Lnc-ITM2C-1 and GPR55 Are Proviral Host Factors for Hepatitis C Virus

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmid and In Vitro Transcription
2.3. Infectious HCV in Cell Culture
2.4. Oligonucleotides (Oligos)
- GmR Negative Control A (Neg. ctr. GmR): 5′-AACACGTCTATACGC-3′;
- GmR 1 for lncR 3/LINC00222 (lncR 3-GmR 1): 5′-GCGTGATTAAATGGAT-3′;
- GmR 2 for lncR 3/LINC00222 (lncR 3-GmR 2): 5′-GACGATAAGAGGTAAC-3′;
- GmR 1 for lncR 7/Lnc-SLC12A7-4 (lncR 7-GmR 1): 5′-TGATTAACAGAACGGA-3′;
- GmR 2 for lncR 7/Lnc-SLC12A7-4 (lncR 7-GmR 2): 5′-ATAAGTGTCTAGTTAG-3′;
- GmR 1 for lncR 8/Lnc-ITM2C-1(lncR 8-GmR 1): 5′-GTTACCAGTGAAGCGG-3′;
- GmR 2 for lncR 8/Lnc-ITM2C-1 (lncR 8-GmR 2): 5′-TCGGATTGGTCACATG-3′;
- GmR 1 for lncR 10/ZNF252P-AS1 (lncR 10-GmR 1): 5′-GTTAATCTGATCTTGC-3′;
- GmR 2 for lncR 10/ZNF252P-AS1 (lncR 10-GmR 2): 5′-TCTGAGCTTGATCACT-3′;
- GmR 1 for GPR55 (GPR55-GmR 1): 5′-GGCGAATCAGATTAAT-3′;
- GmR 2 for GPR55 (GPR55-GmR 2): 5′-AGGACCATCTTGAATG-3′;
2.5. Cell Treatment
2.6. RNA Samples, DNA Removal, and cDNA Preparation
2.7. Microarrays
2.8. Quantitative Real Time-PCR (qRT-PCR)
2.9. Immunofluorescence
2.10. Western Blot
2.11. Protein-Coding Potential
2.12. Statistical Analysis
3. Results
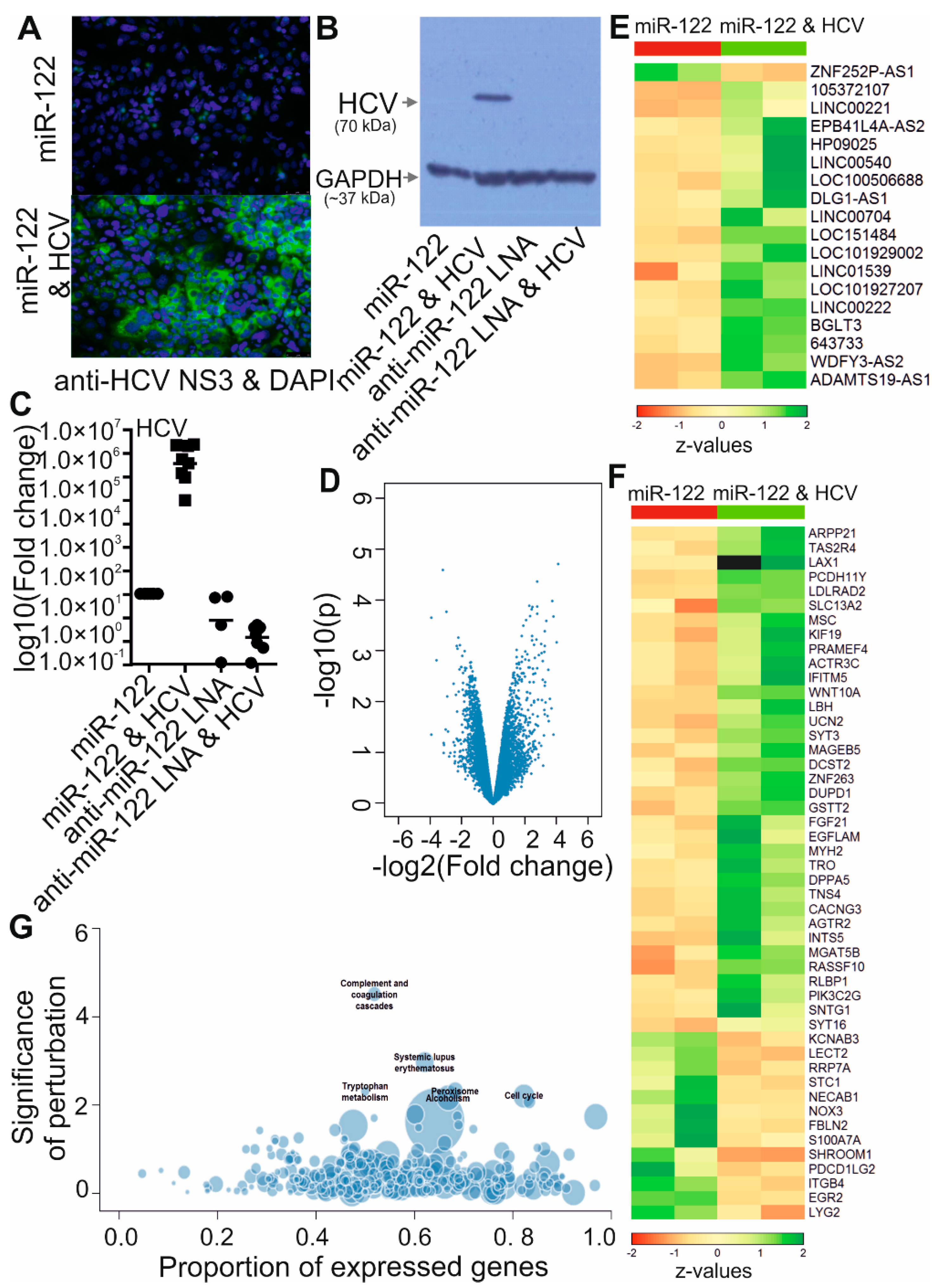
3.1. Identification of lncRNAs Deregulated by HCV Replication
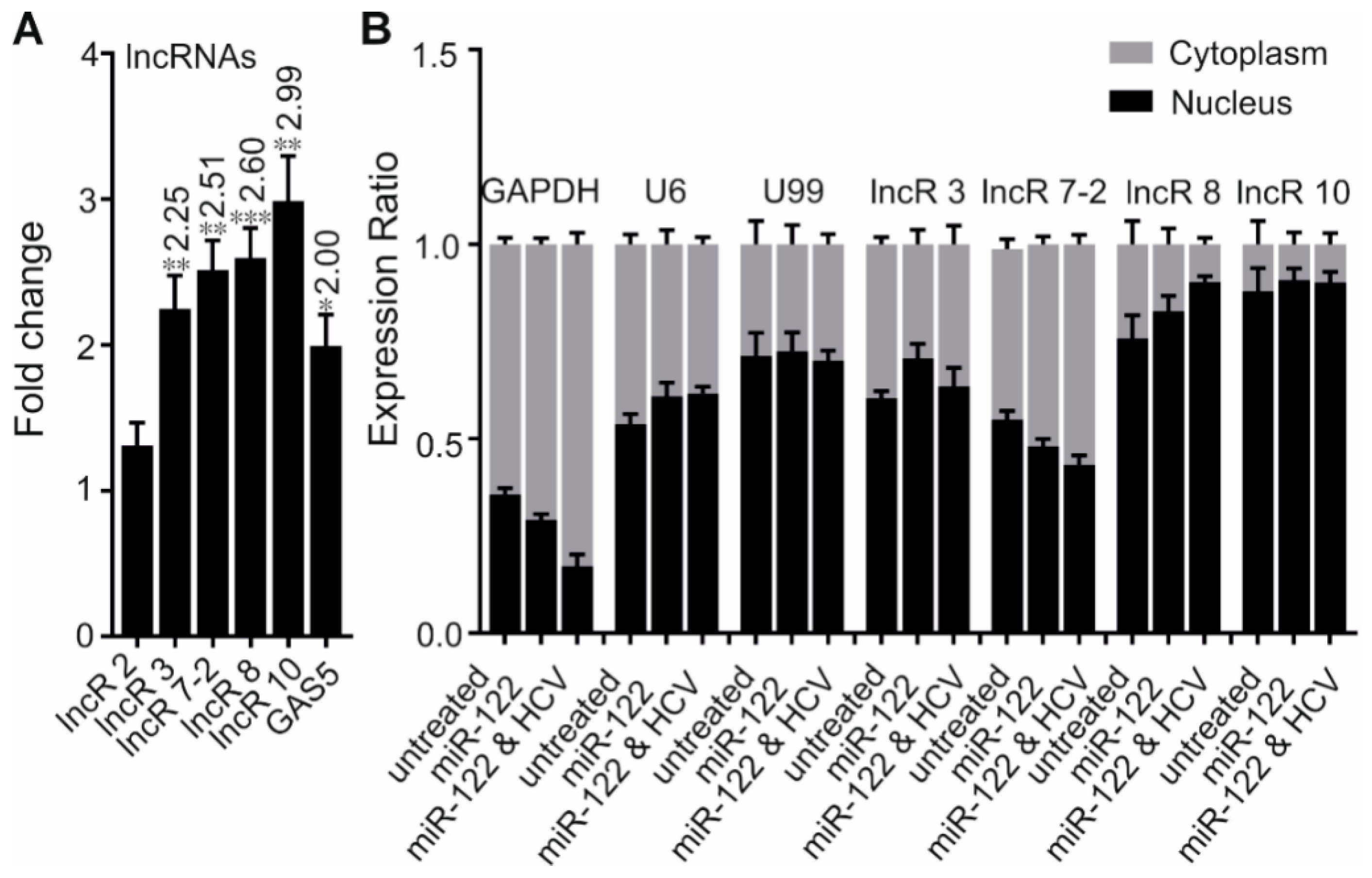
3.2. HCV Replication Increases the Expression of Four lncRNAs
3.3. Low Protein Coding Potential and Subcellular Localization of lncRNAs
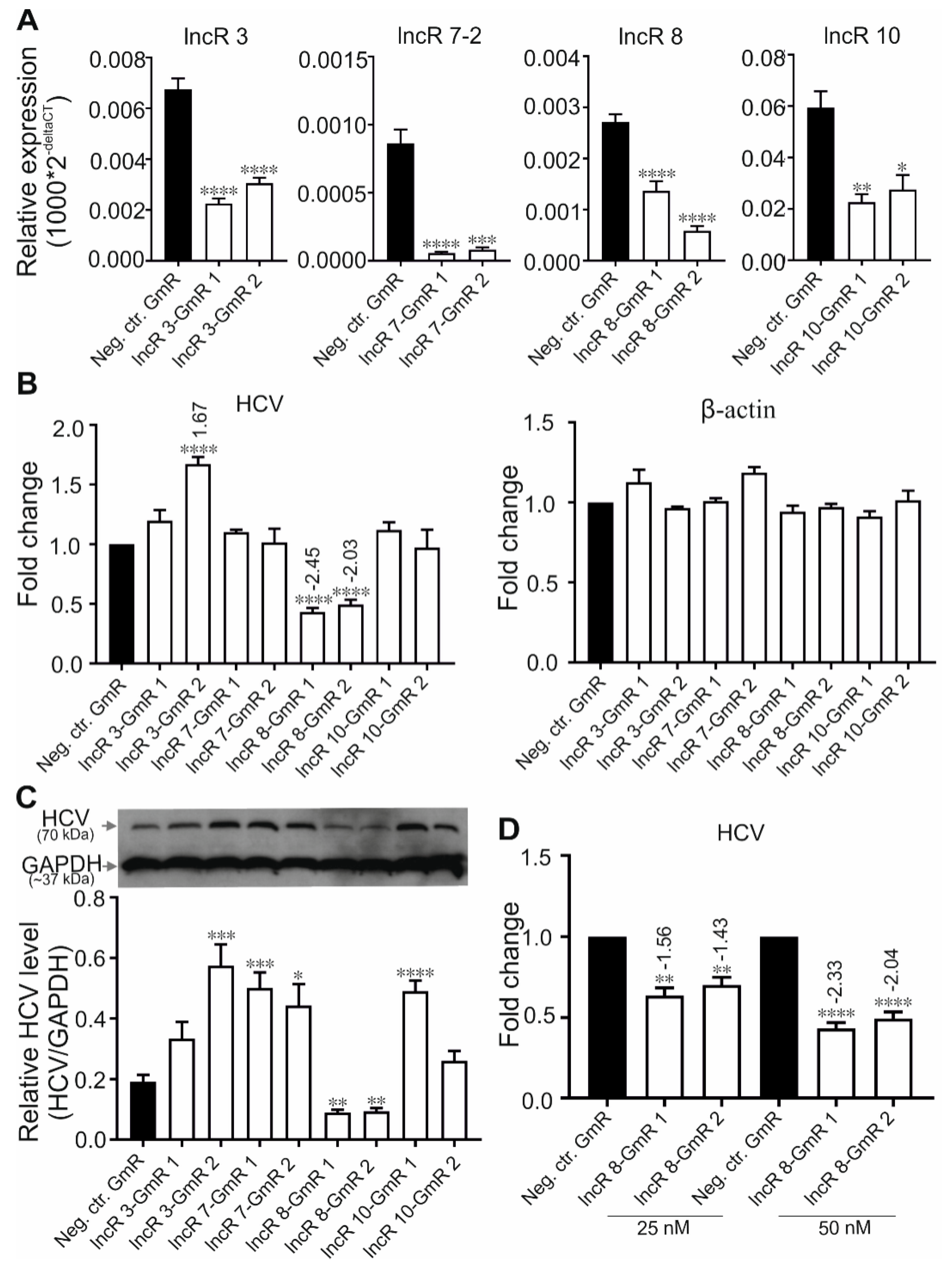
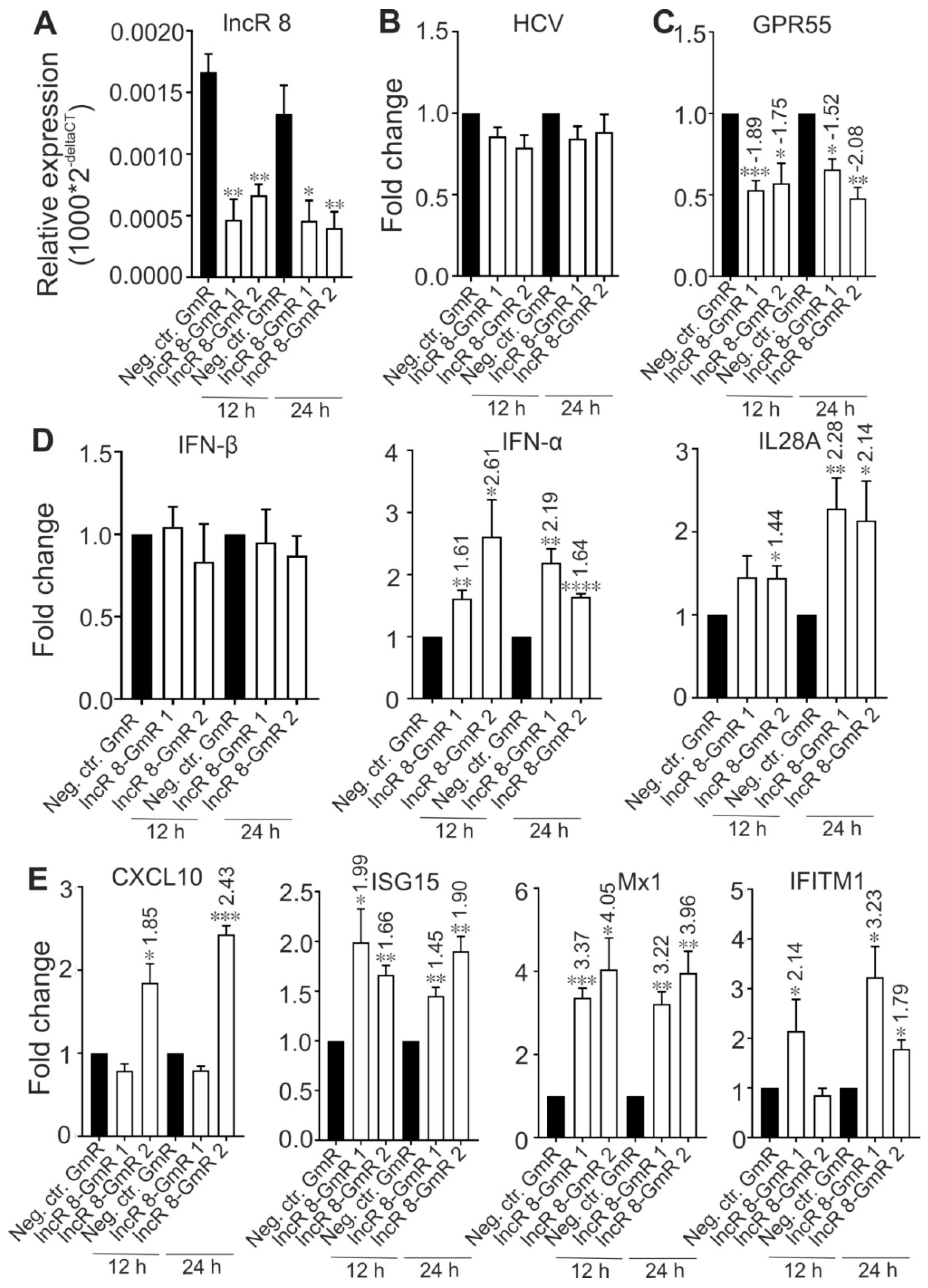
3.4. LncR 8/Lnc-ITM2C-1 Favors HCV Viral Replication
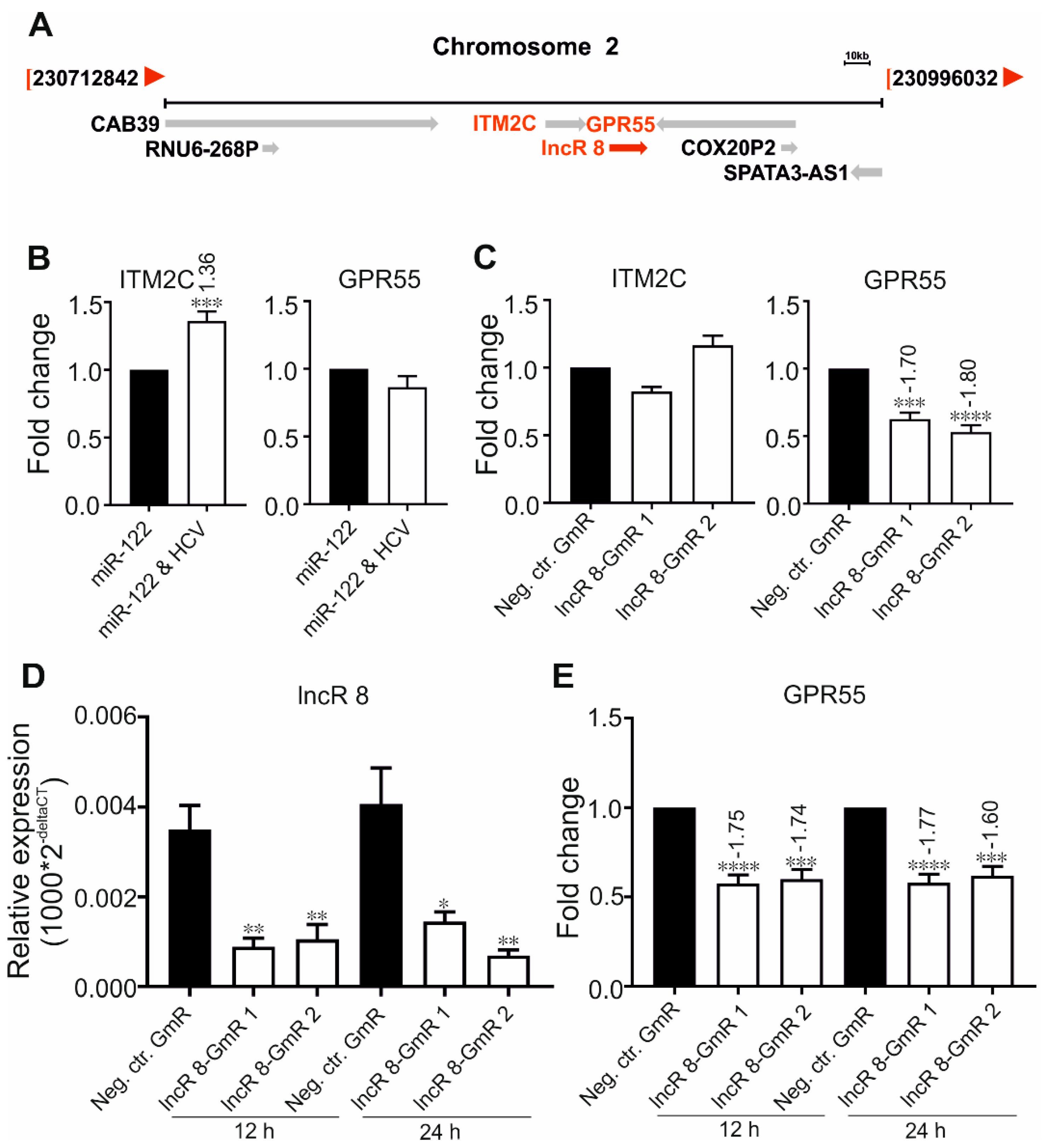
3.5. LncR 8/Lnc-ITM2C-1 Is a Short-Term Cis-Acting Regulator of Its Neighbor GPR55
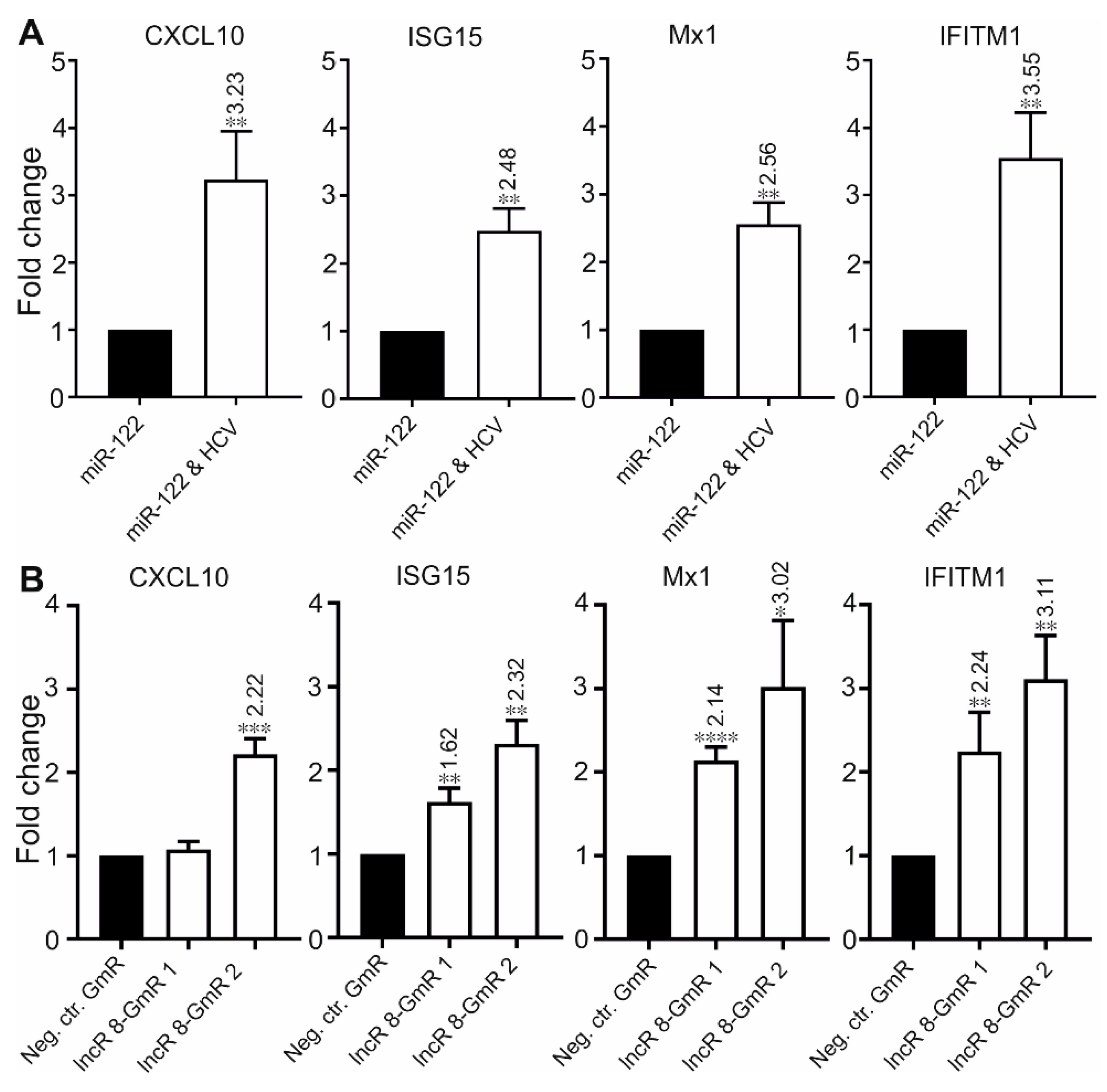
3.6. LncR 8/Lnc-ITM2C-1 Is a Negative regulator of the Antiviral Response
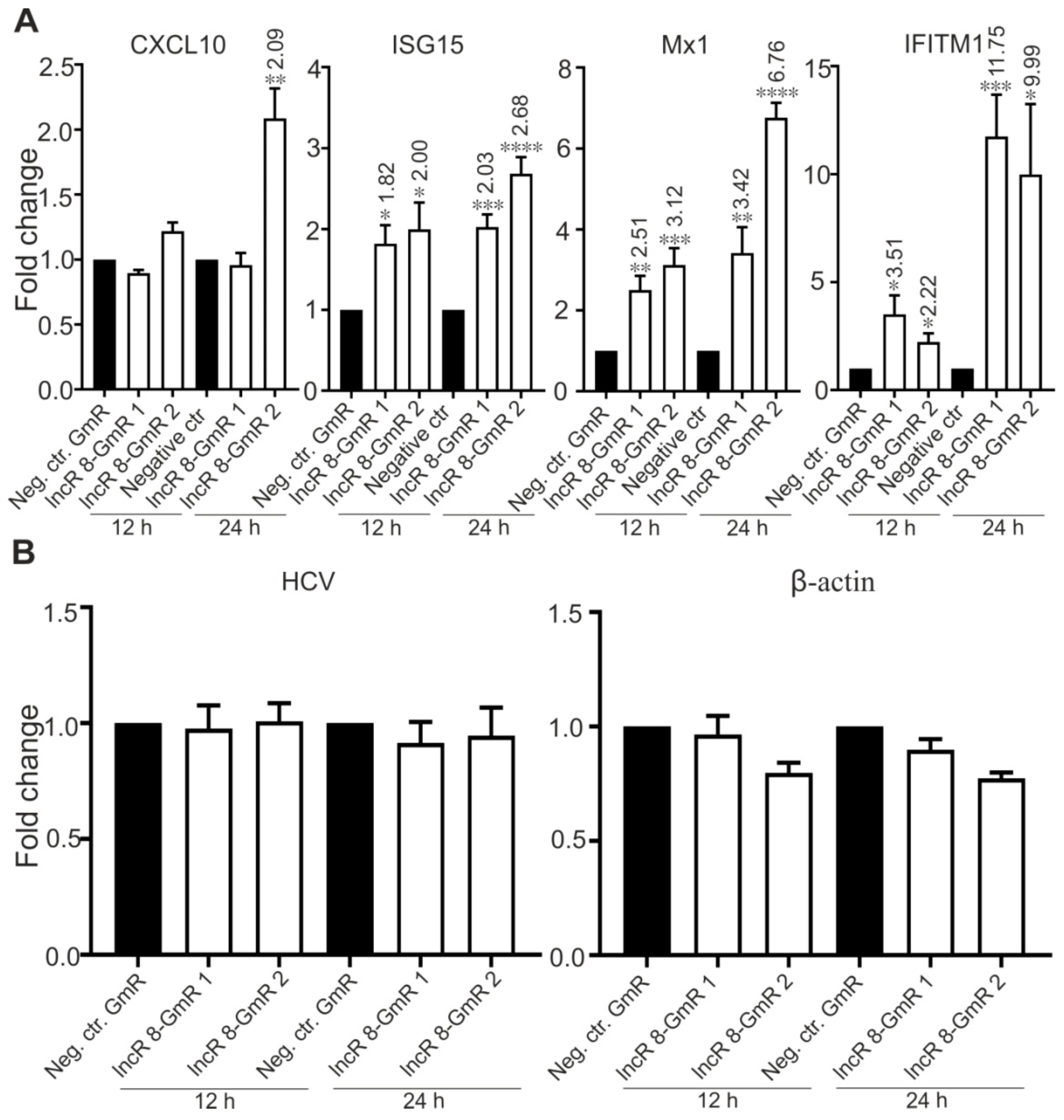
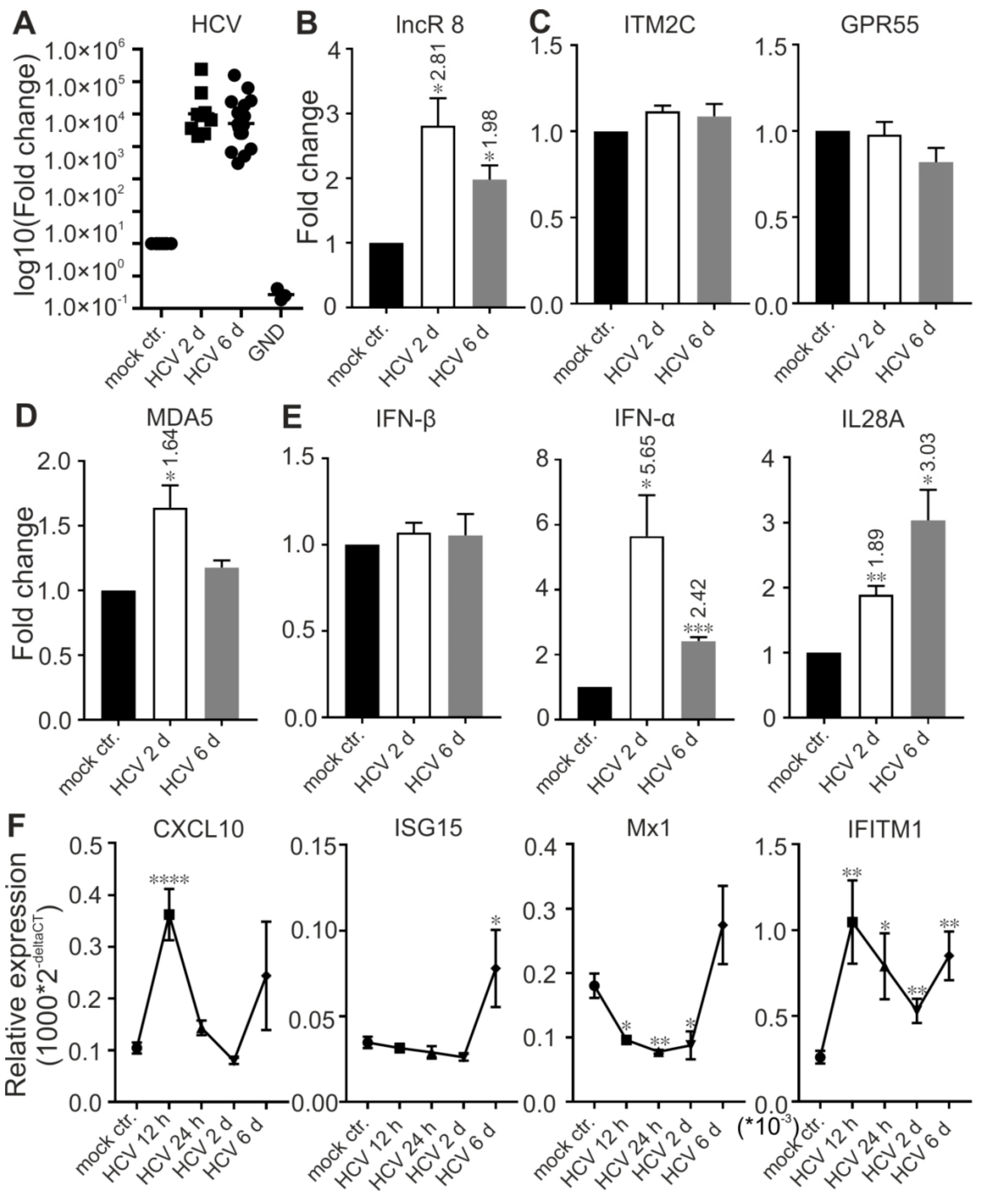
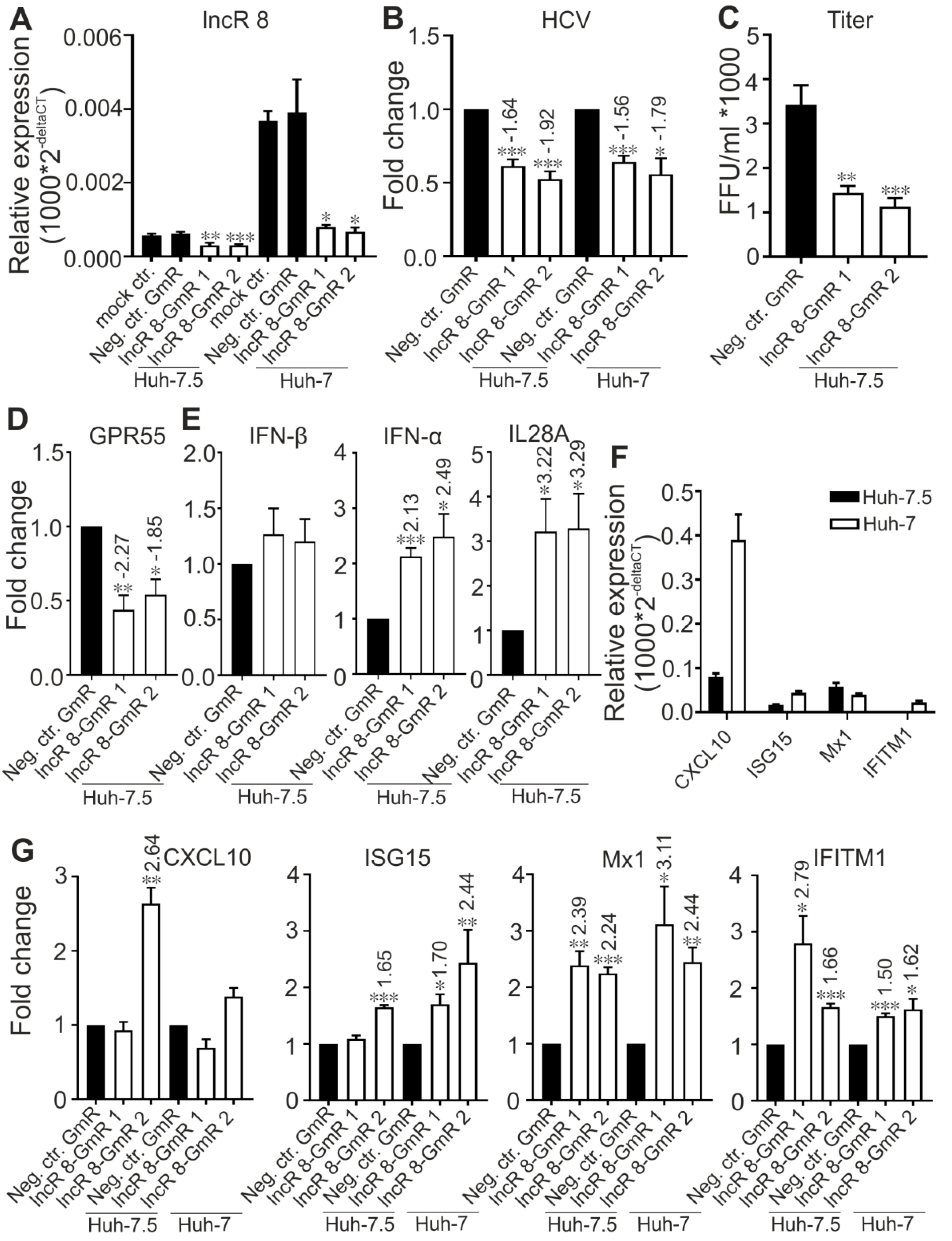
3.7. LncR 8/Lnc-ITM2C-1 Is Upregulated by HCVcc Infection and Facilitates HCV Infection
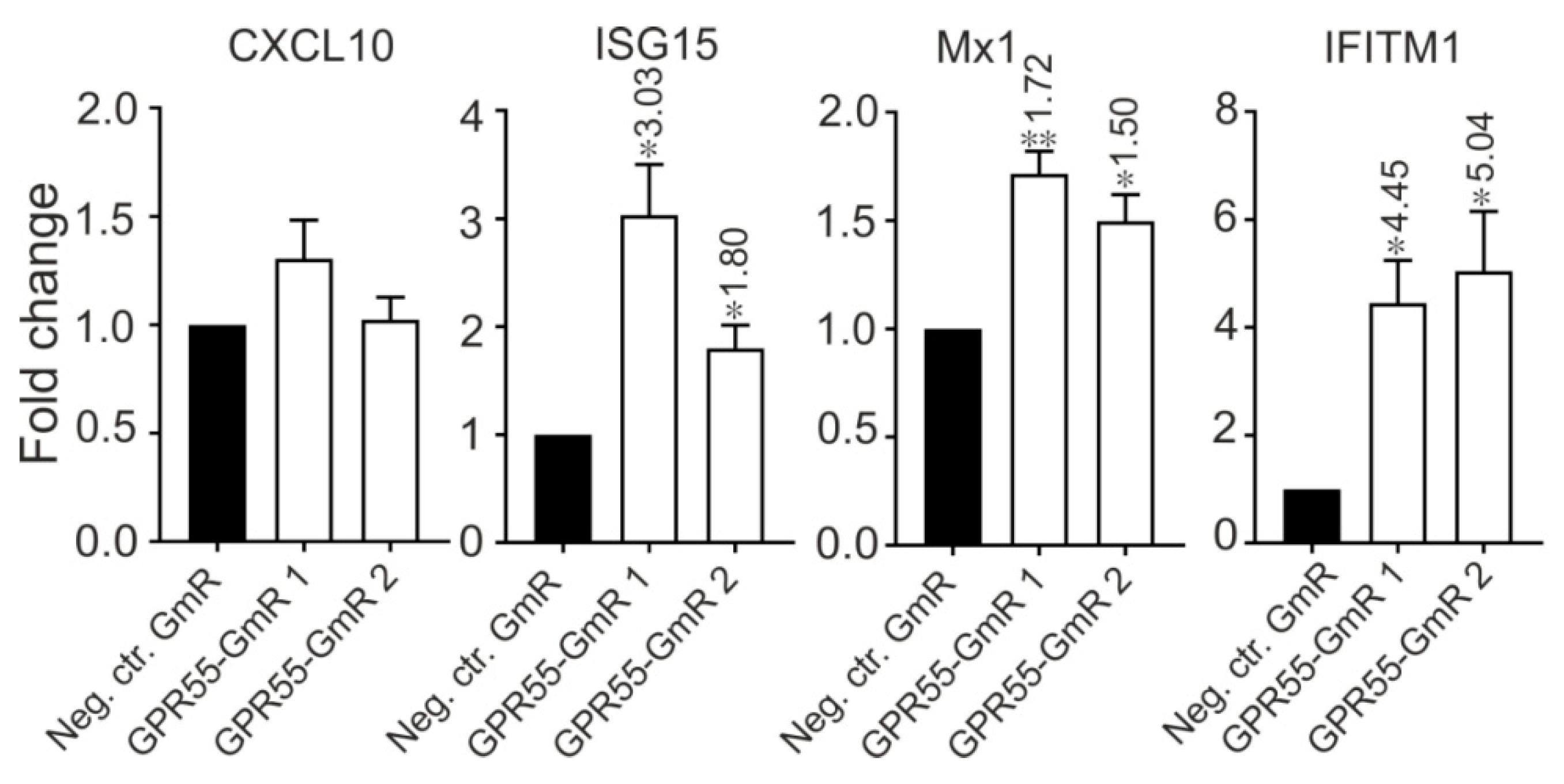
3.8. GPR55 Negatively Regulates ISGs
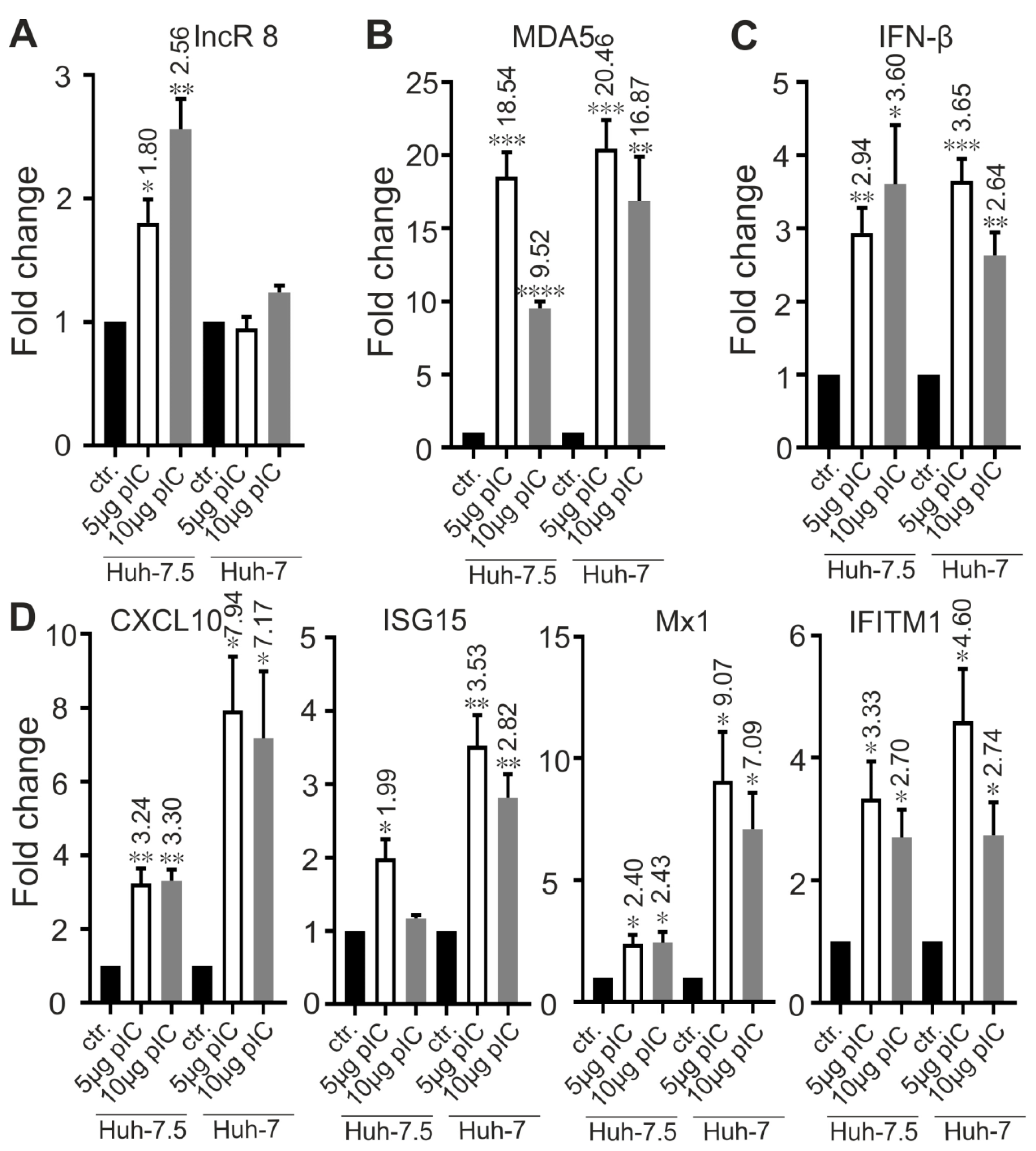
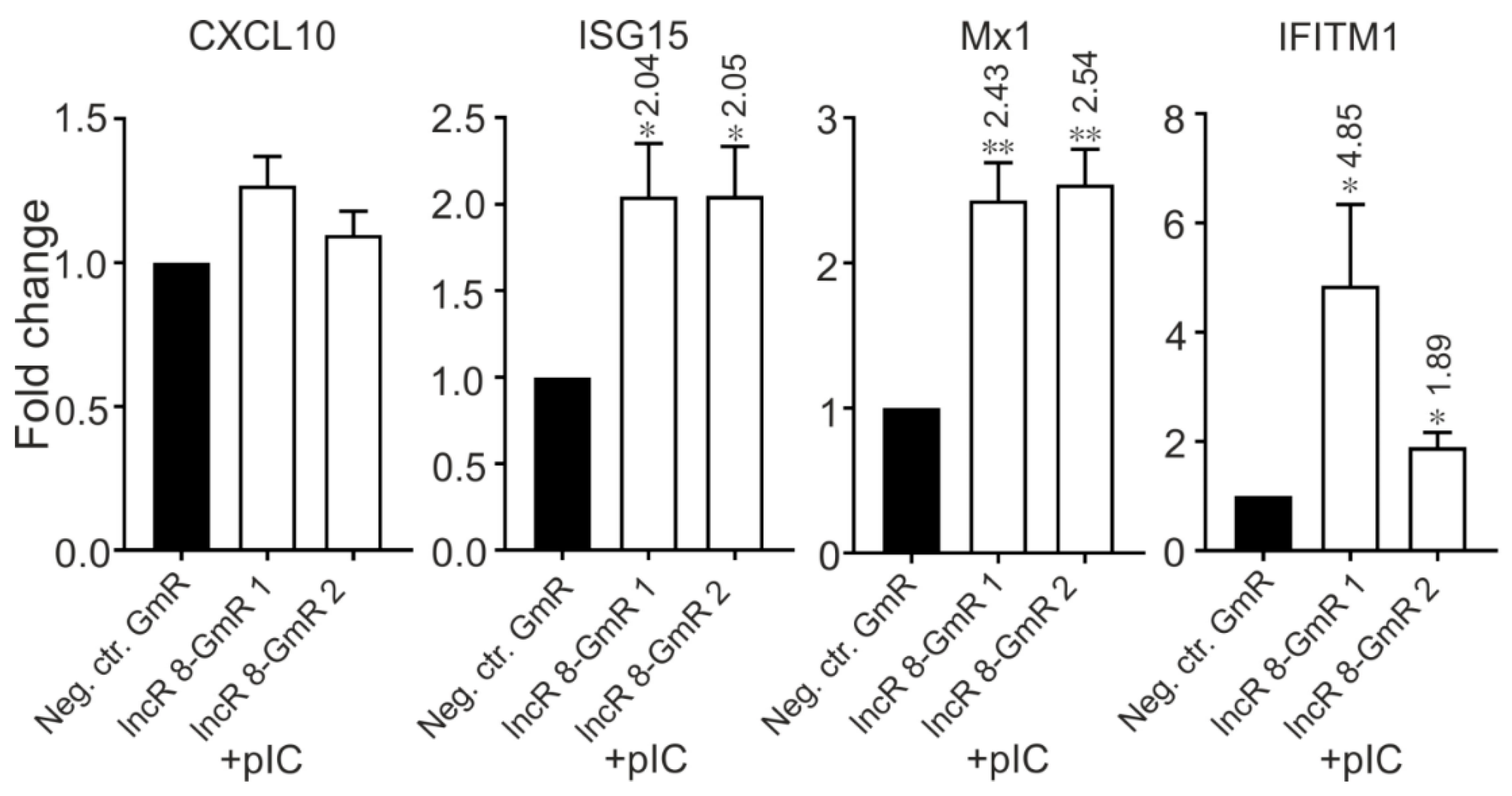
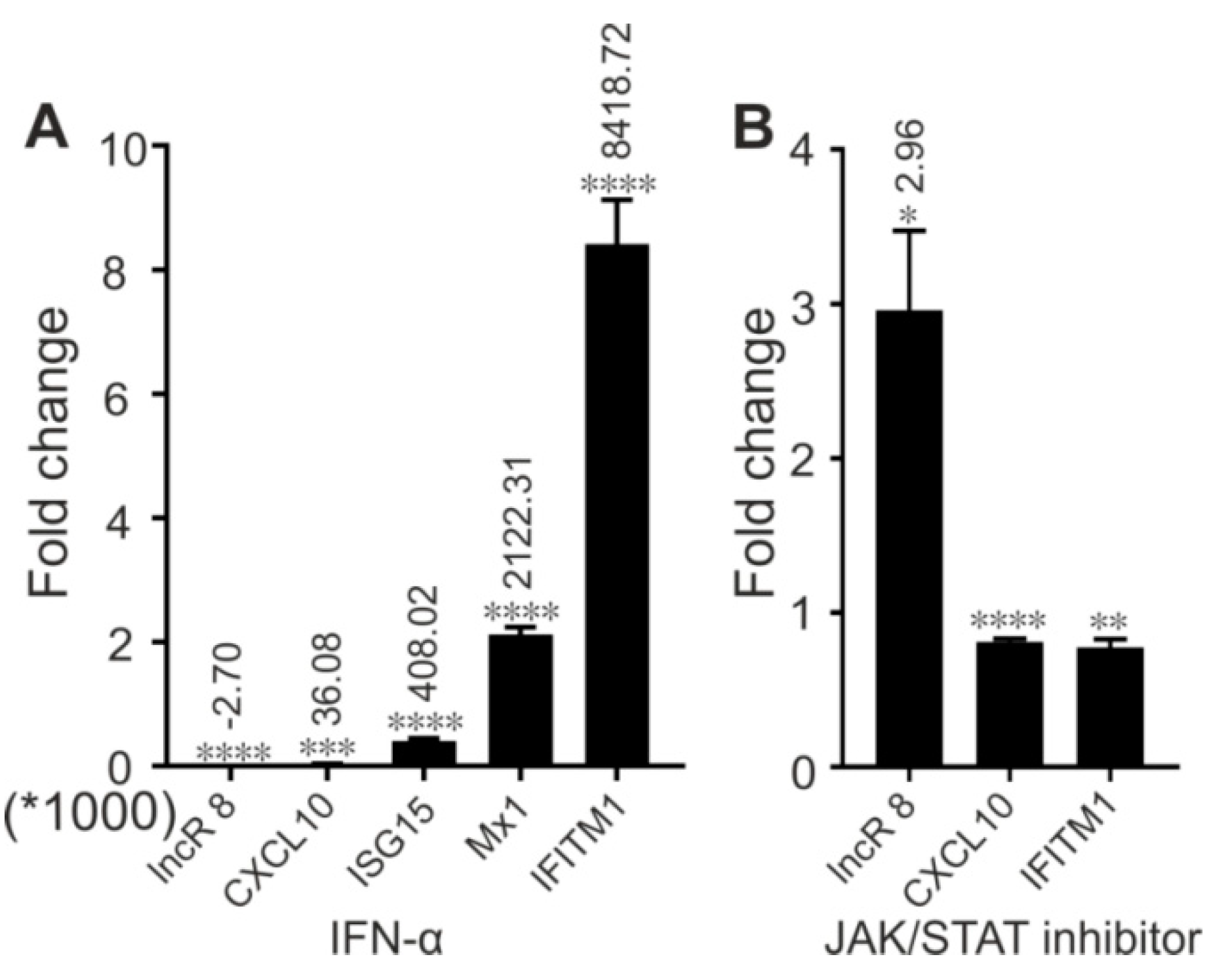
3.9. LncR 8/Lnc-ITM2C-1 Is Induced by polyIC
3.10. LncR 8/Lnc-ITM2C-1 Is Downregulated by JAK/STAT Pathway
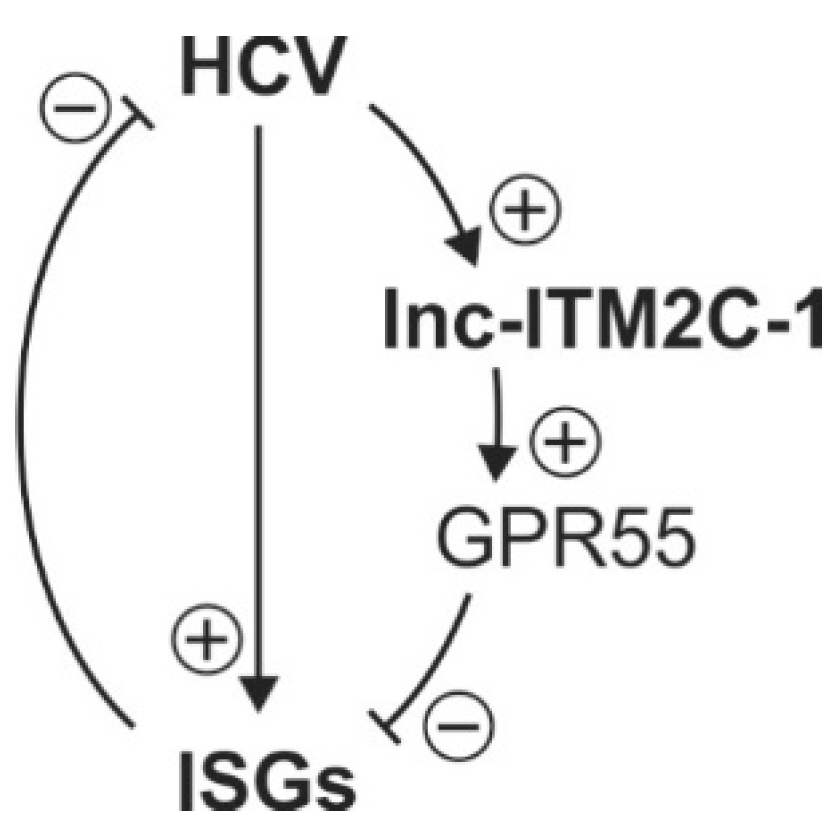
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nouroz, F.; Shaheen, S.; Mujtaba, G.; Noreen, S. An overview on hepatitis C virus genotypes and its control. Egypt. J. Med. Hum. Genet. 2015, 16, 291–298. [Google Scholar] [CrossRef] [Green Version]
