Starvation-Induced Differential Virotherapy Using an Oncolytic Measles Vaccine Virus

 and
and 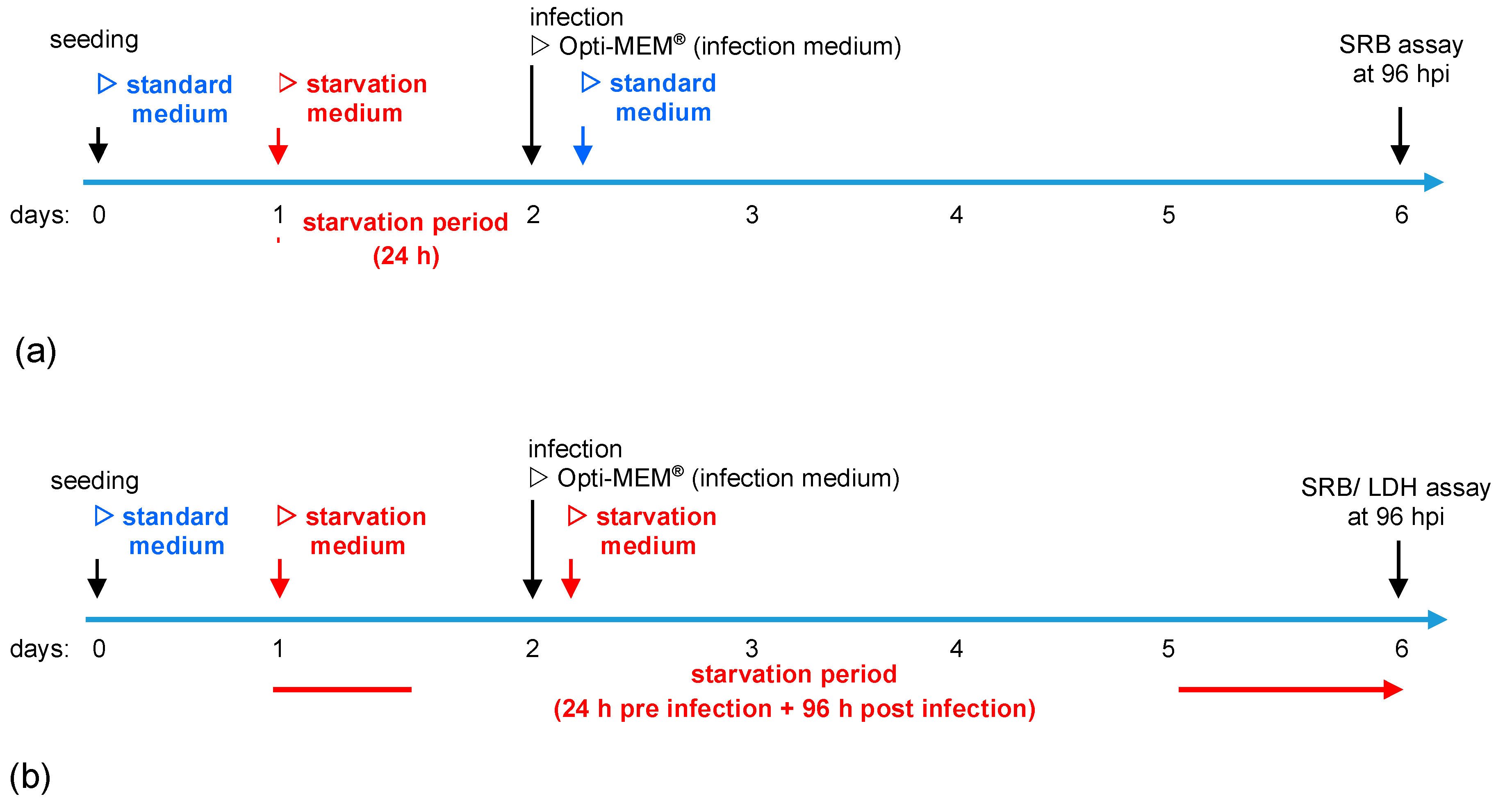{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Experimental Setup
2.3. Oncolytic Virotherapeutic MeV-GFP
2.4. Virus Infection
2.5. Sulforhodamine B (SRB) Cell Viability Assay
2.6. Lactate Dehydrogenase (LDH) Assay
2.7. Viral Growth Curve
2.8. Statistics
3. Results
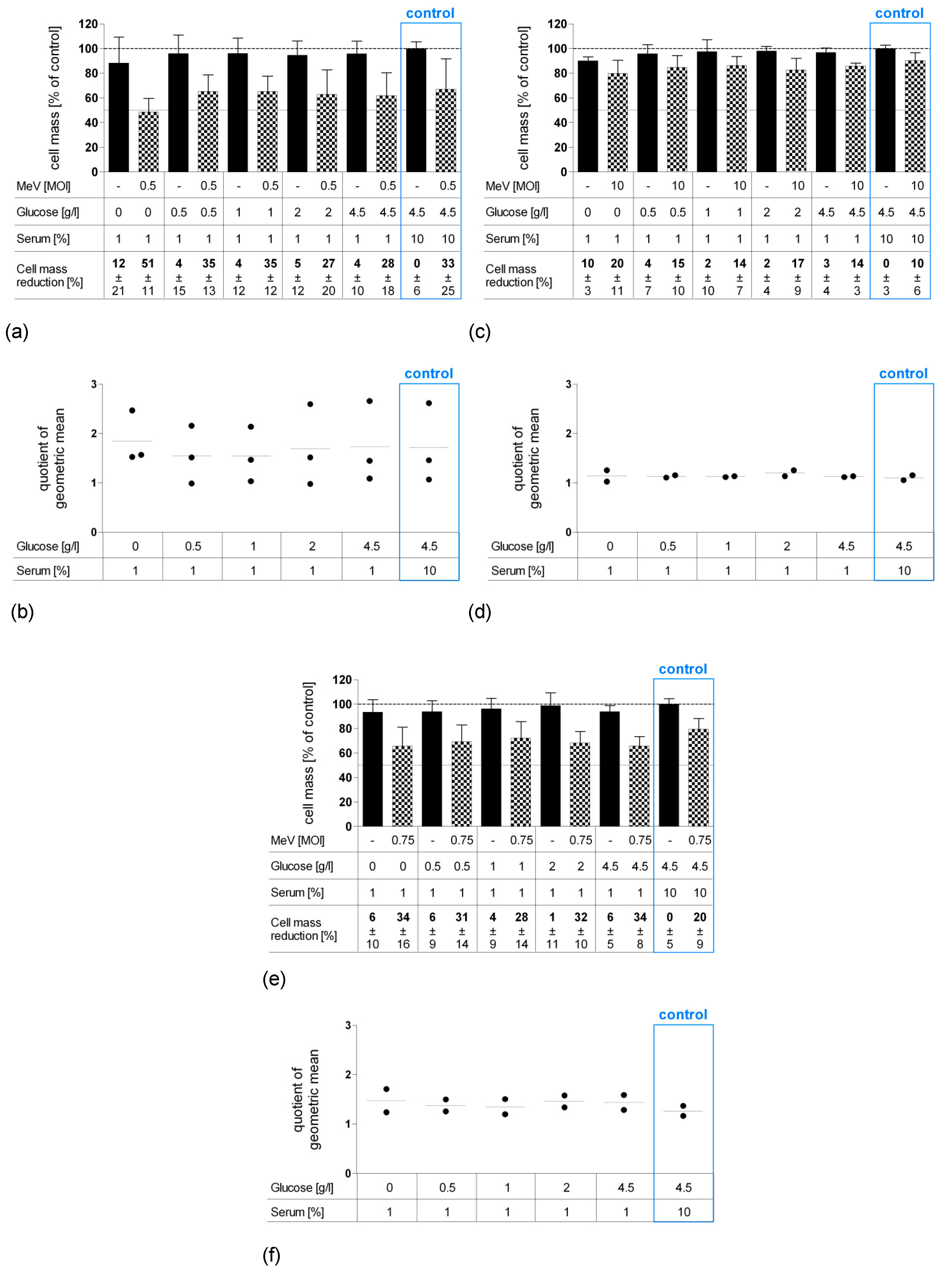
3.1. Determination of Adjusted MOIs in Colon Carcinoma and Non-Malignant Colon Cell Lines
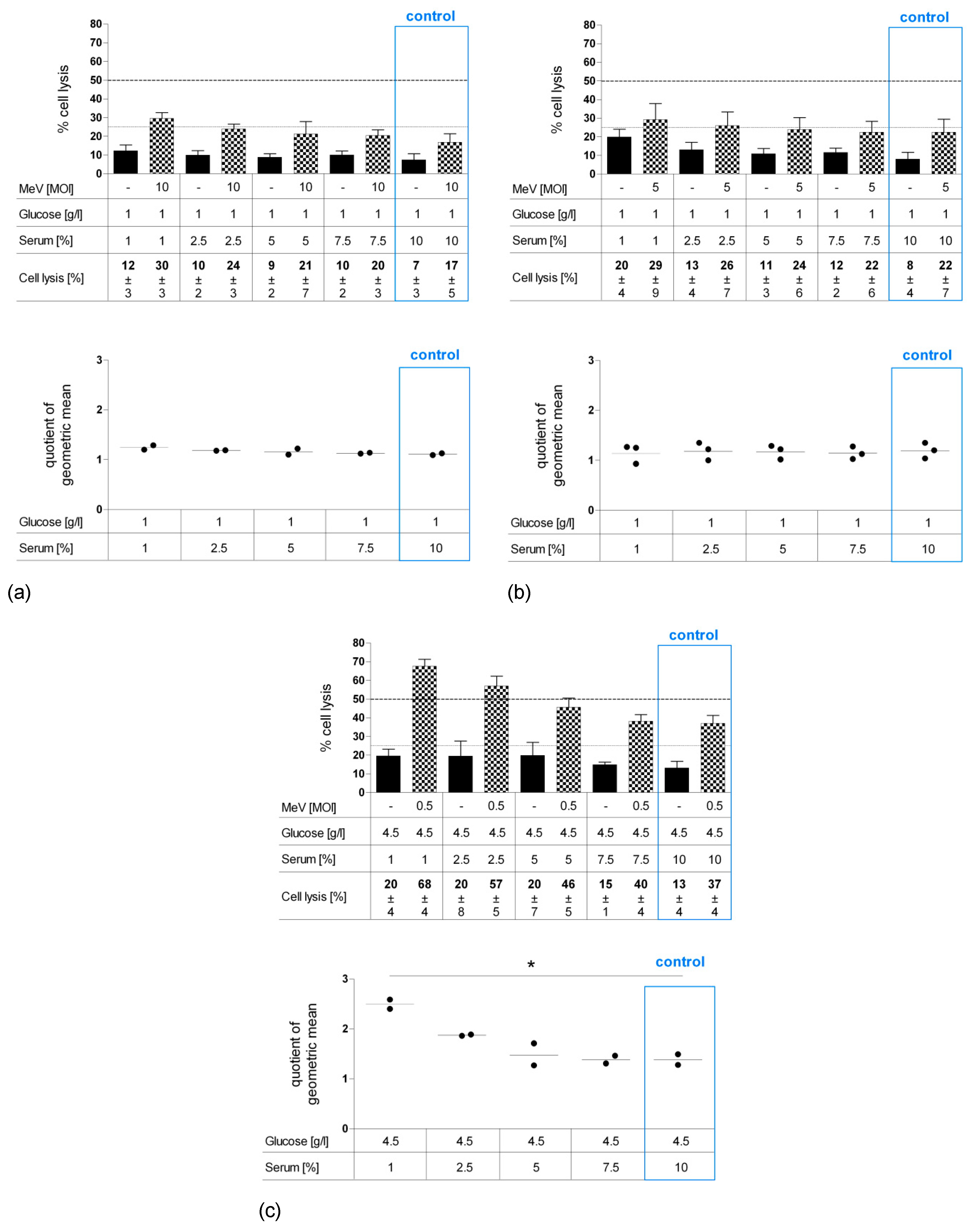
3.2. Short-Term Starvation (24 h) Decelerated Tumor Cell Growth and Kept MeV-GFP-Mediated Oncolysis Intact
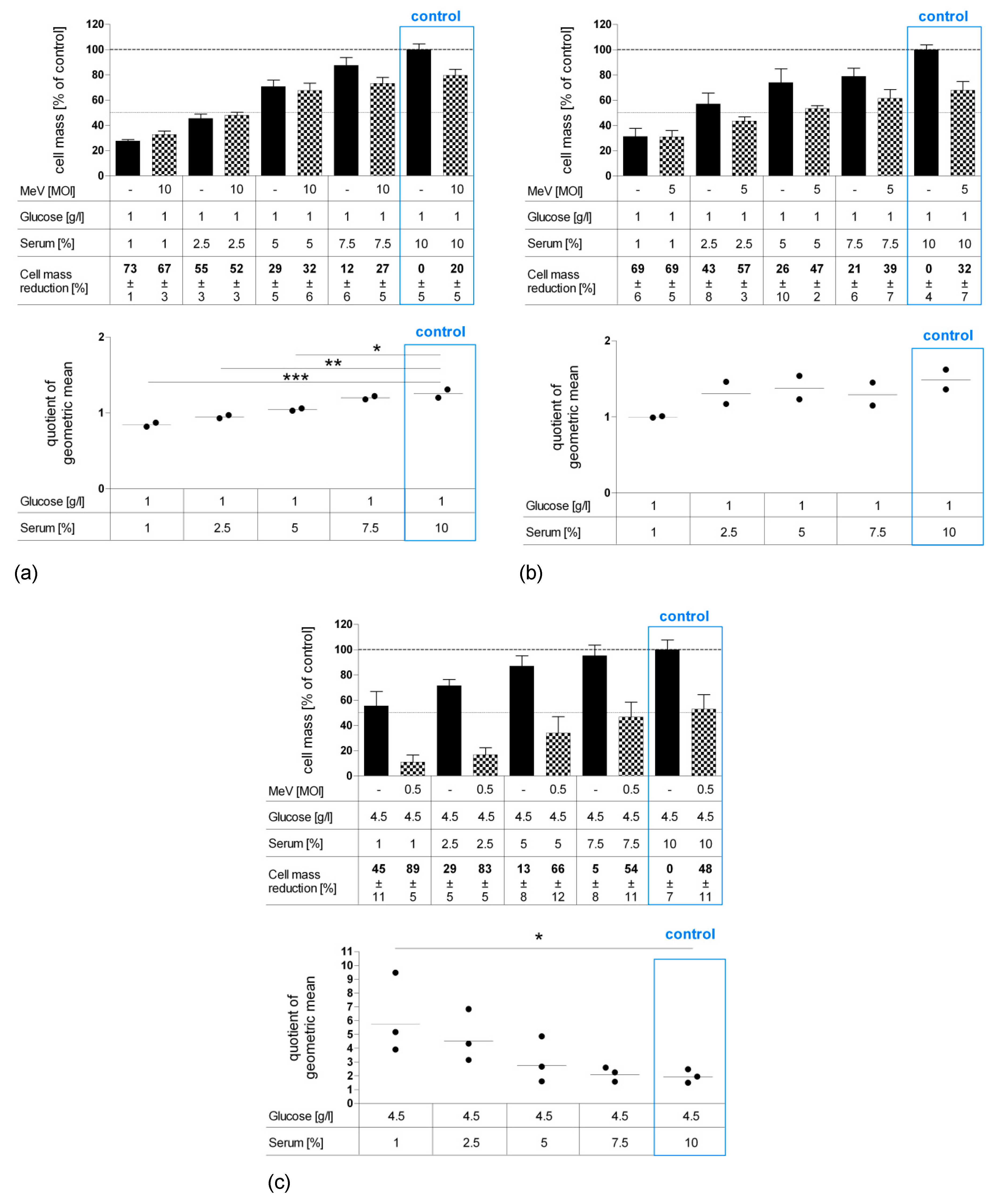
3.3. Long-Term Starvation Substantially Inhibited Tumor Cell Growth and Enhanced the Efficacy of MeV-GFP-Mediated Oncolysis for HT-29 Cells Cultured in Low-Glucose, Low-Serum But Not in Low-Glucose, Standard Serum Medium
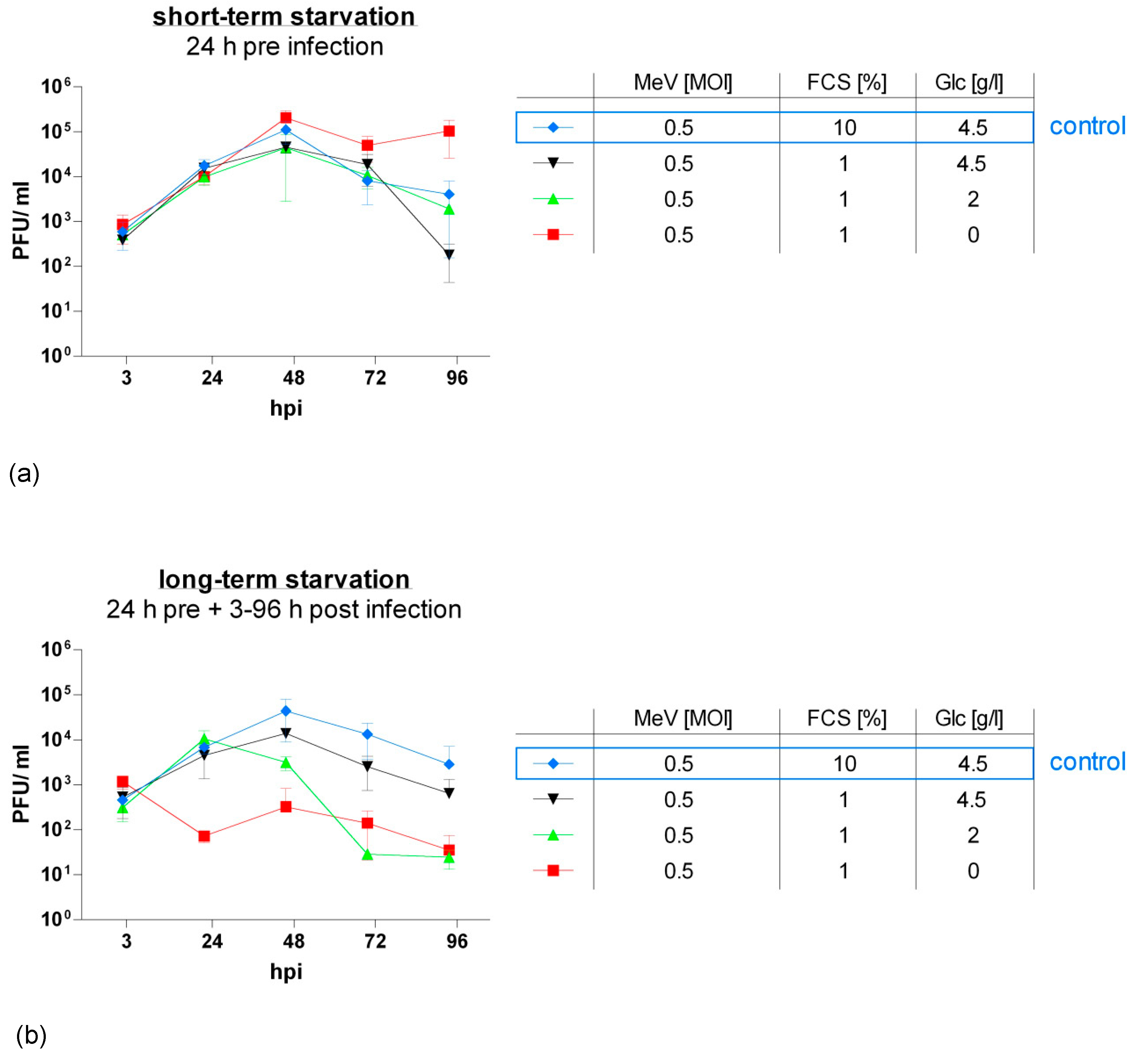
3.4. MeV-GFP Replication was Impaired by Long-Term Low-Glucose, Lo-Serum Starvation, But Widely Unaffected by Long-Term Standard Glucose, Low-Serum Starvation, and Increased in Short-Term Low-Glucose, Low-Serum-Starved HT-29 Cells
3.5. Long-Term Serum Starvation Increased MeV-GFP-Mediated Oncolysis in Human Colon Carcinoma HT-29 Cells, But Not in Normal Human Colon Cells CCD-18 Co and CCD-841 CoN
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lee, C.; Safdie, F.M.; Raffaghello, L.; Wei, M.; Madia, F.; Parrella, E.; Hwang, D.; Cohen, P.; Bianchi, G.; Longo, V.D. Reduced levels of IGF-I mediate differential protection of normal and cancer cells in response to fasting and improve chemotherapeutic index. Cancer Res. 2010, 70, 1564–1572. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Pollak, M.N.; Schernhammer, E.S.; Hankinson, S.E. Insulin-like growth factors and neoplasia. Nat. Rev. Cancer 2004, 4, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Longo, V.D. Fasting vs dietary restriction in cellular protection and cancer treatment: From model organisms to patients. Oncogene 2011, 30, 3305–3316. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Raffaghello, L.; Brandhorst, S.; Safdie, F.M.; Bianchi, G.; Martin-Montalvo, A.; Pistoia, V.; Wei, M.; Hwang, S.; Merlino, A.; et al. Fasting Cycles Retard Growth of Tumors and Sensitize a Range of Cancer Cell Types to Chemotherapy. Sci. Transl. Med. 2012, 4, 124ra27. [Google Scholar] [CrossRef] [PubMed]
