Effects of Arbovirus Multi-Host Life Cycles on Dinucleotide and Codon Usage Patterns

Abstract
:1. Introduction
2. Evolutionary Pressures on Arboviruses Resulting from Replication in Multiple Hosts
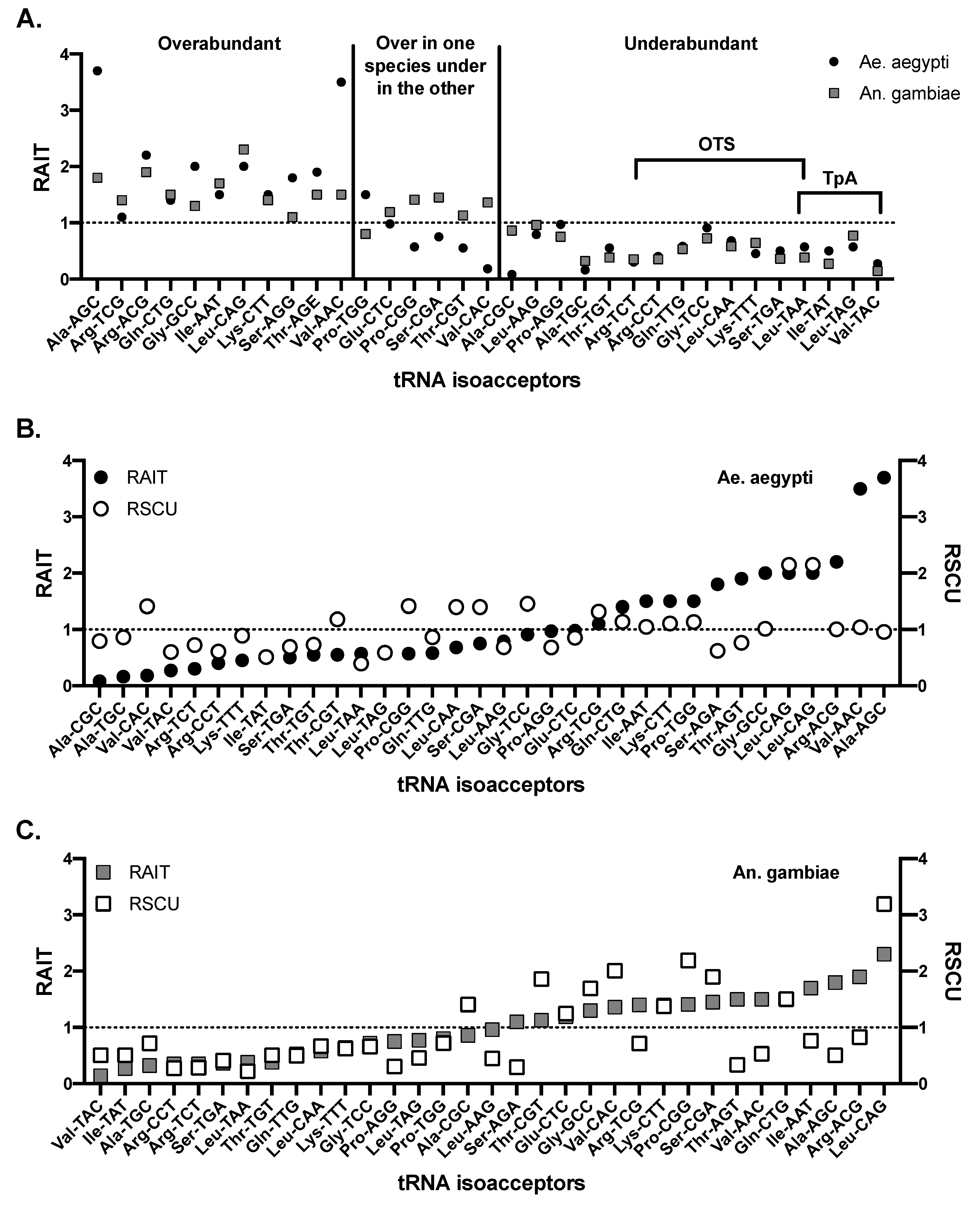
3. Codon Usage Bias in Hematophagous Arthropods
- Box 1. Frequently used measures and scores for nucleotide and tRNA usages. The usage of nucleotides across genomes is not random. Organisms show specific patterns of use for combinations of total nucleotides, pairs of nucleotides (dinucleotides), and for the specific codons used to select for amino acids. Additionally, these different nucleotide measures are intricately linked to each other. To better understand these biases many different measures have been developed. Those referenced in this review are summarized here:
- Relative abundance of isoacceptor tRNA (RAIT). RAIT scores demonstrate preferences for specific isoacceptor tRNAs in a genome. RAIT scores are the ratio of observed isoacceptor tRNA gene copies over the expected copy number for a specific amino acid encoding tRNA group. Expected copy number is determined by the sum of all tRNA genes that result in identical amino acids, divided by the number of different tRNA isoacceptors resulting in that amino acid [60].