- Choo, Q.-L.; Kuo, G.; Weiner, A.J.; Overby, L.R.; Bradley, D.W.; Houghton, M. Isolation of a cDNA Clone Derived from a Blood-Borne Non-A, Non-B Viral Hepatitis Genome. Science 1989, 244, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Pezacki, J.P.; Singaravelu, R.; Lyn, R.K. Host-virus interactions during hepatitis C virus infection: A complex and dynamic molecular biosystem. Mol. Biosyst. 2010, 6, 1131–1142. [Google Scholar] [CrossRef] [PubMed]
- Barriocanal, M.; Fortes, P. Long Non-coding RNAs in Hepatitis C Virus-Infected Cells. Front. Microbiol. 2017, 8, 1833. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Rice, C.M. The ins and outs of hepatitis C virus entry and assembly. Nat. Rev. Microbiol. 2013, 11, 688–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubuisson, J.; Cosset, F.L. Virology and cell biology of the hepatitis C virus life cycle: An update. J. Hepatol. 2014, 61 (Suppl. 1), S3–S13. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Madan, V.; Bartenschlager, R. Hepatitis C virus RNA replication and assembly: Living on the fat of the land. Cell Host Microbe 2014, 16, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Appel, N.; Schaller, T.; Penin, F.; Bartenschlager, R. From structure to function: New insights into hepatitis C virus RNA replication. J. Biol. Chem. 2006, 281, 9833–9836. [Google Scholar] [CrossRef] [PubMed]
- Niepmann, M. Hepatitis C virus RNA translation. Curr. Top. Microbiol. Immunol. 2013, 369, 143–166. [Google Scholar] [CrossRef] [PubMed]
- Heim, M.H.; Thimme, R. Innate and adaptive immune responses in HCV infections. J. Hepatol. 2014, 61 (Suppl. 1), S14–S25. [Google Scholar] [CrossRef] [Green Version]
- Gokhale, N.S.; Vazquez, C.; Horner, S.M. Hepatitis C Virus. Strategies to Evade Antiviral Responses. Future Virol. 2014, 9, 1061–1075. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Rajsbaum, R.; Yi, M. Immune and non-immune responses to hepatitis C virus infection. World J. Gastroenterol. 2015, 21, 10739–10748. [Google Scholar] [CrossRef]
- Barriocanal, M.; Carnero, E.; Segura, V.; Fortes, P. Long Non-Coding RNA BST2/BISPR is Induced by IFN and Regulates the Expression of the Antiviral Factor Tetherin. Front. Immunol. 2014, 5, 655. [Google Scholar] [CrossRef] [PubMed]
- Valadkhan, S.; Fortes, P. Regulation of the Interferon Response by lncRNAs in HCV Infection. Front. Microbiol. 2018, 9, 181. [Google Scholar] [CrossRef] [PubMed]
- Kambara, H.; Niazi, F.; Kostadinova, L.; Moonka, D.K.; Siegel, C.T.; Post, A.B.; Carnero, E.; Barriocanal, M.; Fortes, P.; Anthony, D.D.; et al. Negative regulation of the interferon response by an interferon-induced long non-coding RNA. Nucleic Acids Res. 2014, 42, 10668–10680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, M.T.; Chen, S.S. Emerging roles of interferon-stimulated genes in the innate immune response to hepatitis C virus infection. Cell. Mol. Immunol. 2016, 13, 11–35. [Google Scholar] [CrossRef]