- Raffaghello, L.; Lee, C.; Safdie, F.M.; Wei, M.; Madia, F.; Bianchi, G.; Longo, V.D. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc. Natl. Acad. Sci. USA 2008, 105, 8215–8220. [Google Scholar] [CrossRef] [Green Version]
- Safdie, F.M.; Dorff, T.; Quinn, D.; Fontana, L.; Wei, M.; Lee, C.; Cohen, P.; Longo, V.D. Fasting and cancer treatment in humans: A case series report. Aging 2009, 1, 988–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorff, T.B.; Groshen, S.; Garcia, A.; Shah, M.; Tsao-Wei, D.; Pham, H.; Cheng, C.-W.; Brandhorst, S.; Cohen, P.; Wei, M.; et al. Safety and feasibility of fasting in combination with platinum-based chemotherapy. BMC Cancer 2016, 16, 677. [Google Scholar] [CrossRef]
- De Groot, S.; Vreeswijk, M.P.; Welters, M.J.; Gravesteijn, G.; Boei, J.J.; Jochems, A.; Houtsma, D.; Putter, H.; van der Hoeven, J.J.; Nortier, J.W.; et al. The effects of short-term fasting on tolerance to (neo) adjuvant chemotherapy in HER2-negative breast cancer patients: a randomized pilot study. BMC Cancer 2015, 15, 652. [Google Scholar] [CrossRef]
- Bauersfeld, S.P.; Kessler, C.S.; Wischnewsky, M.; Jaensch, A.; Steckhan, N.; Stange, R.; Kunz, B.; Brückner, B.; Sehouli, J.; Michalsen, A. The effects of short-term fasting on quality of life and tolerance to chemotherapy in patients with breast and ovarian cancer: a randomized cross-over pilot study. BMC Cancer 2018, 18, 476. [Google Scholar] [CrossRef]
- Russell, S.J.; Federspiel, M.J.; Peng, K.-W.; Tong, C.; Dingli, D.; Morice, W.G.; Lowe, V.; O’Connor, M.K.; Kyle, R.A.; Leung, N.; et al. Remission of Disseminated Cancer After Systemic Oncolytic Virotherapy. Mayo Clin. Proc. 2014, 89, 926–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poh, A. First Oncolytic Viral Therapy for Melanoma. Cancer Discov. 2016, 6, 6. [Google Scholar] [PubMed]
- Vacchelli, E.; Eggermont, A.; Sautès-Fridman, C.; Galon, J.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Oncolytic viruses for cancer therapy. Oncoimmunology 2013, 2, e24612. [Google Scholar] [CrossRef] [PubMed]
