1. Recognition by Pathogen-Recognition Receptors in HEV Host Cells
2.2. Innate Immune Cells on HEV Infection
2.3. IFN and Inflammatory Responses to HEV Infection
2.4. The Implications of Innate Immune Response in Patient Outcome and Therapeutic Development
3. Viral Strategies to Counteract Innate Immune Response
4. Conclusion and Perspectives
Funding
Conflicts of Interest
References
- Debing, Y.; Moradpour, D.; Neyts, J.; Gouttenoire, J. Update on hepatitis E virology: Implications for clinical practice. J. Hepatol. 2016, 65, 200–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melgaco, J.G.; Gardinali, N.R.; de Mello, V.D.M.; Leal, M.; Lewis-Ximenez, L.L.; Pinto, M.A. Hepatitis E: Update on prevention and control. Biomed. Res. Int. 2018, 2018, 5769201. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, R.; Patra, S.; David, P.; Vyas, A.; Khanam, A.; Hissar, S.; Gupta, E.; Kumar, G.; Kottilil, S.; Maiwall, R.; et al. Impaired monocyte-macrophage functions and defective toll-like receptor signaling in hepatitis E virus-infected pregnant women with acute liver failure. Hepatology 2015, 62, 1683–1696. [Google Scholar] [CrossRef] [PubMed]
- Beniwal, M.; Kumar, A.; Kar, P.; Jilani, N.; Sharma, J.B. Prevalence and severity of acute viral hepatitis and fulminant hepatitis during pregnancy: A prospective study from North India. Indian J. Med. Microbiol. 2003, 21, 184–185. [Google Scholar] [PubMed]
- Kamar, N.; Selves, J.; Mansuy, J.M.; Ouezzani, L.; Peron, J.M.; Guitard, J.; Cointault, O.; Esposito, L.; Abravanel, F.; Danjoux, M.; et al. Hepatitis E virus and chronic hepatitis in organ-transplant recipients. N. Engl. J. Med. 2008, 358, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; de Man, R.A.; de Knegt, R.J.; Metselaar, H.J.; Peppelenbosch, M.P.; Pan, Q. Epidemiology and management of chronic Hepatitis E infection in solid organ transplantation: A comprehensive literature review. Rev. Med. Virol. 2013, 23, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, G.; Pan, Q.; Zhao, J. Chronic Hepatitis E in a renal transplant recipient: The first report of genotype 4 hepatitis E virus caused chronic infection in organ recipient. Gastroenterology 2018, 154, 1199–1201. [Google Scholar] [CrossRef] [PubMed]
- Hakim, M.S.; Ikram, A.; Zhou, J.; Wang, W.; Peppelenbosch, M.P.; Pan, Q. Immunity against hepatitis E virus infection: Implications for therapy and vaccine development. Rev. Med. Virol. 2018, 28. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, L.; Su, J.; Peppelenbosch, M.P.; Pan, Q. Transcriptional regulation of antiviral interferon-stimulated genes. Trends Microbiol. 2017, 25, 573–584. [Google Scholar] [CrossRef]
- Xu, L.; Wang, W.; Peppelenbosch, M.P.; Pan, Q. Noncanonical antiviral mechanisms of ISGS: Dispensability of inducible interferons. Trends Immunol. 2017, 38, 1–2. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol. 1989, 54 Pt. 1, 1–13. [Google Scholar] [CrossRef]
- Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-Kappab by toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Ohba, Y.; Yanai, H.; Negishi, H.; Mizutani, T.; Takaoka, A.; Taya, C.; Taniguchi, T. Spatiotemporal regulation of myd88-irf-7 signalling for robust type-I interferon induction. Nature 2005, 434, 1035–1040. [Google Scholar] [CrossRef] [PubMed]
- Xagorari, A.; Chlichlia, K. Toll-like receptors and viruses: Induction of innate antiviral immune responses. Open Microbiol. J. 2008, 2, 49–59. [Google Scholar] [CrossRef]