- Sumpter, R., Jr.; Loo, Y.M.; Foy, E.; Li, K.; Yoneyama, M.; Fujita, T.; Lemon, S.M.; Gale, M., Jr. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 2005, 79, 2689–2699. [Google Scholar] [CrossRef]
- Thimme, R.; Binder, M.; Bartenschlager, R. Failure of innate and adaptive immune responses in controlling hepatitis C virus infection. FEMS Microbiol. Rev. 2012, 36, 663–683. [Google Scholar] [CrossRef] [Green Version]
- Messina, J.P.; Humphreys, I.; Flaxman, A.; Brown, A.; Cooke, G.S.; Pybus, O.G.; Barnes, E. Global Distribution and Prevalence of Hepatitis C Virus Genotypes. Hepatology 2015, 61, 77–87. [Google Scholar] [CrossRef]
- Ramakrishnaiah, V.; Thumann, C.; Fofana, I.; Habersetzer, F.; Pan, Q.; de Ruiter, P.E.; Willemsen, R.; Demmers, J.A.; Stalin Raj, V.; Jenster, G.; et al. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc. Natl. Acad. Sci. USA 2013, 110, 13109–13113. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Xu, J.F.; Wang, Y.J.; Cao, X.T. An interferon-independent lncRNA promotes viral replication by modulating cellular metabolism. Science 2017, 358, 1051–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, X.; Xu, C.; Zhao, P.; Qi, Z. Long non-coding RNA GAS5 inhibited hepatitis C virus replication by binding viral NS3 protein. Virology 2016, 492, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Zheng, J.; Mao, Y.; Dong, P.; Lu, Z.; Li, G.; Guo, C.; Liu, Z.; Fan, X. Long Non-coding RNA Growth Arrest-specific Transcript 5 (GAS5) Inhibits Liver Fibrogenesis through a Mechanism of Competing Endogenous RNA. J. Biol. Chem. 2015, 290, 28286–28298. [Google Scholar] [CrossRef] [Green Version]
- Xie, Q.; Chen, S.; Tian, R.; Huang, X.; Deng, R.; Xue, B.; Qin, Y.; Xu, Y.; Wang, J.; Guo, M.; et al. Long Noncoding RNA ITPRIP-1 Positively Regulates the Innate Immune Response through Promotion of Oligomerization and Activation of MDA5. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Nishitsujia, H.; Ujinoa, S.; Yoshioa, S.; Sugiyamaa, M.; Mizokamia, M.; Kantoa, T.; Shimotohnoa, K. Long noncoding RNA #32 contributes to antiviral responses by controlling interferon-stimulated gene expression. Proc. Natl. Acad. Sci. USA 2016, 113, 10388–10393. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Li, Y.; Lu, Z.; Che, Y.; Sun, S.; Mao, S.; Lei, Y.; Zang, R.; Li, N.; Sun, N.; et al. Long non-coding RNA GAS5 is induced by interferons and plays an antitumor role in esophageal squamous cell carcinoma. Cancer Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Sur, S.; Sasaki, R.; Devhare, P.; Steele, R.; Ray, R.; Ray, R.B. Association between microRNA-373 and long non-coding RNA NORAD in hepatitis C virus infected hepatocytes impairs Wee1 expression for growth promotion. J. Virol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Pietschmann, T.; Kaul, A.; Koutsoudakis, G.; Shavinskaya, A.; Kallis, S.; Steinmann, E.; Abid, K.; Negro, F.; Dreux, M.; Cosset, F.L.; et al. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. USA 2006, 103, 7408–7413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carnero, E.; Barriocanal, M.; Prior, C.; Pablo Unfried, J.; Segura, V.; Guruceaga, E.; Enguita, M.; Smerdou, C.; Gastaminza, P.; Fortes, P. Long noncoding RNA EGOT negatively affects the antiviral response and favors HCV replication. EMBO Rep. 2016, 17, 1013–1028. [Google Scholar] [CrossRef]
- Wang, N.; Wang, Q.; Shen, D.; Sun, X.; Cao, X.; Wu, D. Downregulation of microRNA-122 promotes proliferation, migration, and invasion of human hepatocellular carcinoma cells by activating epithelial-mesenchymal transition. Oncotarg. Ther. 2016, 9, 2035–2047. [Google Scholar] [CrossRef]
- R: A Language and Environment for Statistical Computing. Available online: http://www.R-project.org (accessed on 1 March 2016).
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed]
- Silver, J.D.; Ritchie, M.E.; Smyth, G.K. Microarray background correction: Maximum likelihood estimation for the normal-exponential convolution. Biostatistics 2009, 10, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Volders, P.J.; Verheggen, K.; Menschaert, G.; Vandepoele, K.; Martens, L.; Vandesompele, J.; Mestdagh, P. An update on LNCipedia: A database for annotated human lncRNA sequences. Nucleic Acids Res. 2015, 43, 4363–4364. [Google Scholar] [CrossRef] [PubMed]
- Niepmann, M.; Shalamova, L.A.; Gerresheim, G.K.; Rossbach, O. Signals Involved in Regulation of Hepatitis C Virus RNA Genome Translation and Replication. Front. Microbiol. 2018, 9, 395. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L.; Yi, M.K.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of Hepatitis C Virus RNA Abundance by a Liver-Specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henke, J.I.; Goergen, D.; Zheng, J.; Song, Y.; Schuettler, C.G.; Fehr, C.; Juenemann, C.; Niepmann, M. microRNA-122 stimulates translation of hepatitis C virus. EMBO 2008, 27, 3300–3310. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Nicolas, E.; Marks, D.; Sander, C.; Lerro, A.; Buendia, M.A.; Xu, C.; Mason, W.S.; Moloshok, T.; Bort, R.; et al. miR-122, a Mammalian Liver-Specific microRNA, is Processed from hcr mRNA and MayDownregulate the High Affinity Cationic Amino Acid Transporter CAT-1. RNA Biol. 2004, 1, 106–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Fan, D.; Jian, Z.; Chen, G.G.; Lai, P.B. Cancer Specific Long Noncoding RNAs Show Differential Expression Patterns and Competing Endogenous RNA Potential in Hepatocellular Carcinoma. PLoS ONE 2015, 10, e0141042. [Google Scholar] [CrossRef] [PubMed]