- Pol, J.; Bloy, N.; Obrist, F.; Eggermont, A.; Galon, J.; Cremer, I.; Erbs, P.; Limacher, J.-M.; Preville, X.; et al. Trial Watch: Oncolytic viruses for cancer therapy. Oncoimmunology 2014, 3, e28694. [Google Scholar] [CrossRef] [PubMed]
- Ranki, T.; Joensuu, T.; Jäger, E.; Karbach, J.; Wahle, C.; Kairemo, K.; Alanko, T.; Partanen, K.; Turkki, R.; Linder, N.; et al. Local treatment of a pleural mesothelioma tumor with ONCOS-102 induces a systemic antitumor CD8(+) T-cell response, prominent infiltration of CD8(+) lymphocytes and Th1 type polarization. Oncoimmunology 2014, 3, e958937. [Google Scholar] [CrossRef] [PubMed]
- Jonker, D.J.; Tang, P.A.; Kennecke, H.; Welch, S.A.; Cripps, M.C.; Asmis, T.; Chalchal, H.; Tomiak, A.; Lim, H.; Ko, Y.-J.; et al. A Randomized Phase II Study of FOLFOX6/Bevacizumab With or Without Pelareorep in Patients With Metastatic Colorectal Cancer: IND.210, a Canadian Cancer Trials Group Trial. Clin. Color. Cancer 2018, 17, 231–239.e7. [Google Scholar] [CrossRef]
- Bradbury, P.A.; Morris, D.G.; Nicholas, G.; Tu, D.; Tehfe, M.; Goffin, J.R.; Shepherd, F.A.; Gregg, R.W.; Rothenstein, J.; Lee, C.; et al. Canadian Cancer Trials Group (CCTG) IND211: A randomized trial of pelareorep (Reolysin) in patients with previously treated advanced or metastatic non-small cell lung cancer receiving standard salvage therapy. Lung Cancer 2018, 120, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Goel, S.; Aparo, S.; Arora, S.P.; Noronha, N.; Tran, H.; Chakrabarty, R.; Selvaggi, G.; Gutierrez, A.; Coffey, M.; et al. A Phase II Study of Pelareorep (REOLYSIN®) in Combination with Gemcitabine for Patients with Advanced Pancreatic Adenocarcinoma. Cancers 2018, 10, 160. [Google Scholar] [CrossRef]
- Mahalingam, D.; Fountzilas, C.; Moseley, J.; Noronha, N.; Tran, H.; Chakrabarty, R.; Selvaggi, G.; Coffey, M.; Thompson, B.; Sarantopoulos, J. A phase II study of REOLYSIN® (pelareorep) in combination with carboplatin and paclitaxel for patients with advanced malignant melanoma. Cancer Chemother. Pharmacol. 2017, 79, 697–703. [Google Scholar] [CrossRef]