- Relative synonymous codon usage (RSCU). RSCU was developed by Sharp and Li [64] and is used to determine how much bias is present in the use of codons for a particular amino acid. To calculate this, the observed number of a particular codon is divided by the expected number of codons if there was no bias. The expected value is calculated as the total number of codons that result in the amino acid of the codon being investigated divided by the codon degeneracy (number of codons resulting in that amino acid).A value of 1 represents no bias. In this review we consider values above 1.38 and below 0.62 to represent over and underabundant codons. RSCU can be calculated using CodonW [65] or ANACONDA software [60,66].
- Dinucleotide odds ratios. Similar to RSCU, the dinucleotide odds ratios provide numerical values to the amount of bias for or against a specific dinucleotide within a genome. It is measured as the observed number of a specific dinucleotide pair divided by the number of each individual nucleotide that make up that pair multiplied together.Here, fk (where k = x, y, z or w) represent frequency of mononucleotides in a sequence; z and w represent complementary nucleotides to x/y. Similarly, fxy and fzw represent the frequency of dinucleotides in the same sequence. Again, 1 represents no bias; values above 1.25 or below 0.75 are considered to be significantly over or underrepresented [21].
- Effective number of codons (ENC). The ENC was introduced by Wright [67] and is a measure of how equally synonymous codons are being used.F2, F3, F4, and F6 represent the probability that randomly chosen codons for a specific amino acid are identical. ENC calculation results in a value between 20, indicating only one codon is used per amino acid, and 61 indicating that all synonymous codons are being utilized equally. CodonW software can be used to calculate ENC [65].
4. Dinucleotide Preferences in Arboviruses, Comparisons with Single Host Viruses
5. Arbovirus Codon Usage is Driven by Host Association, but Does Not Mimic Host Codon Usage
6. Altering Codon Position and Pair Bias Results in Attenuation of Arthropod-Borne Viruses
7. Genetic Robustness (or Lack Thereof?) in the Context of Alternating Hosts
8. Conclusions and Future Directions
Supplementary Materials
Funding
Conflicts of Interest
References
- Laksono, B.M.; de Vries, R.D.; McQuaid, S.; Duprex, W.P.; de Swart, R.L. Measles Virus Host Invasion and Pathogenesis. Viruses 2016, 8, 210. [Google Scholar] [CrossRef] [PubMed]
- Kuno, G.; Mackenzie, J.S.; Junglen, S.; Hubálek, Z.; Plyusnin, A.; Gubler, D.J. Vertebrate Reservoirs of Arboviruses: Myth, Synonym of Amplifier, or Reality? Viruses 2017, 9, 185. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Reisen, W.K. Present and future arboviral threats. Antiviral Res. 2010, 85, 328–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paixão, E.S.; Teixeira, M.G.; Rodrigues, L.C. Zika, chikungunya and dengue: The causes and threats of new and re-emerging arboviral diseases. BMJ Glob. Health 2018, 3, e000530. [Google Scholar] [CrossRef] [PubMed]
- Maclachlan, N.J.; Zientara, S.; Wilson, W.C.; Richt, J.A.; Savini, G. Bluetongue and epizootic hemorrhagic disease viruses: Recent developments with these globally re-emerging arboviral infections of ruminants. Curr. Opin. Virol. 2019, 34, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Linthicum, K.J.; Britch, S.C.; Anyamba, A. Rift Valley Fever: An Emerging Mosquito-Borne Disease. Annu. Rev. Entomol. 2016, 61, 395–415. [Google Scholar] [CrossRef] [PubMed]
- Lednicky, J.; De Rochars, V.M.B.; Elbadry, M.; Loeb, J.; Telisma, T.; Chavannes, S.; Anilis, G.; Cella, E.; Ciccozzi, M.; Okech, B.; et al. Mayaro Virus in Child with Acute Febrile Illness, Haiti, 2015. Emerg. Infect. Dis. 2016, 22, 2000–2002. [Google Scholar] [CrossRef] [PubMed]
- Peterson, K.J.; Lyons, J.B.; Nowak, K.S.; Takacs, C.M.; Wargo, M.J.; McPeek, M.A. Estimating metazoan divergence times with a molecular clock. Proc. Natl. Acad. Sci. USA 2004, 101, 6536–6541. [Google Scholar] [CrossRef] [Green Version]
- Lobo, F.P.; Mota, B.E.F.; Pena, S.D.J.; Azevedo, V.; Macedo, A.M.; Tauch, A.; Machado, C.R.; Franco, G.R. Virus-Host Coevolution: Common Patterns of Nucleotide Motif Usage in Flaviviridae and Their Hosts. PLoS ONE 2009, 4, e6282-14. [Google Scholar] [CrossRef]