- Morlando, M.; Ballarino, M.; Fatica, A. Long Non-Coding RNAs: New Players in Hematopoiesis and Leukemia. Front. Med. 2015, 2, 23. [Google Scholar] [CrossRef] [PubMed]
- Pessa, H.K.; Will, C.L.; Meng, X.; Schneider, C.; Watkins, N.J.; Perala, N.; Nymark, M.; Turunen, J.J.; Luhrmann, R.; Frilander, M.J. Minor spliceosome components are predominantly localized in the nucleus. Proc. Natl. Acad. Sci. USA 2008, 105, 8655–8660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennox, K.A.; Behlke, M.A. Mini-review:current strategies to knockdown long non-coding RNAs. J. Rare Dis. Res. Treat. 2016, 1, 66–70. [Google Scholar]
- Chan, J.H.; Lim, S.H.; Wong, W.F. Antisense oligonucleotides from design to therapeutic application. Clin. Exp. Pharmacol. Physiol. 2006, 33, 533–540. [Google Scholar] [CrossRef]
- Multiplex qPCR—How to Get Started. Available online: https://www.idtdna.com/pages/education/decoded/article/multiplex-qpcr-how-to-get-started (accessed on 4 January 2018).
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Gao, D.Z. Distinction immune genes of hepatitis-induced heptatocellular carcinoma. Bioinformatics 2012, 28, 3191–3194. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, J.; Zhu, X.; Chen, Y.; Wei, H.; Chen, Q.; Chi, X.; Qi, B.; Zhang, L.; Zhao, Y.; Gao, G.F.; et al. NRAV, a long noncoding RNA, modulates antiviral responses through suppression of interferon-stimulated gene transcription. Cell Host Microbe 2014, 16, 616–626. [Google Scholar] [CrossRef]
- Cao, X.; Ding, Q.; Lu, J.; Tao, W.; Huang, B.; Zhao, Y.; Niu, J.; Liu, Y.J.; Zhong, J. MDA5 plays a critical role in interferon response during hepatitis C virus infection. J. Hepatol. 2015, 62, 771–778. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Z.; Tang, J.; Tang, R.; Shan, X.; Zhang, W.; Chen, Q.; Zhou, F.; Chen, K.; Huang, A.; et al. Hepatitis C virus core protein activates Wnt/beta-catenin signaling through multiple regulation of upstream molecules in the SMMC-7721 cell line. Arch. Virol. 2011, 156, 1013–1023. [Google Scholar] [CrossRef]
- Kukla, M.; Berdowska, A.; Stygar, D.; Gabriel, A.; Mazur, W.; Logiewa-Bazger, B.; Sobala-Szczygiel, B.; Buldak, R.J.; Rokitka, M.; Zajecki, W.; et al. Serum FGF21 and RBP4 levels in patients with chronic hepatitis C. Scand. J. Gastroenterol. 2012, 47, 1037–1047. [Google Scholar] [CrossRef]