- Eigl, B.J.; Chi, K.; Tu, D.; Hotte, S.J.; Winquist, E.; Booth, C.M.; Canil, C.; Potvin, K.; Gregg, R.; North, S.; et al. A randomized phase II study of pelareorep and docetaxel or docetaxel alone in men with metastatic castration resistant prostate cancer: CCTG study IND 209. Oncotarget 2018, 9, 8155–8164. [Google Scholar] [CrossRef]
- Bernstein, V.; Ellard, S.L.; Dent, S.F.; Tu, D.; Mates, M.; Dhesy-Thind, S.K.; Panasci, L.; Gelmon, K.A.; Salim, M.; Song, X.; et al. A randomized phase II study of weekly paclitaxel with or without pelareorep in patients with metastatic breast cancer: final analysis of Canadian Cancer Trials Group IND.213. Breast Cancer Res. Treat. 2018, 167, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Lampe, J.; Bossow, S.; Weiland, T.; Smirnow, I.; Lehmann, R.; Neubert, W.; Bitzer, M.; Lauer, U.M. An armed oncolytic measles vaccine virus eliminates human hepatoma cells independently of apoptosis. Gene Ther. 2013, 20, 1033–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Msaouel, P.; Opyrchal, M.; Domingo-Musibay, E.; Galanis, E. Oncolytic Measles Virus Strains as Novel Anticancer Agents. Expert Opin. Boil. Ther. 2013, 13, 483–502. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Opyrcha, M.; Aderca, I.; Schroeder, M.A.; Sarkaria, J.N.; Domingo, E.; Federspiel, M.J.; Galanis, E. Oncolytic measles virus strains have significant antitumor activity against glioma stem cells. Gene Ther. 2013, 20, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Hartkopf, A.; Bossow, S.; Lampe, J.; Zimmermann, M.; Taran, F.-A.; Wallwiener, D.; Fehm, T.; Bitzer, M.; Lauer, U. Enhanced killing of ovarian carcinoma using oncolytic measles vaccine virus armed with a yeast cytosine deaminase and uracil phosphoribosyltransferase. Gynecol. Oncol. 2013, 130, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-P.; Steele, M.B.; Suksanpaisan, L.; Federspiel, M.J.; Russel, S.J.; Peng, K.W.; Bakkum-Gamez, J.N. Oncolytic measles and vesicular stomatitis virotherapy for endometrial cancer. Gynecol. Oncol. 2014, 132, 194–202. [Google Scholar] [CrossRef] [Green Version]
- Patel, M.R.; Jacobson, B.A.; Belgum, H.; Raza, A.; Sadiq, A.; Drees, J.; Wang, H.; Jay-Dixon, J.; Etchison, R.; Federspiel, M.J.; et al. Measles vaccine strains for virotherapy of non-small cell lung carcinoma. J. Thorac. Oncol. 2014, 9, 1101–1110. [Google Scholar] [CrossRef]