- Velazquez-Salinas, L.; Zarate, S.; Eschbaumer, M.; Pereira Lobo, F.; Gladue, D.P.; Arzt, J.; Novella, I.S.; Rodriguez, L.L. Selective Factors Associated with the Evolution of Codon Usage in Natural Populations of Arboviruses. PLoS ONE 2016, 11, e0159943-17. [Google Scholar] [CrossRef]
- Mavian, C.; Rife, B.D.; Dollar, J.J.; Cella, E.; Ciccozzi, M.; Prosperi, M.C.F.; Lednicky, J.; Morris, J.G.; Capua, I.; Salemi, M. Emergence of recombinant Mayaro virus strains from the Amazon basin. Sci. Rep. 2017, 7, 8718. [Google Scholar] [CrossRef] [PubMed]
- Enard, D.; Cai, L.; Gwennap, C.; Petrov, D.A. Viruses are a dominant driver of protein adaptation in mammals. Elife 2016, 5, e12469. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, M.D.; Malik, H.S. Rules of engagement: Molecular insights from host-virus arms races. Annu. Rev. Genet. 2012, 46, 677–700. [Google Scholar] [CrossRef] [PubMed]
- Lawrie, D.S.; Messer, P.W.; Hershberg, R.; Petrov, D.A. Strong Purifying Selection at Synonymous Sites in D. melanogaster. PLoS Genet. 2013, 9, e1003527. [Google Scholar] [CrossRef] [PubMed]
- Takata, M.A.; Gonçalves-Carneiro, D.; Zang, T.M.; Soll, S.J.; York, A.; Blanco-Melo, D.; Bieniasz, P.D. CG dinucleotide suppression enables antiviral defence targeting non-self RNA. Nature 2017, 550, 124–127. [Google Scholar] [CrossRef]
- Smyth, R.P.; Negroni, M.; Lever, A.M.; Mak, J.; Kenyon, J.C. RNA Structure—A Neglected Puppet Master for the Evolution of Virus and Host Immunity. Front. Immunol. 2018, 9, 2097. [Google Scholar] [CrossRef]
- Hershberg, R.; Petrov, D.A. Selection on Codon Bias. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef] [Green Version]
- Moratorio, G.; Henningsson, R.; Barbezange, C.; Carrau, L.; Bordería, A.V.; Blanc, H.; Beaucourt, S.; Poirier, E.Z.; Vallet, T.; Boussier, J.; et al. Attenuation of RNA viruses by redirecting their evolution in sequence space. Nat. Microbiol. 2017, 2, 1–12. [Google Scholar] [CrossRef]
- Shen, S.H.; Stauft, C.B.; Gorbatsevych, O.; Song, Y.; Ward, C.B.; Yurovsky, A.; Mueller, S.; Futcher, B.; Wimmer, E. Large-scale recoding of an arbovirus genome to rebalance its insect versus mammalian preference. Proc. Natl. Acad. Sci. USA 2015, 112, 4749–4754. [Google Scholar] [CrossRef] [Green Version]
- de Fabritus, L.; Nougairède, A.; Aubry, F.; Gould, E.A.; de Lamballerie, X. Attenuation of Tick-Borne Encephalitis Virus Using Large-Scale Random Codon Re-encoding. Plos Pathog. 2015, 11, e1004738-18. [Google Scholar] [CrossRef]
- Karlin, S.; Mrázek, J. Compositional differences within and between eukaryotic genomes. Proc. Natl. Acad. Sci. USA 1997, 94, 10227–10232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlin, S.; Ladunga, I.; Blaisdell, B.E. Heterogeneity of genomes: Measures and values. Proc. Natl. Acad. Sci. USA 1994, 91, 12837–12841. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, J.B.; Kudla, G. Synonymous but not the same: The causes and consequences of codon bias. Nat. Rev. Genet. 2011, 12, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Hanson, G.; Coller, J. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 2017, 19, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Supek, F. The Code of Silence: Widespread Associations Between Synonymous Codon Biases and Gene Function. J. Mol. Evol. 2015, 82, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Xu, Y.; Wang, X.; Gu, J.; Yan, G.; Chen, X.-G. Antiviral systems in vector mosquitoes. Dev. Comp. Immunol. 2018, 83, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Vabret, N.; Bhardwaj, N.; Greenbaum, B.D. Sequence-Specific Sensing of Nucleic Acids. Trends Immunol. 2017, 38, 53–65. [Google Scholar] [CrossRef]
- Netea, M.G.; Schlitzer, A.; Placek, K.; Joosten, L.A.B.; Schultze, J.L. Innate and Adaptive Immune Memory: An Evolutionary Continuum in the Host’s Response to Pathogens. Cell Host Microbe 2019, 25, 13–26. [Google Scholar] [CrossRef]
- Merkling, S.H.; van Rij, R.P. Beyond RNAi: Antiviral defense strategies in Drosophila and mosquito. J. Insect. Physiol. 2013, 59, 159–170. [Google Scholar] [CrossRef]
- Fensterl, V.; Chattopadhyay, S.; Sen, G.C. No Love Lost Between Viruses and Interferons. Annu. Rev. Virol. 2015, 2, 549–572. [Google Scholar] [CrossRef]
- Halbach, R.; Junglen, S.; van Rij, R.P. Mosquito-specific and mosquito-borne viruses: Evolution, infection, and host defense. Curr. Opin. Insect. Sci. 2017, 22, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Brackney, D.E.; Beane, J.E.; Ebel, G.D. RNAi targeting of West Nile virus in mosquito midguts promotes virus diversification. PLoS Pathog. 2009, 5, e1000502. [Google Scholar] [CrossRef] [PubMed]
