Genetic Analysis of the Full-Length gag Gene from the Earliest Korean Subclade B of HIV-1: An Outbreak among Korean Hemophiliacs



Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Patients and Samples
2.2. RNA Preparation and gag Gene Amplification
2.3. Phylogenetic Tree Analysis
2.4. Viral Signature Pattern Analysis (VESPA)
2.5. Statistical Analysis
2.6. Nucleotide Sequence Data
3. Results
3.1. Origin of the KSB of Subtype B
3.2. Molecular Epidemiologic Data on the gag Gene
3.3. Nucleotide/Amino Acid Signature Patterns
3.4. Korean Signature Pattern Amino Acid Residues and Duplication of the PTAP Motif in the p6 Gag Protein
3.5. Comparison of the Frequency of 100%-Specific Nucleotides among the Nine Genes
3.6. Sequence Identities of Donors O and P
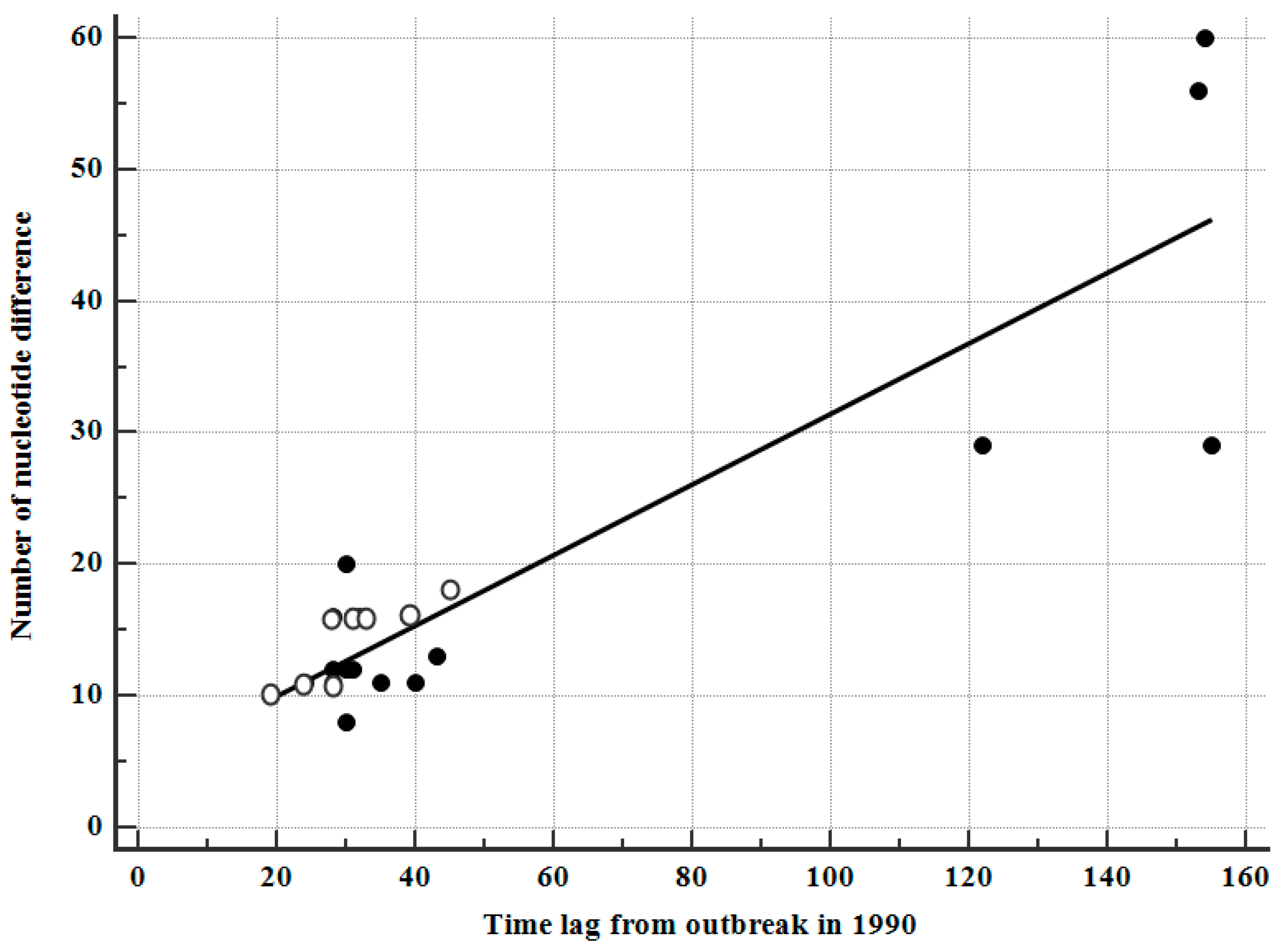
3.7. Correlation between Sampling Intervals and Number of Nucleotide Differences
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cho, Y.K.; Foley, B.T.; Sung, H.; Kim, Y.B.; Kim, J.H. Molecular epidemiologic study on HIV-1 outbreak in hemophiliacs B through clotting factor 9 after 1990. Vox Sang. 2007, 92, 113–120. [Google Scholar] [CrossRef]
- Cho, Y.K.; Jung, Y.S.; Foley, B.T. Phylogenetic analysis of full-length pol gene from Korean hemophiliacs and plasma donors infected with Korean subclade B of HIV-1. AIDS Res. Hum. Retrovir. 2011, 27, 613–621. [Google Scholar] [CrossRef]
- Cho, Y.K.; Jung, Y.; Lee, J.S.; Foley, B.T. Molecular evidence of HIV-1 transmission in 20 Korean hemophiliacs; phylogenetic analysis of vif gene. Haemophilia 2012, 18, 291–299. [Google Scholar] [CrossRef]
- Cho, Y.K.; Kim, J.E.; Foley, B.T. Phylogenetic analysis of the earliest nef gene from hemophiliacs and local controls in Korea. BioRes. Open Access 2012, 1, 41–49. [Google Scholar] [CrossRef]
- Cho, Y.K.; Kim, J.E.; Jeong, D.; Foley, B.T. Signature pattern analysis for the full-length env gene of the earliest Korean subclade B of HIV-1: Outbreak among Korean hemophiliacs. Virus Genes 2017, 53, 789–796. [Google Scholar] [CrossRef]
- Junqueira, D.M.; Almeida, S.E. HIV-1 subtype B: Traces of a pandemic. Virology 2016, 495, 173–184. [Google Scholar] [CrossRef]