- Liu, C.; Hasegawa, K.; Russell, S.J.; Sadelain, M.; Peng, K.-W. Prostate-Specific Membrane Antigen Retargeted Measles Virotherapy for the Treatment of Prostate Cancer. Prostate 2009, 69, 1128–1141. [Google Scholar] [CrossRef]
- Boisgerault, N.; Guillerme, J.-B.; Pouliquen, D.; Mesel-Lemoine, M.; Achard, C.; Combredet, C.; Fonteneau, J.-F.; Tangy, F.; Grégoire, M. Natural Oncolytic Activity of Live-Attenuated Measles Virus against Human Lung and Colorectal Adenocarcinomas. BioMed Res. Int. 2013, 2013, 387362. [Google Scholar] [CrossRef]
- Noll, M.; Berchtold, S.; Lampe, J.; Malek, N.P.; Bitzer, M.; Lauer, U.M. Primary resistance phenomena to oncolytic measles vaccine viruses. Int. J. Oncol. 2013, 43, 103–112. [Google Scholar] [CrossRef]
- Esaki, S.; Rabkin, S.D.; Martuza, R.L.; Wakimoto, H. Transient fasting enhances replication of oncolytic herpes simplex virus in glioblastoma. Am. J. Cancer Res. 2016, 6, 300–311. [Google Scholar] [PubMed]
- Berchtold, S.; Lampe, J.; Weiland, T.; Smirnow, I.; Schleicher, S.; Handgretinger, R.; Kopp, H.-G.; Reiser, J.; Stubenrauch, F.; Mayer, N.; et al. Innate Immune Defense Defines Susceptibility of Sarcoma Cells to Measles Vaccine Virus-Based Oncolysis. J. Virol. 2013, 87, 3484–3501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spearman, C. The method of ‘right and wrong cases’ (‘constant stimuli’) without Gauss’s formulae. Br. J. Psychol. 1908, 2, 227–242. [Google Scholar] [CrossRef]
- Kärber, G. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1931, 162, 480–483. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- A Parato, K.; Breitbach, C.J.; Le Boeuf, F.; Wang, J.; Storbeck, C.; Ilkow, C.; Diallo, J.-S.; Falls, T.; Burns, J.; García, V.; et al. The Oncolytic Poxvirus JX-594 Selectively Replicates in and Destroys Cancer Cells Driven by Genetic Pathways Commonly Activated in Cancers. Mol. Ther. 2012, 20, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567. [Google Scholar] [CrossRef]
- Chiang, C.; Hiscott, J.; Beljanski, V. The intersection between viral oncolysis, drug resistance, and autophagy. Boil. Chem. 2015, 396, 1269–1280. [Google Scholar]