- Grubaugh, N.D.; Smith, D.R.; Brackney, D.E.; Bosco-Lauth, A.M.; Fauver, J.R.; Campbell, C.L.; Felix, T.A.; Romo, H.; Duggal, N.K.; Dietrich, E.A.; et al. Experimental evolution of an RNA virus in wild birds: Evidence for host-dependent impacts on population structure and competitive fitness. PLoS Pathog. 2015, 11, e1004874. [Google Scholar] [CrossRef] [PubMed]
- Grubaugh, N.D.; Weger-Lucarelli, J.; Murrieta, R.A.; Fauver, J.R.; Garcia Luna, S.M.; Prasad, A.N.; Black, W.C.; Ebel, G.D. Genetic Drift during Systemic Arbovirus Infection of Mosquito Vectors Leads to Decreased Relative Fitness during Host Switching. Cell Host Microbe 2016, 19, 481–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubaugh, N.D.; Ebel, G.D. Dynamics of West Nile virus evolution in mosquito vectors. Curr. Opin. Virol. 2016, 21, 132–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brackney, D.E.; Schirtzinger, E.E.; Harrison, T.D.; Ebel, G.D.; Hanley, K.A. Modulation of flavivirus population diversity by RNA interference. J. Virol. 2015, 89, 4035–4039. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.W.; Sibley, S.D.; Kolokotronis, S.-O.; Hamer, G.L.; Newman, C.M.; Anderson, T.K.; Walker, E.D.; Kitron, U.D.; Brawn, J.D.; Ruiz, M.O.; et al. Selective constraint and adaptive potential of West Nile virus within and among naturally infected avian hosts and mosquito vectors. Virus Evolut. 2018, 4, vey013. [Google Scholar] [CrossRef] [PubMed]
- Villordo, S.M.; Filomatori, C.V.; Sánchez-Vargas, I.; Blair, C.D.; Gamarnik, A.V. Dengue Virus RNA Structure Specialization Facilitates Host Adaptation. PLoS Pathog. 2015, 11, e1004604-22. [Google Scholar] [CrossRef]
- Li, F.; Zheng, Q.; Ryvkin, P.; Dragomir, I.; Desai, Y.; Aiyer, S.; Valladares, O.; Yang, J.; Bambina, S.; Sabin, L.R.; et al. Global Analysis of RNA Secondary Structure in Two Metazoans. Cell Rep. 2012, 1, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Coffey, L.L.; Vignuzzi, M. Host alternation of chikungunya virus increases fitness while restricting population diversity and adaptability to novel selective pressures. J. Virol. 2011, 85, 1025–1035. [Google Scholar] [CrossRef]
- Vasilakis, N.; Deardorff, E.R.; Kenney, J.L.; Rossi, S.L.; Hanley, K.A.; Weaver, S.C. Mosquitoes put the brake on arbovirus evolution: Experimental evolution reveals slower mutation accumulation in mosquito than vertebrate cells. PLoS Pathog. 2009, 5, e1000467. [Google Scholar] [CrossRef] [PubMed]
- Ciota, A.T.; Lovelace, A.O.; Jones, S.A.; Payne, A.; Kramer, L.D. Adaptation of two flaviviruses results in differences in genetic heterogeneity and virus adaptability. J. Gen. Virol. 2007, 88, 2398–2406. [Google Scholar] [CrossRef] [PubMed]
- Jerzak, G.V.S.; Brown, I.; Shi, P.-Y.; Kramer, L.D.; Ebel, G.D. Genetic diversity and purifying selection in West Nile virus populations are maintained during host switching. Virology 2008, 374, 256–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, I.P.; Wang, E.; Deardorff, E.R.; Milleron, R.; Domingo, E.; Weaver, S.C. Effect of alternating passage on adaptation of sindbis virus to vertebrate and invertebrate cells. J. Virol. 2005, 79, 14253–14260. [Google Scholar] [CrossRef] [PubMed]
- Moutailler, S.; Roche, B.; Thiberge, J.-M.; Caro, V.; Rougeon, F.; Failloux, A.-B. Host alternation is necessary to maintain the genome stability of rift valley fever virus. PLoS Negl. Trop. Dis. 2011, 5, e1156. [Google Scholar] [CrossRef] [PubMed]
- Novella, I.S.; Clarke, D.K.; Quer, J.; Duarte, E.A.; Lee, C.H.; Weaver, S.C.; Elena, S.F.; Moya, A.; Domingo, E.; Holland, J.J. Extreme fitness differences in mammalian and insect hosts after continuous replication of vesicular stomatitis virus in sandfly cells. J. Virol. 1995, 69, 6805–6809. [Google Scholar] [PubMed]
- Novella, I.S.; Hershey, C.L.; Escarmis, C.; Domingo, E.; Holland, J.J. Lack of evolutionary stasis during alternating replication of an arbovirus in insect and mammalian cells. J. Mol. Biol. 1999, 287, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Brault, A.C.; Kang, W.; Holland, J.J. Genetic and fitness changes accompanying adaptation of an arbovirus to vertebrate and invertebrate cells. J. Virol. 1999, 73, 4316–4326. [Google Scholar] [PubMed]
- Ciota, A.T.; Kramer, L.D. Insights into arbovirus evolution and adaptation from experimental studies. Viruses 2010, 2, 2594–2617. [Google Scholar] [CrossRef]