- Ahmad, W.; Ijaz, B.; Hassan, S. Gene expression profiling of HCV genotype 3a initial liver fibrosis and cirrhosis patients using microarray. J. Transl. Med. 2012, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Berger, K.L.; Cooper, J.D.; Heaton, N.S.; Yoon, R.; Oakland, T.E.; Jordan, T.X.; Mateu, G.; Grakoui, A.; Randall, G. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 7577–7582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okabe, H.; Delgado, E.; Lee, J.M.; Yang, J.; Kinoshita, H.; Hayashi, H.; Tsung, A.; Behari, J.; Beppu, T.; Baba, H.; et al. Role of leukocyte cell-derived chemotaxin 2 as a biomarker in hepatocellular carcinoma. PLoS ONE 2014, 9, e98817. [Google Scholar] [CrossRef] [PubMed]
- Ovejero, C.; Cavard, C.; Perianin, A.; Hakvoort, T.; Vermeulen, J.; Godard, C.; Fabre, M.; Chafey, P.; Suzuki, K.; Romagnolo, B.; et al. Identification of the leukocyte cell-derived chemotaxin 2 as a direct target gene of beta-catenin in the liver. Hepatology 2004, 40, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Jaksik, R.; Iwanaszko, M.; Rzeszowska-Wolny, J.; Kimmel, M. Microarray experiments and factors which affect their reliability. Biol. Direct 2015, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Stretch, C.; Khan, S.; Asgarian, N.; Eisner, R.; Vaisipour, S.; Damaraju, S.; Graham, K.; Bathe, O.F.; Steed, H.; Greiner, R.; et al. Effects of sample size on differential gene expression, rank order and prediction accuracy of a gene signature. PLoS ONE 2013, 8, e65380. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Hsueh, H.M.; Delongchamp, R.R.; Lin, C.J.; Tsai, C.A. Reproducibility of microarray data: A further analysis of microarray quality control (MAQC) data. BMC Bioinform. 2007, 8, 412. [Google Scholar] [CrossRef]
- Gerresheim, G.K.; Bathke, J.; Michel, A.M.; Andreev, D.E.; Shalamova, L.A.; Rossbach, O.; Hu, P.; Glebe, D.; Fricke, M.; Marz, M.; et al. Cellular Gene Expression during Hepatitis C Virus Replication as Revealed by Ribosome Profiling. Int. J. Mol. Sci. 2019, 20, 1321. [Google Scholar] [CrossRef]
- Durinck, K.; Wallaert, A.; Van de Walle, I.; Van Loocke, W.; Volders, P.J.; Vanhauwaert, S.; Geerdens, E.; Benoit, Y.; Van Roy, N.; Poppe, B.; et al. The Notch driven long non-coding RNA repertoire in T-cell acute lymphoblastic leukemia. Haematologica 2014, 99, 1808–1816. [Google Scholar] [CrossRef] [Green Version]
- Iwai, A.; Takegami, T.; Shiozaki, T.; Miyazaki, T. Hepatitis C virus NS3 protein can activate the Notch-signaling pathway through binding to a transcription factor, SRCAP. PLoS ONE 2011, 6, e20718. [Google Scholar] [CrossRef]