- Li, K.; Li, N.L.; Wei, D.; Pfeffer, S.R.; Fan, M.; Pfeffer, L.M. Activation of chemokine and inflammatory cytokine response in hepatitis C virus-infected hepatocytes depends on toll-like receptor 3 sensing of hepatitis C virus double-stranded RNA intermediates. Hepatology 2012, 55, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Negash, A.A.; Ramos, H.J.; Crochet, N.; Lau, D.T.; Doehle, B.; Papic, N.; Delker, D.A.; Jo, J.; Bertoletti, A.; Hagedorn, C.H.; et al. Il-1beta production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 2013, 9, e1003330. [Google Scholar] [CrossRef]
- Real, C.I.; Lu, M.; Liu, J.; Huang, X.; Trippler, M.; Hossbach, M.; Deckert, J.; Jahn-Hofmann, K.; Ickenstein, L.M.; John, M.J.; et al. Hepatitis b virus genome replication triggers toll-like receptor 3-dependent interferon responses in the absence of hepatitis B surface antigen. Sci. Rep. 2016, 6, 24865. [Google Scholar] [CrossRef]
- Sepehri, Z.; Kiani, Z.; Alavian, S.M.; Arababadi, M.K.; Kennedy, D. The link between TLR7 signaling and hepatitis B virus infection. Life Sci. 2016, 158, 63–69. [Google Scholar] [CrossRef]
- Devhare, P.B.; Desai, S.; Lole, K.S. Innate immune responses in human hepatocyte-derived cell lines alter genotype 1 hepatitis E virus replication efficiencies. Sci. Rep. 2016, 6, 26827. [Google Scholar] [CrossRef] [Green Version]
- Devhare, P.B.; Chatterjee, S.N.; Arankalle, V.A.; Lole, K.S. Analysis of antiviral response in human epithelial cells infected with hepatitis E virus. PLoS ONE 2013, 8, e63793. [Google Scholar] [CrossRef]
- Nan, Y.; Ma, Z.; Wang, R.; Yu, Y.; Kannan, H.; Fredericksen, B.; Zhang, Y.J. Enhancement of interferon induction by ORF3 product of hepatitis E virus. J. Virol. 2014, 88, 8696–8705. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, M.; Ratho, R.K.; Chawla, Y.; Singh, M.P. Role of tlr gene expression and cytokine profiling in the immunopathogenesis of viral hepatitis E. J. Clin. Virol. 2015, 73, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of mda5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Kato, H.; Kumagai, Y.; Yoneyama, M.; Sato, S.; Matsushita, K.; Tsujimura, T.; Fujita, T.; Akira, S.; Takeuchi, O. Lgp2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc. Natl. Acad. Sci. USA 2010, 107, 1512–1517. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-i and melanoma differentiation-associated gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and MDA5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of mavs, a mitochondrial antiviral signaling protein that activates NF-Kappab and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. Visa is an adapter protein required for virus-triggered IFN-Beta signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the rig-i antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef]
- Lu, H.L.; Liao, F. Melanoma differentiation-associated gene 5 senses hepatitis B virus and activates innate immune signaling to suppress virus replication. J. Immunol. 2013, 191, 3264–3276. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Li, K.; Kameyama, T.; Hayashi, T.; Ishida, Y.; Murakami, S.; Watanabe, T.; Iijima, S.; Sakurai, Y.; Watashi, K.; et al. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 2015, 42, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Owen, D.M.; Jiang, F.; Marcotrigiano, J.; Gale, M., Jr. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 2008, 454, 523–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Ding, Q.; Lu, J.; Tao, W.; Huang, B.; Zhao, Y.; Niu, J.; Liu, Y.J.; Zhong, J. MDA5 plays a critical role in interferon response during hepatitis C virus infection. J. Hepatol. 2015, 62, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Pan, T.; Xu, J.; Zhang, Y.; Song, W.; Yi, Z.; Yuan, Z. Hepatitis C virus replicative double-stranded RNA is a potent interferon inducer that triggers interferon production through MDA5. J. Gen. Virol 2016, 97, 2868–2882. [Google Scholar] [CrossRef] [PubMed]