- Novella, I.S.; Presloid, J.B.; Smith, S.D.; Wilke, C.O. Specific and Nonspecific Host Adaptation during Arboviral Experimental Evolution. J. Mol. Microbiol. Biotechnol. 2011, 21, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, G.N.; Clark, P.L. Quality over quantity: Optimizing co-translational protein folding with non-“optimal” synonymous codons. Curr. Opin. Struct. Biol. 2016, 38, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Křížek, M.; Křížek, P. Why has nature invented three stop codons of DNA and only one start codon? J. Theor. Biol. 2012, 304, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Burow, D.A.; Martin, S.; Quail, J.F.; Alhusaini, N.; Coller, J.; Cleary, M.D. Attenuated Codon Optimality Contributes to Neural-Specific mRNA Decay in Drosophila. Cell Rep. 2018, 24, 1704–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldsmith, M.; i5K Consortium. i5K Consortium The i5K Initiative: Advancing arthropod genomics for knowledge, human health, agriculture, and the environment. J. Hered. 2013, 104, 595–600. [Google Scholar]
- Rodriguez, O.; Singh, B.K.; Severson, D.W.; Behura, S.K. Translational selection of genes coding for perfectly conserved proteins among three mosquito vectors. Infect. Genet. Evol. 2012, 12, 1535–1542. [Google Scholar] [CrossRef] [Green Version]
- Whittle, C.A.; Extavour, C.G. Codon and Amino Acid Usage Are Shaped by Selection Across Divergent Model Organisms of the Pancrustacea. Genes Genomes Genet. 2015, 5, 2307–2321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abascal, F.; Posada, D.; Knight, R.D.; Zardoya, R. Parallel Evolution of the Genetic Code in Arthropod Mitochondrial Genomes. PLoS Biol. 2006, 4, e127. [Google Scholar] [CrossRef]
- Novoa, E.M.; Pavon-Eternod, M.; Pan, T.; Ribas de Pouplana, L. A role for tRNA modifications in genome structure and codon usage. Cell 2012, 149, 202–213. [Google Scholar] [CrossRef]
- Tuorto, F.; Lyko, F. Genome recoding by tRNA modifications. Open Biol. 2016, 6, 160287. [Google Scholar] [CrossRef]
- Behura, S.K.; Severson, D.W. Coadaptation of isoacceptor tRNA genes and codon usage bias for translation efficiency in Aedes aegypti and Anopheles gambiae. Insect Mol. Biol. 2010, 20, 177–187. [Google Scholar] [CrossRef] [Green Version]
- Parisien, M.; Wang, X.; Pan, T. Diversity of human tRNA genes from the 1000-genomes project. RNA Biol. 2013, 10, 1853–1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giallonardo, F.; Schlub, T.E.; Shi, M.; Holmes, E.C. Dinucleotide Composition in Animal RNA Viruses Is Shaped More by Virus Family than by Host Species. J. Virol. 2017, 91, e02381-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behura, S.K.; Severson, D.W. Codon usage bias: Causative factors, quantification methods and genome-wide patterns: With emphasis on insect genomes. Biol. Rev. Camb. Philos. Soc. 2013, 88, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Tuohy, T.M.; Mosurski, K.R. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986, 14, 5125–5143. [Google Scholar] [CrossRef] [PubMed]
- Peden, J.F. Analysis of Codon Usage. Ph.D. Thesis, University of Nottingham, Nottingham, UK, 1999; pp. 1–226. [Google Scholar]
- Pinheiro, M.; Afreixo, V.; Moura, G.; Freitas, A.; Santos, M.A.S.; Oliveira, J.L. Statistical, computational and visualization methodologies to unveil gene primary structure features. Methods Inf. Med. 2006, 45, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Wright, F. The “effective number of codons” used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Zaborske, J.M.; Bauer DuMont, V.L.; Wallace, E.W.J.; Pan, T.; Aquadro, C.F.; Drummond, D.A. A Nutrient-Driven tRNA Modification Alters Translational Fidelity and Genome-wide Protein Coding across an Animal Genus. PLoS Biol. 2014, 12, e1002015-13. [Google Scholar] [CrossRef]
- Schimmel, P. The emerging complexity of the tRNA world: Mammalian tRNAs beyond protein synthesis. Nat. Rev. Mol. Cell Biol. 2018, 19, 45. [Google Scholar] [CrossRef]
- Ran, W.; Higgs, P.G. The Influence of Anticodon–Codon Interactions and Modified Bases on Codon Usage Bias in Bacteria. Mol. Biol. Evol. 2010, 27, 2129–2140. [Google Scholar] [CrossRef]
- Smith, B.L.; Chen, G.; Wilke, C.O.; Krug, R.M. Avian Influenza Virus PB1 Gene in H3N2 Viruses Evolved in Humans To Reduce Interferon Inhibition by Skewing Codon Usage toward Interferon-Altered tRNA Pools. MBio 2018, 9, e01222-18. [Google Scholar] [CrossRef] [Green Version]