- Levine, M.E.; Suarez, J.A.; Brandhorst, S.; Balasubramanian, P.; Cheng, C.-W.; Madia, F.; Fontana, L.; Mirisola, M.G.; Guevara-Aguirre, J.; Wan, J.; et al. Low Protein Intake is Associated with a Major Reduction in IGF-1, Cancer, and Overall Mortality in the 65 and Younger but Not Older Population. Cell Metab. 2014, 19, 407–417. [Google Scholar] [CrossRef]
- Cox, M.E.; Gleave, M.E.; Zakikhani, M.; Bell, R.H.; Piura, E.; Vickers, E.; Cunningham, M.; Larsson, O.; Fazli, L.; Pollak, M. Insulin receptor expression by human prostate cancers. Prostate 2009, 69, 33–40. [Google Scholar] [CrossRef]
- Law, J.H.; Habibi, G.; Hu, K.; Masoudi, H.; Wang, M.Y.; Stratford, A.L.; Park, E.; Gee, J.M.; Finlay, P.; Jones, H.E.; et al. Phosphorylated Insulin-Like Growth Factor-I/Insulin Receptor Is Present in All Breast Cancer Subtypes and Is Related to Poor Survival. Cancer Res. 2008, 68, 10238–10246. [Google Scholar] [CrossRef] [PubMed]
- Tandon, R.; Kapoor, S.; Vali, S.; Senthil, V.; Nithya, D.; Venkataramanan, R.; Sharma, A.; Talwadkar, A.; Ray, A.; Bhatnagar, P.K.; et al. Dual epidermal growth factor receptor (EGFR)/insulin-like growth factor-1 receptor (IGF-1R) inhibitor: A novel approach for overcoming resistance in anticancer treatment. Eur. J. Pharmacol. 2011, 667, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-C.; Hou, S.-C.; Hung, C.-M.; Lin, J.-N.; Chen, W.-C.; Ho, C.-T.; Kuo, S.-C.; Way, T.-D. Inhibition of the insulin-like growth factor 1 receptor by CHM-1 blocks proliferation of glioblastoma multiforme cells. Chem. Interact. 2015, 231, 119–126. [Google Scholar] [CrossRef] [PubMed]
- E Merrick, A.; Ilett, E.J.; A Melcher, A. JX-594, a targeted oncolytic poxvirus for the treatment of cancer. Curr. Opin. Investig. Drugs 2009, 10, 1372–1382. [Google Scholar]
- Grégoire, I.P.; Richetta, C.; Meyniel-Schicklin, L.; Borel, S.; Pradezynski, F.; Diaz, O.; Deloire, A.; Azocar, O.; Baguet, J.; Le Breton, M.; et al. IRGM Is a Common Target of RNA Viruses that Subvert the Autophagy Network. PLOS Pathog. 2011, 7, e1002422. [Google Scholar] [CrossRef] [PubMed]
- Richetta, C.; Grégoire, I.P.; Verlhac, P.; Azocar, O.; Baguet, J.; Flacher, M.; Tangy, F.; Rabourdin-Combe, C.; Faure, M. Sustained Autophagy Contributes to Measles Virus Infectivity. PLOS Pathog. 2013, 9, e1003599. [Google Scholar] [CrossRef] [PubMed]