- Jagya, N.; Varma, S.P.; Thakral, D.; Joshi, P.; Durgapal, H.; Panda, S.K. RNA-seq based transcriptome analysis of hepatitis E virus (HEV) and hepatitis B virus (HBV) replicon transfected HUH-7 cells. PLoS ONE 2014, 9, e87835. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, Y.; Qu, C.; Wang, S.; Zhou, J.; Cao, W.; Xu, L.; Ma, B.; Hakim, M.S.; Yin, Y.; et al. The RNA genome of hepatitis E virus robustly triggers an antiviral interferon response. Hepatology 2018, 67, 2096–2112. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Bendall, R.P.; Peron, J.M.; Cintas, P.; Prudhomme, L.; Mansuy, J.M.; Rostaing, L.; Keane, F.; Ijaz, S.; Izopet, J.; et al. Hepatitis E virus and neurologic disorders. Emerg. Infect. Dis. 2011, 17, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Cheung, M.C.; Maguire, J.; Carey, I.; Wendon, J.; Agarwal, K. Review of the neurological manifestations of hepatitis E infection. Ann. Hepatol. 2012, 11, 618–622. [Google Scholar]
- Wang, Y.; Wang, S.; Wu, J.; Jiang, Y.; Zhang, H.; Li, S.; Liu, H.; Yang, C.; Tang, H.; Guo, N.; et al. Hepatitis E virus infection in acute non-traumatic neuropathy: A large prospective case-control study in china. EBioMedicine 2018, 36, 122–130. [Google Scholar] [CrossRef]
- Kamar, N.; Mansuy, J.M.; Esposito, L.; Legrand-Abravanel, F.; Peron, J.M.; Durand, D.; Rostaing, L.; Izopet, J. Acute hepatitis and renal function impairment related to infection by hepatitis E virus in a renal allograft recipient. Am. J. Kidney Dis. 2005, 45, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Jha, R.; Lakhtakia, S.; Narayan, G. Acute pancreatitis following kidney transplantation - role of viral infections. Clin. Transplant. 2003, 17, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Deniel, C.; Coton, T.; Brardjanian, S.; Guisset, M.; Nicand, E.; Simon, F. Acute pancreatitis: A rare complication of acute hepatitis E. J. Clin. Virol 2011, 51, 202–204. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Huang, F.; Xu, L.; Lin, Z.; de Vrij, F.M.S.; Ayo-Martin, A.C.; van der Kroeg, M.; Zhao, M.; Yin, Y.; Wang, W.; et al. Hepatitis E virus infects neurons and brains. J. Infect. Dis. 2017, 215, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Shi, R.; Liu, T.; She, R.; Wu, Q.; An, J.; Hao, W.; Soomro, M.H. Brain infection by hepatitis E virus probably via damage of the blood-brain barrier due to alterations of tight junction proteins. Front. Cell Infect. Microbiol. 2019, 9, 52. [Google Scholar] [CrossRef]
- Huang, F.; Yang, C.; Zhou, X.; Yu, W.; Pan, Q. Rhesus macaques persistently infected with hepatitis E shed virus into Urine. J. Hepatol. 2016, 64, 1446–1447. [Google Scholar] [CrossRef]
- Gouilly, J.; Chen, Q.; Siewiera, J.; Cartron, G.; Levy, C.; Dubois, M.; Al-Daccak, R.; Izopet, J.; Jabrane-Ferrat, N.; El Costa, H. Genotype specific pathogenicity of hepatitis E virus at the human mateRNAl-fetal interface. Nat. Commun. 2018, 9, 4748. [Google Scholar] [CrossRef]
- Rouse, B.T.; Sehrawat, S. Immunity and immunopathology to viruses: What decides the outcome? Nat. Rev. Immunol. 2010, 10, 514–526. [Google Scholar] [CrossRef]
- Cooper, M.A.; Fehniger, T.A.; Turner, S.C.; Chen, K.S.; Ghaheri, B.A.; Ghayur, T.; Carson, W.E.; Caligiuri, M.A. Human natural killer cells: A unique innate immunoregulatory role for the CD56(bright) subset. Blood 2001, 97, 3146–3151. [Google Scholar] [CrossRef]