- Daniels, R.S.; Kang, C.; Patel, D.; Xiang, Z.; Douglas, N.W.; Zheng, N.N.; Cho, H.W.; Lee, J.S. An HIV type 1 subtype B founder effect in Korea: gp160 signature patterns infer circulation of CTL-escape strains at the population level. AIDS Res. Hum. Retrovir. 2003, 19, 631–641. [Google Scholar] [CrossRef]
- Kim, M.S.; Jang, S.Y.; Park, C.S.; Lee, K.M.; Lee, D.H.; Lee, C.H. Timing and evolution of the most recent common ancestor of the Korean clade HIV subtype B based on nef and vif sequences. J. Microbiol. 2009, 47, 85–90. [Google Scholar] [CrossRef]
- Korber, B.; Myers, G. Signature pattern analysis: A method for assessing viral sequence relatedness. AIDS Res. Hum. Retrovir. 1992, 8, 1549–1560. [Google Scholar] [CrossRef]
- Leitner, T.; Escanilla, D.; Franzén, C.; Uhlén, M.; Albert, J. Accurate reconstruction of a known HIV-1 transmission history by phylogenetic tree analysis. Proc. Natl. Acad. Sci. USA 1996, 93, 10864–10869. [Google Scholar] [CrossRef]
- Jee, Y.M.; Go, U.; Cheon, D.; Kang, Y.; Yoon, J.D.; Lee, S.W.; Shin, Y.H.; Kim, K.S.; Lee, J.K.; Jeong, E.K.; et al. Detection of hepatitis A virus from clotting factors implicated as a source of HAV infection among haemophilia patients in Korea. Epidemiol. Infect. 2006, 134, 87–93. [Google Scholar] [CrossRef]
- Stephenson, K.E.; Barouch, D.H. A global approach to HIV-1 vaccine development. Immunol. Rev. 2013, 254, 295–304. [Google Scholar] [CrossRef]
- Cho, Y.K.; Kim, J.E.; Foley, B.T. Phylogenetic analysis of near full-length HIV-1 genomic sequences from 21 Korean individuals. Aids Res. Hum. Retrovir. 2013, 29, 738–743. [Google Scholar] [CrossRef]
- Cho, Y.K.; Sung, H.; Bae, I.G.; Oh, H.B.; Kim, N.J.; Woo, J.H.; Kim, Y.B. Full sequence of HIV type 1 Korean subtype B in an AIDS case with atypical seroconversion: TAAAA at TATA box. AIDS Res. Hum. Retrovir. 2005, 21, 961–964. [Google Scholar] [CrossRef]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef]
- Ou, C.Y.; Ciesielski, C.A.; Myers, G.; Bandea, C.I.; Luo, C.C.; Korber, B.T.; Mullins, J.I.; Schochetman, G.; Berkelman, R.L.; Economou, A.N.; et al. Molecular epidemiology of HIV transmission in a dental practice. Science 1992, 256, 1165–1171. [Google Scholar] [CrossRef]