- Wienert, B.; Shin, J.; Zelin, E.; Pestal, K.; Corn, J.E. In vitro-transcribed guide RNAs trigger an innate immune response via the RIG-I pathway. PLoS Biol. 2018, 16, e2005840. [Google Scholar] [CrossRef] [PubMed]
- Loo, Y.M.; Fornek, J.; Crochet, N.; Bajwa, G.; Perwitasari, O.; Martinez-Sobrido, L.; Akira, S.; Gill, M.A.; Garcia-Sastre, A.; Katze, M.G.; et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 2008, 82, 335–345. [Google Scholar] [CrossRef]
- Zhou, J.; Burkovskiy, I.; Yang, H.; Sardinha, J.; Lehmann, C. CB2 and GPR55 Receptors as Therapeutic Targets for Systemic Immune Dysregulation. Front. Pharmacol. 2016, 7, 264. [Google Scholar] [CrossRef]
- Chiurchiu, V.; Lanuti, M.; De Bardi, M.; Battistini, L.; Maccarrone, M. The differential characterization of GPR55 receptor in human peripheral blood reveals a distinctive expression in monocytes and NK cells and a proinflammatory role in these innate cells. Int. Immunol. 2015, 27, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhou, J.; Lehmann, C. GPR55—A putative “type 3” cannabinoid receptor in inflammation. J. Basic Clin. Physiol. Pharmacol. 2016, 27, 297–302. [Google Scholar] [CrossRef]
- Pesce, M.; D’Alessandro, A.; Borrelli, O.; Gigli, S.; Seguella, L.; Cuomo, R.; Esposito, G.; Sarnelli, G. Endocannabinoid-related compounds in gastrointestinal diseases. J. Cell. Mol. Med. 2018, 22, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Patsenker, E.; Sachse, P.; Chicca, A.; Gachet, M.S.; Schneider, V.; Mattsson, J.; Lanz, C.; Worni, M.; de Gottardi, A.; Semmo, M.; et al. Elevated levels of endocannabinoids in chronic hepatitis C may modulate cellular immune response and hepatic stellate cell activation. Int. J. Mol. Sci. 2015, 16, 7057–7076. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Primer Sequences (5′-3′) | Amplicon Size (bp) |
|---|---|---|
| LncR 2 | F: CTCCCAGAACCTATCGGCAT | 130 |
| R: CACAAAGCCTGCGTTCATTC | ||
| LncR 3 | F: AGGATGTGACTGCCAGGTAATG | 100 |
| R: CAGACCCAGCCTAGCACACAG | ||
| LncR 33′ | F: GTGACCCAACTAGAGCCAATAGG | 135 |
| R: CTCAAATCAGCTCATGACCATAAG | ||
| LncR 7-1 | F: AGGCTACAGGAGGCACTGAGGG | 144 |
| R: GGAGCCATCTGGGAGAATGAAATAC | ||
| LncR 7-2 | F: GAGGCTACAGGAGGCACTCTTTG | 79 |
| R: GGAGCCATCTGGGAGAATGAAATAC | ||
| LncR 73′ | F: TCGGGTTCTTGATTTGATTCTC | 142 |
| R: TGGACCAAGTATCCTCTAAAAATG | ||
| LncR 8 | F: GGTTTTTTGACCTTGGCAATG | 102 |
| R: GTGACCCTTGGTGGCTGTTTAT | ||
| LncR 83′ | F: GATTCTGTCTCATCCAATCAAGACT | 123 |
| R: GTTGTGCTGAGGATTCTGGGT | ||
| LncR 10 | F: CGGAAATGCCTAATCTGAACTT | 80 |
| R: TAGAGCGGACCCACGAAAC | ||
| LncR 103′ | F: CCCCTGATGCTTCATAATGG | 111 |
| R: AGTTCTAACCTAATTTCCCATCAC |
| Target Gene | Primer Sequences (5′-3′) | Amplicon Size (bp) |
|---|---|---|
| GAPDH | F: GAGTCAACGGATTTGGTCGT | 224 |