- Racanelli, V.; Rehermann, B. The liver as an immunological organ. Hepatology 2006, 43, S54–S62. [Google Scholar] [CrossRef]
- Rehermann, B.; Nascimbeni, M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat. Rev. Immunol. 2005, 5, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Alvarez, F. Role of nk and nkt cells in the immunopathogenesis of HCV-induced hepatitis. J. Leukoc. Biol. 2004, 76, 743–759. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.; Aggarwal, R.; Jameel, S.; Puri, P.; Gupta, V.K.; Ramesh, V.S.; Bhatia, S.; Naik, S. Cellular immune responses in acute hepatitis E virus infection to the viral open reading frame 2 protein. Viral. Immunol. 2007, 20, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.; Aggarwal, R.; Bhagat, M.R.; Chowdhury, A.; Naik, S. Alterations in natural killer cells and natural killer T cells during acute viral hepatitis E. J. Viral. Hepat. 2008, 15, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Baley, J.E.; Schacter, B.Z. Mechanisms of diminished natural killer cell activity in pregnant women and neonates. J. Immunol. 1985, 134, 3042–3048. [Google Scholar] [PubMed]
- Prabhu, S.B.; Gupta, P.; Durgapal, H.; Rath, S.; Gupta, S.D.; Acharya, S.K.; Panda, S.K. Study of cellular immune response against hepatitis E virus (HEV). J. Viral. Hepat. 2011, 18, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Abravanel, F.; Barrague, H.; Dorr, G.; Saune, K.; Peron, J.M.; Alric, L.; Kamar, N.; Izopet, J.; Champagne, E. Conventional and innate lymphocytes response at the acute phase of HEV infection in transplanted patients. J. Infect. 2016, 72, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Clausen, J.; Vergeiner, B.; Enk, M.; Petzer, A.L.; Gastl, G.; Gunsilius, E. Functional significance of the activation-associated receptors CD25 and CD69 on human nk-cells and NK-like T-cells. Immunobiology 2003, 207, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Xiao, P.; Li, R.; She, R.; Tian, J.; Wang, J.; Mao, J.; Yin, J.; Shi, R. Increased mast cell activation in mongolian gerbils infected by hepatitis E virus. Front. Microbiol. 2018, 9, 2226. [Google Scholar] [CrossRef] [PubMed]
- Mesev, E.V.; LeDesma, R.A.; Ploss, A. Decoding type i and iii interferon signalling during viral infection. Nat. Microbiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type i interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yin, Y.; Xu, L.; Su, J.; Huang, F.; Wang, Y.; Boor, P.P.C.; Chen, K.; Wang, W.; Cao, W.; et al. Unphosphorylated ISGF3 drives constitutive expression of interferon-stimulated genes to protect against viral infections. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Li, X.; Ambardekar, C.; Hu, Z.; Lhomme, S.; Feng, Z. Hepatitis E virus persists in the presence of a type iii interferon response. PLoS Pathog. 2017, 13, e1006417. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Tanaka, T.; Takahashi, H.; Hoshino, Y.; Nagashima, S.; Jirintai; Mizuo, H.; Yazaki, Y.; Takagi, T.; Azuma, M.; et al. Hepatitis E virus (HEV) strains in serum samples can replicate efficiently in cultured cells despite the coexistence of HEV antibodies: Characterization of hev virions in blood circulation. J. Clin. Microbiol. 2010, 48, 1112–1125. [Google Scholar] [PubMed]
- Xu, L.; Wang, W.; Li, Y.; Zhou, X.; Yin, Y.; Wang, Y.; de Man, R.A.; van der Laan, L.J.W.; Huang, F.; Kamar, N.; et al. Rig-i is a key antiviral interferon-stimulated gene against hepatitis E virus regardless of interferon production. Hepatology 2017, 65, 1823–1839. [Google Scholar] [CrossRef]