- Cho, Y.K.; Kim, J.E.; Woo, J.H. Genetic defects in the nef gene are associated with Korean Red Ginseng intake: Monitoring of nef sequence polymorphisms over 20 years. J. Ginseng Res. 2017, 41, 144–150. [Google Scholar] [CrossRef]
- Cho, Y.K.; Kim, J.E.; Woo, J.H. Long-term follow up of HIV-1-infected Korean haemophiliacs, after infection from a common source of virus. Haemophilia 2015, 21, e1–e11. [Google Scholar]
- Cho, Y.K.; Kim, J.E.; Lee, S.H.; Foley, B.T.; Choi, B.S. Impact of HIV-1 subtype and Korean Red Ginseng on AIDS progression: Comparison of subtype B and subtype D. J. Ginseng Res. 2019, 43, 312–319. [Google Scholar] [CrossRef]
- Sharma, S.; Arunachalam, P.S.; Menon, M.; Ragupathy, V.; Satya, R.V.; Jebaraj, J.; Aralaguppe, S.G.; Rao, C.; Pal, S.; Saravanan, S.; et al. PTAP motif duplication in the p6 Gag protein confers a replication advantage on HIV-1 subtype C. J. Biol. Chem. 2018, 293, 11687–11708. [Google Scholar] [CrossRef] [Green Version]
- Schmalen, A.; Karius-Fischer, J.; Rauch, P.; Setz, C.; Korn, K.; Henklein, P.; Fossen, T.; Schubert, U. The N-terminus of the HIV-1 p6 Gag protein regulates susceptibility to degradation by IDE. Viruses 2018, 10, 710. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| 14 Signature Nucleotide Positions | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide positions in HXB2 | 807 | 823 | 940 | 1012 | 1151 | 1264 | 1281 | 1674 | 1707 | 1722 | 1897 | 1899 | 2155 | 2190 |
| Nucleotide in HXB2 | A | A | T | C | A | A | C | C | C | G | G | A | A | A |
| Signature nucleotide | G | T | T | T | A | A | G | T | G | |||||
| Cluster-O (n = 9) | 1.0a | 1.0 | 1.0 | 1.0 | 1.0 | 0.89 | 1.0 | 1.0 | 1.0 | |||||
| Other KSB (n = 85) | 0.04b | 0.13 | 0.05 | 0.11 | 0.05 | 0.02 | 0.00 | 0.13 | 0.13 | |||||
| Signature nucleotide | G | C | A | G | G | |||||||||
| Cluster-P (n = 13) | 0.85 | 1.0 | 0.92 | 1.0 | 0.92 | |||||||||
| Other KSB (n = 81) | 0.25 | 0.04 | 0.12 | 0.30 | 0.40 | |||||||||
| Signature Pattern Amino Acids at 12 Residues | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Position in HXB2 | 30 | 34 | 62 | 94 | 102 | 114 | 119 | 138 | 389 | 403 | 483 | 490 |
| KSB (n = 94) | R83 | L78 | G86 | I89 | E77 | K93 | T89 | L93 | T74 | K89 | L88 | R84 |
| k11 | i14 | e5 | v5 | d14 | n1 | a5 | m1 | i10 | r5 | p3 | k20 | |
| v2 | a2, r1 | k1, h1 | p10 | q2, k2, h1 | ||||||||