- Petkova, D.S.; Verlhac, P.; Rozières, A.; Baguet, J.; Claviere, M.; Kretz-Remy, C.; Mahieux, R.; Viret, C.; Faure, M. Distinct Contributions of Autophagy Receptors in Measles Virus Replication. Viruses 2017, 9, 123. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nature 2013, 15, 713–720. [Google Scholar] [CrossRef]
- Gomes, L.C.; Dikic, I. Autophagy in Antimicrobial Immunity. Mol. Cell 2014, 54, 224–233. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.C.; Yuan, H.X.; Guan, K.L. Autophagy regulation by nutrient signaling. Cell Res. 2014, 24, 42–57. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; da Silva, S.R.; He, M.; Liang, Q.; Lu, C.; Feng, P.; Jung, J.U.; Gao, S.-J. An Oncogenic Virus Promotes Cell Survival and Cellular Transformation by Suppressing Glycolysis. PLoS Pathog. 2016, 12, 1005648. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V. Cellular Fatty Acid Metabolism and Cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Pópulo, H.; Lopes, J.M.; Soares, P. The mTOR Signalling Pathway in Human Cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef] [PubMed]
- Sticz, T.; Molnár, A.; Márk, A.; Hajdu, M.; Nagy, N.; Végső, G.; Micsik, T.; Kopper, L.; Sebestyén, A. mTOR activity and its prognostic significance in human colorectal carcinoma depending on C1 and C2 complex-related protein expression. J. Clin. Pathol. 2017, 70, 410–416. [Google Scholar] [CrossRef]
- Longo, V.D.; Fontana, L. Intermittent supplementation with rapamycin as a dietary restriction mimetic. Aging 2011, 3, 1039–1040. [Google Scholar] [CrossRef] [Green Version]
- Apontes, P.; Leontieva, O.V.; Demidenko, Z.N.; Li, F.; Blagosklonny, M.V. Exploring long-term protection of normal human fibroblasts and epithelial cells from chemotherapy in cell culture. Oncotarget 2011, 2, 222–233. [Google Scholar] [CrossRef] [Green Version]
- Lun, X.; Chan, J.; Zhou, H.; Sun, B.; Kelly, J.J.; Stechishin, O.O.; Bell, J.C.; Parato, K.; Hu, K.; Vaillant, D.; et al. Efficacy and Safety/Toxicity Study of Recombinant Vaccinia Virus JX-594 in Two Immunocompetent Animal Models of Glioma. Mol. Ther. 2010, 18, 1927–1936. [Google Scholar] [CrossRef] [PubMed]