| R: GATCTCGCTCCTGGAAGATG (= RT) | ||
| U6 | F: CTCGCTTCGGCAGCACA | 94 |
| R: AACGCTTCACGAATTTGCGT | ||
| U99 | F: CCTCCTTTTCTTGGCGGGGA | 138 |
| R: CGTTTGAGGATAGAACCAGC | ||
| β-actin | F: CATGTACGTTGCTATCCAGGC | 250 |
| R: CTCCTTAATGTCACGCACGAT | ||
| Jc1-NS3 | RT: GTATGCCACGGCATTCAAG | 190 |
| F: GATATAGGTCGACGGCTCCA | ||
| R: TTCCTCGGAACAACCATCTC | ||
| GAS5 | F: CCTGTGAGGTATGGTGCTGG | 383 |
| R: GGTCCAGGCAAGTTGGACTC | ||
| ITM2C | F: GTGGTGTGCTGTATGAGGACT | 93 |
| R: CGTAGTTCTCGTCGAGGTAGAT | ||
| GPR55 | F: GAAAACCCTACAGTTTGCAGTCC | 123 |
| R: GAGGTGGCAGCATAATCGGG | ||
| CXCL10 | F: GTGGCATTCAAGGAGTACCTC R: TGATGGCCTTCGATTCTGGATT | 198 |
| ISG15 | F: ACTCATCTTTGCCAGTACAGGAG R: CAGCATCTTCACCGTCAGGTC | 88 |
| Mx1 | F: TGCATCGACCTCATTGACTC R: ACCTTGCCTCTCCACTTATC | 218 |
| IFITM1 | F: ACTCCGTGAAGTCTAGGGACA R: AGAGCCGAATACCAGTAACAG | 149 |
| MDA5 | F:TCGAATGGGTATTCCACAGACG | 152 |
| R:GTGGCGACTGTCCTCTGAA | ||
| IFN-β | F:GCTTGGATTCCTACAAAGAAGCA | 166 |
| R:ATAGATGGTCAATGCGGCGTC | ||
| IFN-α | F: GGAGGTTGTCAGAGCAGA | 150 |
| R: AATGACAGAATTCATGAAAGCGT | ||
| IL28A | F: CAGCCTCAGAGTGTTTCTTCT | 117 |
| R: TCCAGTCACGGTCAGCA |
| lncRNA | Ref. | Chr | Length(bp) | Gene Symbol | Name |
|---|---|---|---|---|---|
| lncR 3 | NR_033376.1 | 6 | 1753 | lincRNA 222 | LINC00222 |
| lncR 7-2 | NR_104615.1 | 5 | 3451 | LOC100506688 | Lnc-SLC12A7-4:5 |
| lncR 8 | NR_038238.1 | 2 | 1893 | LOC151484 | Lnc-ITM2C-1 |
| lncR 10 | NR_026974.1 | 8 | 3250 | ZNF252P antisense RNA 1 | ZNF252P-AS1 |
| Metric | lncR 3 | lncR 7-2 | lncR 8 | lncR 10 |
|---|---|---|---|---|
| CPAT coding probability | 1.33% | 10.83% | 69.31% | 80.45% |
| PhyloCSF score | −67.4569 | 13.6639 | −112.1426 | 11.7381 |
| PRIDE reprocessing 2.0 | 0 | 0 | 0 | 0 |
| Lee translation initiation sites | 0 | 0 | 0 | 0 |
| Bazzini small ORFs | 0 | 0 | 0 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, P.; Wilhelm, J.; Gerresheim, G.K.; Shalamova, L.A.; Niepmann, M. Lnc-ITM2C-1 and GPR55 Are Proviral Host Factors for Hepatitis C Virus. Viruses 2019, 11, 549. https://doi.org/10.3390/v11060549
Hu P, Wilhelm J, Gerresheim GK, Shalamova LA, Niepmann M. Lnc-ITM2C-1 and GPR55 Are Proviral Host Factors for Hepatitis C Virus. Viruses. 2019; 11(6):549. https://doi.org/10.3390/v11060549
Chicago/Turabian StyleHu, Pan, Jochen Wilhelm, Gesche K. Gerresheim, Lyudmila A. Shalamova, and Michael Niepmann. 2019. "Lnc-ITM2C-1 and GPR55 Are Proviral Host Factors for Hepatitis C Virus" Viruses 11, no. 6: 549. https://doi.org/10.3390/v11060549
APA StyleHu, P., Wilhelm, J., Gerresheim, G. K., Shalamova, L. A., & Niepmann, M. (2019). Lnc-ITM2C-1 and GPR55 Are Proviral Host Factors for Hepatitis C Virus. Viruses, 11(6), 549. https://doi.org/10.3390/v11060549