- Drolet, B.S.; van Rijn, P.; Howerth, E.W.; Beer, M.; Mertens, P.P. A Review of Knowledge Gaps and Tools for Orbivirus Research. Vector Borne Zoonotic Dis. 2015, 15, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Simón, D.; Fajardo, A.; Sóñora, M.; Delfraro, A.; Musto, H. Host influence in the genomic composition of flaviviruses: A multivariate approach. Biochem. Biophys. Res. Commun. 2017, 492, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Blitvich, B.J.; Firth, A.E. A Review of Flaviviruses that Have No Known Arthropod Vector. Viruses 2017, 9, 154. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, G.M.; Pagel, M.; Gould, E.A.; Paolo MD, A.; Holmes, E.C. Evolution of Base Composition and Codon Usage Bias in the Genus Flavivirus. J. Mol. Evol. 2001, 52, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.K.; Tyagi, A. A detailed analysis of codon usage patterns and influencing factors in Zika virus. Arch. Virol. 2017, 162, 1963–1973. [Google Scholar] [CrossRef] [PubMed]
- Moratorio, G.; Iriarte, A.; Moreno, P.; Musto, H.; Cristina, J. A detailed comparative analysis on the overall codon usage patterns in West Nile virus. Infect. Genet. Evol. 2013, 14, 396–400. [Google Scholar] [CrossRef]
- Stephanie Chan; Jing-hsiung Ou Hepatitis C Virus-Induced Autophagy and Host Innate Immune Response. Viruses 2017, 9, 224. [CrossRef]
- Sharma, G.; Raheja, H.; Das, S. Hepatitis C virus: Enslavement of host factors. IUBMB Life 2018, 70, 41–49. [Google Scholar] [CrossRef]
- Chiari, Y.; Dion, K.; Colborn, J.; Parmakelis, A.; Powell, J.R. On the Possible Role of tRNA Base Modifications in the Evolution of Codon Usage: Queuosine and Drosophila. J. Mol. Evol. 2010, 70, 339–345. [Google Scholar] [CrossRef]
- Butt, A.M.; Nasrullah, I.; Qamar, R.; Tong, Y. Evolution of codon usage in Zika virus genomes is host and vector specific. Emerg. Microbes Infect. 2016, 5, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Liu, Q.; Xiong, Y.; Ye, C.; Jin, D.; Xu, J. Genome-wide analysis of synonymous codon usage in Huaiyangshan virus and other bunyaviruses. J. Basic Microbiol. 2015, 55, 1374–1383. [Google Scholar] [CrossRef] [PubMed]
- Gumpper, R.H.; Li, W.; Luo, M. Constraints of viral RNA synthesis on codon usage of negative strand RNA virus. J. Virol. 2018, 93, e01775-18. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, G.M.; Holmes, E.C. The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res. 2003, 92, 1–7. [Google Scholar] [CrossRef]
- Xu, X.; Fei, D.; Han, H.; Liu, H.; Zhang, J.; Zhou, Y.; Xu, C.; Wang, H.; Cao, H.; Zhang, H. Comparative characterization analysis of synonymous codon usage bias in classical swine fever virus. Microb. Pathog. 2017, 107, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, J.; Zhou, J.-H.; Chen, H.-T.; Ma, L.-N.; Ding, Y.-Z.; Liu, W.-Q.; Liu, Y.-S. Analysis of codon usage in bovine viral diarrhea virus. Arch. Virol. 2011, 156, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, M.; Peterhans, E. Pestiviruses. Annu. Rev. Anim. Biosci. 2014, 2, 141–163. [Google Scholar] [CrossRef] [PubMed]
- Keller, B.C.; Johnson, C.L.; Erickson, A.K.; Gale, M. Innate immune evasion by hepatitis C virus and West Nile virus. Cytokine Growth Factor Rev. 2007, 18, 535–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.L.; Lee, W.; Hottes, A.K.; Shapiro, L.; McAdams, H.H. Codon usage between genomes is constrained by genome-wide mutational processes. Proc. Natl. Acad. Sci. USA 2004, 101, 3480–3485. [Google Scholar] [CrossRef] [Green Version]
- Knight, R.D.; Freeland, S.J.; Landweber, L.F. A simple model based on mutation and selection explains trends in codon and amino-acid usage and GC composition within and across genomes. Genome Biol. 2001, 2, RESEARCH0010. [Google Scholar]
- Belalov, I.S.; Lukashev, A.N. Causes and Implications of Codon Usage Bias in RNA Viruses. PLoS ONE 2013, 8, e56642-9. [Google Scholar] [CrossRef]
- Burns, C.C.; Shaw, J.; Campagnoli, R.; Jorba, J.; Vincent, A.; Quay, J.; Kew, O. Modulation of poliovirus replicative fitness in HeLa cells by deoptimization of synonymous codon usage in the capsid region. J. Virol. 2006, 80, 3259–3272. [Google Scholar] [CrossRef] [PubMed]