- Xu, L.; Zhou, X.; Wang, W.; Wang, Y.; Yin, Y.; Laan, L.J.; Sprengers, D.; Metselaar, H.J.; Peppelenbosch, M.P.; Pan, Q. IFN regulatory factor 1 restricts hepatitis E virus replication by activating stat1 to induce antiviral ifn-stimulated genes. Faseb J. 2016, 30, 3352–3367. [Google Scholar] [CrossRef]
- Hsiang, T.Y.; Zhao, C.; Krug, R.M. Interferon-induced isg15 conjugation inhibits influenza A virus gene expression and replication in human cells. J. Virol. 2009, 83, 5971–5977. [Google Scholar] [CrossRef]
- Broering, R.; Zhang, X.; Kottilil, S.; Trippler, M.; Jiang, M.; Lu, M.; Gerken, G.; Schlaak, J.F. The interferon stimulated gene 15 functions as a proviral factor for the hepatitis C virus and as a regulator of the ifn response. Gut 2010, 59, 1111–1119. [Google Scholar] [CrossRef]
- Sooryanarain, H.; Rogers, A.J.; Cao, D.; Haac, M.E.R.; Karpe, Y.A.; Meng, X.J. ISG15 modulates type I interferon signaling and the antiviral response during hepatitis E virus replication. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Cao, D.; Cao, Q.M.; Subramaniam, S.; Yugo, D.M.; Heffron, C.L.; Rogers, A.J.; Kenney, S.P.; Tian, D.; Matzinger, S.R.; Overend, C.; et al. Pig model mimicking chronic hepatitis E virus infection in immunocompromised patients to assess immune correlates during chronicity. Proc. Natl. Acad. Sci. USA 2017, 114, 6914–6923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, H.S.; Han, S.H.; Kim, Y.H.; Park, B.J.; Kim, D.H.; Lee, J.B.; Park, S.Y.; Song, C.S.; Lee, S.W.; Choi, C.; et al. Adverse fetal outcomes in pregnant rabbits experimentally infected with rabbit hepatitis E virus. Virology 2017, 512, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Devi, S.G.; Kar, P.; Agarwal, S.; Husain, S.A.; Gupta, R.K.; Sharma, S. Association of cytokines in hepatitis E with pregnancy outcome. Cytokine 2014, 65, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, L.; Brandsma, J.H.; Wang, Y.; Hakim, M.S.; Zhou, X.; Yin, Y.; Fuhler, G.M.; van der Laan, L.J.; van der Woude, C.J.; et al. Convergent transcription of interferon-stimulated genes by tnf-alpha and ifn-alpha augments antiviral activity against hcv and hev. Sci. Rep. 2016, 6, 25482. [Google Scholar] [CrossRef] [PubMed]
- Kraef, C.; Schlein, C.; Hiller, J.; Westholter, D.; Denzer, U.; Horvatits, T.; Peine, S.; Lohse, A.W.; Lutgehetmann, M.; Polywka, S.; et al. Course of hev viremia and anti-HEV igm/igg response in asymptomatic blood donors. J. Clin. Virol. 2018, 105, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Eldin, S.S.; Seddik, I.; Daef, E.A.; Shata, M.T.; Raafat, M.; Abdel Baky, L.; Nafeh, M.A. Risk factors and immune response to hepatitis E viral infection among acute hepatitis patients in assiut, Egypt. Egypt J. Immunol. 2010, 17, 73–86. [Google Scholar]
- Pal, R.; Aggarwal, R.; Naik, S.R.; Das, V.; Das, S.; Naik, S. Immunological alterations in pregnant women with acute hepatitis E. J. Gastroenterol. Hepatol. 2005, 20, 1094–1101. [Google Scholar] [CrossRef]
- Navaneethan, U.; Al Mohajer, M.; Shata, M.T. Hepatitis E and pregnancy: Understanding the pathogenesis. Liver Int. 2008, 28, 1190–1199. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, X.; Debing, Y.; Chen, K.; van Der Laan, L.J.; Neyts, J.; Janssen, H.L.; Metselaar, H.J.; Peppelenbosch, M.P.; Pan, Q. Calcineurin inhibitors stimulate and mycophenolic acid inhibits replication of hepatitis E virus. Gastroenterology 2014, 146, 1775–1783. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, Y.; Metselaar, H.J.; Janssen, H.L.; Peppelenbosch, M.P.; Pan, Q. Rapamycin and everolimus facilitate hepatitis E virus replication: Revealing a basal defense mechanism of PI3K-PKB-mtor pathway. J. Hepatol. 2014, 61, 746–754. [Google Scholar] [CrossRef]