- Werden, S.J.; McFadden, G. Pharmacological Manipulation of the Akt Signaling Pathway Regulates Myxoma Virus Replication and Tropism in Human Cancer Cells. J. Virol. 2010, 84, 3287–3302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Aronzo, M.; Vinciguerra, M.; Mazza, T.; Panebianco, C.; Saracino, C.; Pereira, S.P.; Graziano, P.; Pazienza, V. Fasting cycles potentiate the efficacy of gemcitabine treatment in in vitro and in vivo pancreatic cancer models. Oncotarget 2015, 6, 18545–18557. [Google Scholar] [CrossRef] [PubMed]
- Caccialanza, R.; Aprile, G.; Cereda, E.; Pedrazzoli, P. Fasting in oncology: A word of caution. Nat. Rev. Cancer 2019, 19, 177. [Google Scholar] [CrossRef] [PubMed]
- Caccialanza, R.; de Lorenzo, F.; Gianotti, L.; Zagonel, V.; Gavazzi, C.; Farina, G.; Cotogni, P.; Cinieri, S.; Cereda, E.; Marchetti, P.; et al. Nutritional support for cancer patients: Still a neglected right? Support Care Cancer 2017, 25, 3001–3004. [Google Scholar] [CrossRef] [PubMed]
- Arends, J.; Bachmann, P.; Baracos, V.; Barthelemy, N.; Bertz, H.; Bozzetti, F.; Fearon, K.; Hütterer, E.; Isenring, E.; Kaasa, S.; et al. ESPEN guidelines on nutrition in cancer patients. Clin. Nutr. 2017, 36, 11–48. [Google Scholar] [CrossRef] [PubMed]
- Kalaany, N.Y.; Sabatini, D.M. Tumours with PI3K activation are resistant to dietary restriction. Nature 2009, 458, 725–731. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scheubeck, G.; Berchtold, S.; Smirnow, I.; Schenk, A.; Beil, J.; Lauer, U.M. Starvation-Induced Differential Virotherapy Using an Oncolytic Measles Vaccine Virus. Viruses 2019, 11, 614. https://doi.org/10.3390/v11070614
Scheubeck G, Berchtold S, Smirnow I, Schenk A, Beil J, Lauer UM. Starvation-Induced Differential Virotherapy Using an Oncolytic Measles Vaccine Virus. Viruses. 2019; 11(7):614. https://doi.org/10.3390/v11070614
Chicago/Turabian StyleScheubeck, Gabriel, Susanne Berchtold, Irina Smirnow, Andrea Schenk, Julia Beil, and Ulrich M. Lauer. 2019. "Starvation-Induced Differential Virotherapy Using an Oncolytic Measles Vaccine Virus" Viruses 11, no. 7: 614. https://doi.org/10.3390/v11070614
APA StyleScheubeck, G., Berchtold, S., Smirnow, I., Schenk, A., Beil, J., & Lauer, U. M. (2019). Starvation-Induced Differential Virotherapy Using an Oncolytic Measles Vaccine Virus. Viruses, 11(7), 614. https://doi.org/10.3390/v11070614