- Burns, C.C.; Campagnoli, R.; Shaw, J.; Vincent, A.; Jorba, J.; Kew, O. Genetic inactivation of poliovirus infectivity by increasing the frequencies of CpG and UpA dinucleotides within and across synonymous capsid region codons. J. Virol. 2009, 83, 9957–9969. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Gorbatsevych, O.; Liu, Y.; Mugavero, J.; Shen, S.H.; Ward, C.B.; Asare, E.; Jiang, P.; Paul, A.V.; Mueller, S.; et al. Limits of variation, specific infectivity, and genome packaging of massively recoded poliovirus genomes. Proc. Natl. Acad. Sci. USA 2017, 114, E8731–E8740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velazquez-Salinas, L.; Risatti, G.R.; Holinka, L.G.; O’Donnell, V.; Carlson, J.; Alfano, M.; Rodriguez, L.L.; Carrillo, C.; Gladue, D.P.; Borca, M.V. Recoding structural glycoprotein E2 in classical swine fever virus (CSFV) produces complete virus attenuation in swine and protects infected animals against disease. Virology 2016, 494, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Takata, M.A.; Soll, S.J.; Emery, A.; Blanco-Melo, D.; Swanstrom, R.; Bieniasz, P.D. Global synonymous mutagenesis identifies cis-acting RNA elements that regulate HIV-1 splicing and replication. PLoS Pathog. 2018, 14, e1006824-26. [Google Scholar] [CrossRef] [PubMed]
- Martínez, M.A.; Jordan-Paiz, A.; Franco, S.; Nevot, M. Synonymous Virus Genome Recoding as a Tool to Impact Viral Fitness. Trends Microbiol. 2016, 24, 134–147. [Google Scholar]
- Stauft, C.B.; Shen, S.H.; Song, Y.; Gorbatsevych, O.; Asare, E.; Futcher, B.; Mueller, S.; Payne, A.; Brecher, M.; Kramer, L.; et al. Extensive recoding of dengue virus type 2 specifically reduces replication in primate cells without gain-of-function in Aedes aegypti mosquitoes. PLoS ONE 2018, 13, e0198303. [Google Scholar] [CrossRef]
- Li, P.; Ke, X.; Wang, T.; Tan, Z.; Luo, D.; Miao, Y.; Sun, J.; Zhang, Y.; Liu, Y.; Hu, Q.; et al. Zika Virus Attenuation by Codon Pair Deoptimization Induces Sterilizing Immunity in Mouse Models. J. Virol. 2018, 92, e00701-18. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yang, C.; Tekes, G.; Mueller, S.; Paul, A.; Whelan, S.P.J.; Wimmer, E. Recoding of the vesicular stomatitis virus L gene by computer-aided design provides a live, attenuated vaccine candidate. MBio 2015, 6, e00237-15. [Google Scholar] [CrossRef]
- Nougairede, A.; De Fabritus, L.; Aubry, F.; Gould, E.A.; Holmes, E.C.; de Lamballerie, X. Random Codon Re-encoding Induces Stable Reduction of Replicative Fitness of Chikungunya Virus in Primate and Mosquito Cells. PLoS Pathog. 2013, 9, e1003172-18. [Google Scholar] [CrossRef]
- Kunec, D.; Osterrieder, N. Codon Pair Bias Is a Direct Consequence of Dinucleotide Bias. Cell Rep. 2016, 14, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Tulloch, F.; Evans, D.J.; Ryan, M.D. Attenuation of dengue (and other RNA viruses) with codon pair recoding can be explained by increased CpG/UpA dinucleotide frequencies. Proc. Natl. Acad. Sci. USA 2015, 112, E3633–E3634. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, N.J.; Witteveldt, J.; Evans, D.J.; Simmonds, P. The influence of CpG and UpA dinucleotide frequencies on RNA virus replication and characterization of the innate cellular pathways underlying virus attenuation and enhanced replication. Nucleic Acids Res. 2014, 42, 4527–4545. [Google Scholar] [CrossRef] [PubMed]
- de Visser, J.A.G.M.; Hermisson, J.; Wagner, G.P.; Meyers, L.A.; Chaichian, H.B.; Blanchard, J.L.; Chao, L.; Cheverud, J.M.; Elena, S.F.; Fontana, W.; et al. Perspective: Evolution and Detection of Genetic Robustness. Evolution 2003, 57, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Fares, M.A. The origins of mutational robustness. Trends Genet. 2015, 31, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Lachowiec, J.; Queitsch, C.; Kliebenstein, D.J. Molecular mechanisms governing differential robustness of development and environmental responses in plants. Ann. Bot. 2016, 117, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R. Mutational fitness effects in RNA and single-stranded DNA viruses: Common patterns revealed by site-directed mutagenesis studies. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 1975–1982. [Google Scholar]
- Belshaw, R.; Gardner, A.; Rambaut, A.; Pybus, O.G. Pacing a small cage: Mutation and RNA viruses. Trends Ecol. Evol. 2008, 23, 188–193. [Google Scholar] [CrossRef]
- Ochsenreiter, R.; Hofacker, I.L.; Wolfinger, M.T. Functional RNA Structures in the 3′UTR of Tick-Borne, Insect-Specific and No-Known-Vector Flaviviruses. Viruses 2019, 11, 298. [Google Scholar] [CrossRef]