- Wang, Y.; Metselaar, H.J.; Peppelenbosch, M.P.; Pan, Q. Chronic hepatitis E in solid-organ transplantation: The key implications of immunosuppressants. Curr. Opin. Infect. Dis. 2014, 27, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Xu, L.; Wang, W.; Watashi, K.; Wang, Y.; Sprengers, D.; de Ruiter, P.E.; van der Laan, L.J.; Metselaar, H.J.; Kamar, N.; et al. Disparity of basal and therapeutically activated interferon signalling in constraining hepatitis E virus infection. J. Viral. Hepat. 2016, 23, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Van de Garde, M.D.B.; Pas, S.D.; van Oord, G.W.; Gama, L.; Choi, Y.; de Man, R.A.; Boonstra, A.; Vanwolleghem, T. Interferon-alpha treatment rapidly clears hepatitis E virus infection in humanized mice. Sci. Rep. 2017, 7, 8267. [Google Scholar] [CrossRef] [PubMed]
- Peters van Ton, A.M.; Gevers, T.J.; Drenth, J.P. Antiviral therapy in chronic hepatitis E: A systematic review. J. Viral. Hepat. 2015, 22, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Haagsma, E.B.; Riezebos-Brilman, A.; van den Berg, A.P.; Porte, R.J.; Niesters, H.G. Treatment of chronic hepatitis E in liver transplant recipients with pegylated interferon Alpha-2B. Liver Transpl. 2010, 16, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Abravanel, F.; Garrouste, C.; Cardeau-Desangles, I.; Mansuy, J.M.; Weclawiak, H.; Izopet, J.; Rostaing, L. Three-month pegylated interferon-alpha-2a therapy for chronic hepatitis E virus infection in a haemodialysis patient. Nephrol. Dial. Transplant. 2010, 25, 2792–2795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamar, N.; Rostaing, L.; Abravanel, F.; Garrouste, C.; Esposito, L.; Cardeau-Desangles, I.; Mansuy, J.M.; Selves, J.; Peron, J.M.; Otal, P.; et al. Pegylated interferon-alpha for treating chronic hepatitis E virus infection after liver transplantation. Clin. Infect. Dis. 2010, 50, e30–e33. [Google Scholar] [CrossRef] [PubMed]
- Kasumba, D.M.; Grandvaux, N. Therapeutic targeting of rig-i and mda5 might not lead to the same rome. Trends Pharmacol. Sci. 2019, 40, 116–127. [Google Scholar] [CrossRef]
- Nan, Y.; Yu, Y.; Ma, Z.; Khattar, S.K.; Fredericksen, B.; Zhang, Y.J. Hepatitis E virus inhibits type I interferon induction by ORF1 products. J. Virol. 2014, 88, 11924–11932. [Google Scholar] [CrossRef]
- Bagdassarian, E.; Doceul, V.; Pellerin, M.; Demange, A.; Meyer, L.; Jouvenet, N.; Pavio, N. The amino-terminal region of hepatitis E virus orf1 containing a methyltransferase (Met) and a papain-like cysteine protease (PCP) domain counteracts type i interferon response. Viruses 2018, 10. [Google Scholar] [CrossRef]
- Ojha, N.K.; Lole, K.S. Hepatitis E virus orf1 encoded macro domain protein interacts with light chain subunit of human ferritin and inhibits its secretion. Mol. Cell Biochem. 2016, 417, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Haldipur, B.; Bhukya, P.L.; Arankalle, V.; Lole, K. Positive regulation of hepatitis E virus replication by microRNA-122. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Zhang, J.; Wang, Y.B.; Li, S.W.; Yang, H.J.; Luo, W.X.; Xia, N.S. Selection of a peptide mimic the neutralization epitope of hepatitis E virus with phage peptide display technology. Sheng Wu Gong Cheng Xue Bao 2003, 19, 680–685. [Google Scholar] [PubMed]
- Montpellier, C.; Wychowski, C.; Sayed, I.M.; Meunier, J.C.; Saliou, J.M.; Ankavay, M.; Bull, A.; Pillez, A.; Abravanel, F.; Helle, F.; et al. Hepatitis E virus lifecycle and identification of 3 forms of the ORF2 capsid protein. Gastroenterology 2018, 154, 211–223 e218. [Google Scholar] [CrossRef] [PubMed]