| Non-KSB (n = 25) | k13 | I20 | g15 | V19 | e15 | k13 | A24 | l15 | i9 | r14 | l13 | k15 |
| r9 | l4 | e6 | i5 | d10 | r7 | t1 | m5 | t6 | k11 | m9 | r10 | |
| q3 | i2 | t4 | i3, v2 | n3, s3 | r2 | |||||||
| v1 | n1 | m3, g1 | q1 | |||||||||
| 10 Specific Nucleotides | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | gag | pol | vif | vpr | tat, rev, vpu | env | nef | |||||
| No. of KSB (n) | 94 | 91 [2] | 127 [3] | 52* | 52* | 77 [5] | 143 [4] | |||||
| Nucleotide position | 1899 | 2321 | 4235 | 5070 | 5253 | 5621 | None | 6473 | 8252 | 8670 | 8774 | None |
| Signature | G | G | A | A | T | C | T | |||||
| Cluster-O (n = 9) | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | |||||
| Other KSB (n = 85) | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |||||
| Signature | G | A | G | |||||||||
| Cluster-P (n = 13) | 1.0 | 1.0 | 1.0 | |||||||||
| Other KSB (n = 81) | 0.0 | 0.0 | 0.0 | |||||||||
| Gene | Cluster O (n = 9) | Cluster P (n = 13) | KSB (n = 19) [13] |
|---|---|---|---|
| pol | 99.1 ± 0.5 | 99.2 ± 0.4a | 97.1 |
| gag | 98.9 ± 0.2 | 98.9 ± 0.5 | 95.2 |
| vif | 98.9 ± 0.5 | 98.9 ± 0.7 | 96.2 |
| nef | 97.6 ± 1.6 | 97.3 ± 2.1a, b | 95.6 |
| env | 96.3 ± 1.7 | 94.5 ± 3.2b | 89.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, Y.-K.; Kim, J.-E.; Foley, B.T. Genetic Analysis of the Full-Length gag Gene from the Earliest Korean Subclade B of HIV-1: An Outbreak among Korean Hemophiliacs. Viruses 2019, 11, 545. https://doi.org/10.3390/v11060545
Cho Y-K, Kim J-E, Foley BT. Genetic Analysis of the Full-Length gag Gene from the Earliest Korean Subclade B of HIV-1: An Outbreak among Korean Hemophiliacs. Viruses. 2019; 11(6):545. https://doi.org/10.3390/v11060545
Chicago/Turabian StyleCho, Young-Keol, Jung-Eun Kim, and Brian T. Foley. 2019. "Genetic Analysis of the Full-Length gag Gene from the Earliest Korean Subclade B of HIV-1: An Outbreak among Korean Hemophiliacs" Viruses 11, no. 6: 545. https://doi.org/10.3390/v11060545
APA StyleCho, Y. -K., Kim, J. -E., & Foley, B. T. (2019). Genetic Analysis of the Full-Length gag Gene from the Earliest Korean Subclade B of HIV-1: An Outbreak among Korean Hemophiliacs. Viruses, 11(6), 545. https://doi.org/10.3390/v11060545