- Prostova, M.A.; Gmyl, A.P.; Bakhmutov, D.V.; Shishova, A.A.; Khitrina, E.V.; Kolesnikova, M.S.; Serebryakova, M.V.; Isaeva, O.V.; Agol, V.I. Mutational robustness and resilience of a replicative cis-element of RNA virus: Promiscuity, limitations, relevance. RNA Biol. 2015, 12, 1338–1354. [Google Scholar] [CrossRef]
- Rodrigo, G.; Daròs, J.-A.; Elena, S.F. Virus-host interactome: Putting the accent on how it changes. J. Proteomics 2017, 156, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S.; Acevedo, A.; Cooper, S.B.; Andino, R. Codon usage determines the mutational robustness, evolutionary capacity, and virulence of an RNA virus. Cell Host Microbe 2012, 12, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Burch, C.L.; Chao, L. Evolvability of an RNA virus is determined by its mutational neighbourhood. Nature 2000, 406, 625. [Google Scholar] [CrossRef] [PubMed]
- Dolan, P.T.; Whitfield, Z.J.; Andino, R. Mapping the Evolutionary Potential of RNA Viruses. Cell Host Microbe 2018, 23, 435–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, T.; Bordería, A.V.; Barbezange, C.; Vignuzzi, M.; Louzoun, Y. Long term context dependent genetic adaptation of the viral genetic cloud. Bioinformatics 2018, 35, 1907–1915. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S.; Frydman, J.; Andino, R. The role of mutational robustness in RNA virus evolution. Nat. Rev. Microbiol. 2013, 11, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elena, S.F. RNA virus genetic robustness: Possible causes and some consequences. Curr. Opin. Virol. 2012, 2, 525–530. [Google Scholar] [CrossRef]
- Wilke, C.O.; Wang, J.L.; Ofria, C.; Lenski, R.E.; Adami, C. Evolution of digital organisms at high mutation rates leads to survival of the flattest. Nature 2001, 412, 331–333. [Google Scholar] [CrossRef] [Green Version]
- Rihn, S.J.; Hughes, J.; Wilson, S.J.; Bieniasz, P.D. Uneven genetic robustness of HIV-1 integrase. J. Virol. 2015, 89, 552–567. [Google Scholar] [CrossRef]
- Visher, E.; Whitefield, S.E.; McCrone, J.T.; Fitzsimmons, W.; Lauring, A.S. The Mutational Robustness of Influenza A Virus. PLoS Pathog. 2016, 12, e1005856-25. [Google Scholar] [CrossRef]
- Stern, A.; Bianco, S.; Te Yeh, M.; Wright, C.; Butcher, K.; Tang, C.; Nielsen, R.; Andino, R. Costs and Benefits of Mutational Robustness in RNA Viruses. Cell Rep. 2014, 8, 1026–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuevas, J.M.; Moya, A.; Sanjuan, R. A genetic background with low mutational robustness is associated with increased adaptability to a novel host in an RNA virus. J. Evol. Biol. 2009, 22, 2041–2048. [Google Scholar] [CrossRef] [PubMed]
- Warmbrod, K.L.; Patterson, E.I.; Kautz, T.F.; Stanton, A.; Rockx-Brouwer, D.; Kalveram, B.K.; Khanipov, K.; Thangamani, S.; Fofanov, Y.; Forrester, N.L. Viral RNA-dependent RNA polymerase mutants display an altered mutation spectrum resulting in attenuation in both mosquito and vertebrate hosts. PLoS Pathog. 2019, 15, e1007610. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Virus | Family | Codon Altered Proteins | CpGs Controlled? | Replication Kinetics | Competitive Fitness | Attenuation | Refs | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Invert | Vert | Invert | Vert | Invert | Vert | |||||
| DENV | Flavivirus | Env, Helicase (NS3), RdRP/Mtase (NS5) | No | NC | DECR | - | - | No | Yes | [19,98] |
| ZIKV | Flavivirus | Env, NS1 | No | DECR | DECR | - | - | - | Yes | [99] |
| VSV | Rhabdovirus | RdRP (L gene) | Yes | - | NC | - | - | - | Yes | [100] |
| CHIKV | Togavirus | Env, RdRP (nsp1+4) | Yes | DECR | DECR | DECR | DECR | - | - | [101] |
| TBEV | Flavivirus | RdRP/Mtase (NS5) | - | - | NC | - | DECR | - | Yes | [20] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sexton, N.R.; Ebel, G.D. Effects of Arbovirus Multi-Host Life Cycles on Dinucleotide and Codon Usage Patterns. Viruses 2019, 11, 643. https://doi.org/10.3390/v11070643
Sexton NR, Ebel GD. Effects of Arbovirus Multi-Host Life Cycles on Dinucleotide and Codon Usage Patterns. Viruses. 2019; 11(7):643. https://doi.org/10.3390/v11070643
Chicago/Turabian StyleSexton, Nicole R., and Gregory D. Ebel. 2019. "Effects of Arbovirus Multi-Host Life Cycles on Dinucleotide and Codon Usage Patterns" Viruses 11, no. 7: 643. https://doi.org/10.3390/v11070643
APA StyleSexton, N. R., & Ebel, G. D. (2019). Effects of Arbovirus Multi-Host Life Cycles on Dinucleotide and Codon Usage Patterns. Viruses, 11(7), 643. https://doi.org/10.3390/v11070643