- Kalia, M.; Chandra, V.; Rahman, S.A.; Sehgal, D.; Jameel, S. Heparan sulfate proteoglycans are required for cellular binding of the hepatitis E virus ORF2 capsid protein and for viral infection. J. Virol. 2009, 83, 12714–12724. [Google Scholar] [CrossRef] [PubMed]
- Surjit, M.; Varshney, B.; Lal, S.K. The orf2 glycoprotein of hepatitis E virus inhibits cellular nf-kappab activity by blocking ubiquitination mediated proteasomal degradation of ikappabalpha in human hepatoma cells. BMC Biochem 2012, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Huang, W.; Yang, J.; Wen, Z.; Geng, Y.; Zhao, C.; Zhang, H.; Wang, Y. Systematic identification of hepatitis E virus ORF2 interactome reveals that TMEM134 engages in ORF2-mediated NF-Kappab pathway. Virus Res. 2017, 228, 102–108. [Google Scholar] [CrossRef] [PubMed]
- John, L.; Thomas, S.; Herchenroder, O.; Putzer, B.M.; Schaefer, S. Hepatitis E virus ORF2 protein activates the pro-apoptotic gene chop and anti-apoptotic heat shock proteins. PLoS ONE 2011, 6, e25378. [Google Scholar] [CrossRef]
- Lei, Q.; Li, L.; Zhang, S.; Li, T.; Zhang, X.; Ding, X.; Qin, B. Hev orf3 downregulates tlr7 to inhibit the generation of type i interferon via impairment of multiple signaling pathways. Sci. Rep. 2018, 8, 8585. [Google Scholar] [CrossRef]
- Ding, Q.; Heller, B.; Capuccino, J.M.; Song, B.; Nimgaonkar, I.; Hrebikova, G.; Contreras, J.E.; Ploss, A. Hepatitis E virus ORF3 is a functional ion channel required for release of infectious particles. Proc. Natl. Acad. Sci. USA 2017, 114, 1147–1152. [Google Scholar] [CrossRef] [Green Version]
- Chandra, V.; Kalia, M.; Hajela, K.; Jameel, S. The orf3 protein of hepatitis E virus delays degradation of activated growth factor receptors by interacting with CIN85 and blocking formation of the CBL-CIN85 complex. J. Virol. 2010, 84, 3857–3867. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Zafrullah, M.; Mixson-Hayden, T.; Dai, X.; Liang, J.; Meng, J.; Kamili, S. Suppression of interferon-alpha signaling by hepatitis E virus. Hepatology 2012, 55, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Wang, M.; Huang, Y.; Peng, W.; Zheng, Z.; Xia, N.; Xu, J.; Tian, D. The orf3 protein of genotype 1 hepatitis E virus suppresses TLR3-induced NF-Kappab signaling via tradd and RIP1. Sci. Rep. 2016, 6, 27597. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.P.; Anang, S.; Subramani, C.; Madhvi, A.; Bakshi, K.; Srivastava, A.; Shalimar; Nayak, B.; Ranjith Kumar, C.T.; Surjit, M. Endoplasmic reticulum stress induced synthesis of a novel viral factor mediates efficient replication of genotype-1 hepatitis E virus. PLoS Pathog. 2016, 12, e1005521. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Qu, C.; Yu, P.; Ou, X.; Pan, Q.; Wang, W. The Interplay between Host Innate Immunity and Hepatitis E Virus. Viruses 2019, 11, 541. https://doi.org/10.3390/v11060541
Li Y, Qu C, Yu P, Ou X, Pan Q, Wang W. The Interplay between Host Innate Immunity and Hepatitis E Virus. Viruses. 2019; 11(6):541. https://doi.org/10.3390/v11060541
Chicago/Turabian StyleLi, Yang, Changbo Qu, Peifa Yu, Xumin Ou, Qiuwei Pan, and Wenshi Wang. 2019. "The Interplay between Host Innate Immunity and Hepatitis E Virus" Viruses 11, no. 6: 541. https://doi.org/10.3390/v11060541
APA StyleLi, Y., Qu, C., Yu, P., Ou, X., Pan, Q., & Wang, W. (2019). The Interplay between Host Innate Immunity and Hepatitis E Virus. Viruses, 11(6), 541. https://doi.org/10.3390/v11060541