A Lineage of Begomoviruses Encode Rep and AC4 Proteins of Enigmatic Ancestry: Hints on the Evolution of Geminiviruses in the New World

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Begomovirus Detection
2.2. Cloning of Full-Length Viral Genomes
2.3. Plant Infection Assays
2.4. Phylogenetic Analyses
3. Results
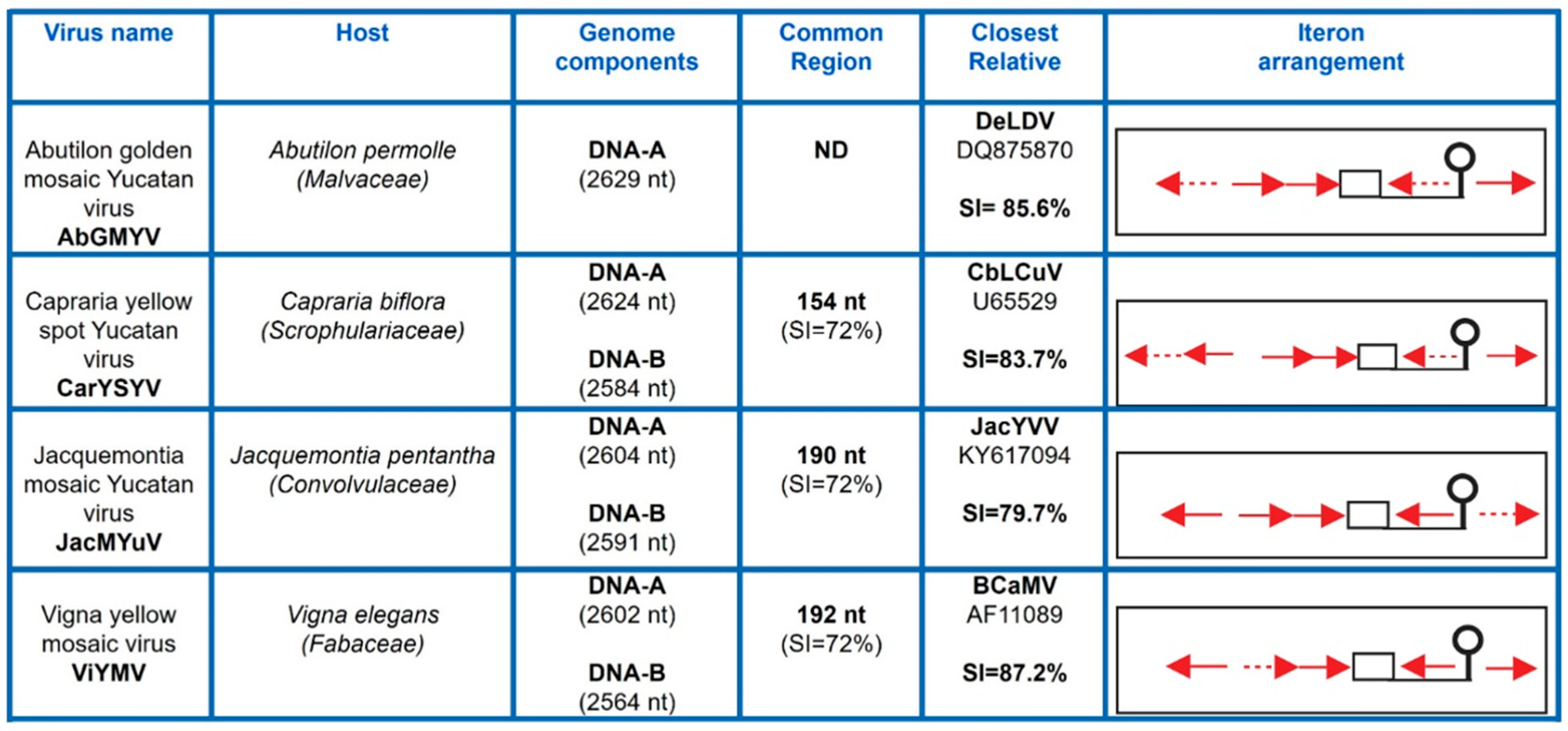
3.1. Isolation and Characterization of Novel SLCV-Lineage (S-Lin) Begomoviruses
3.2. Recombination Analysis
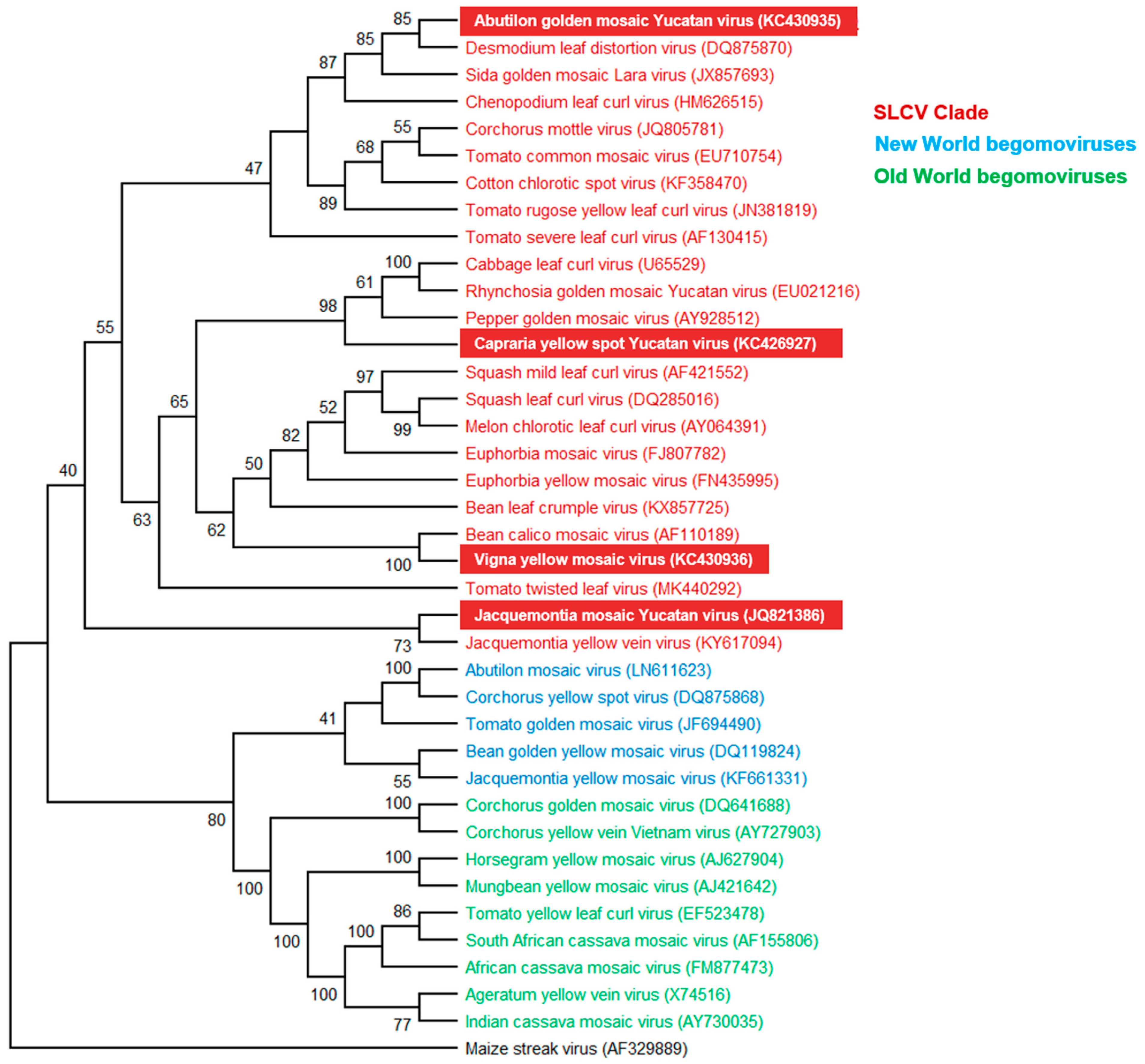
3.3. Phylogenetic Relationships of the New Begomoviruses
3.4. Infectivity of Cloned DNA-A and DNA-B Components
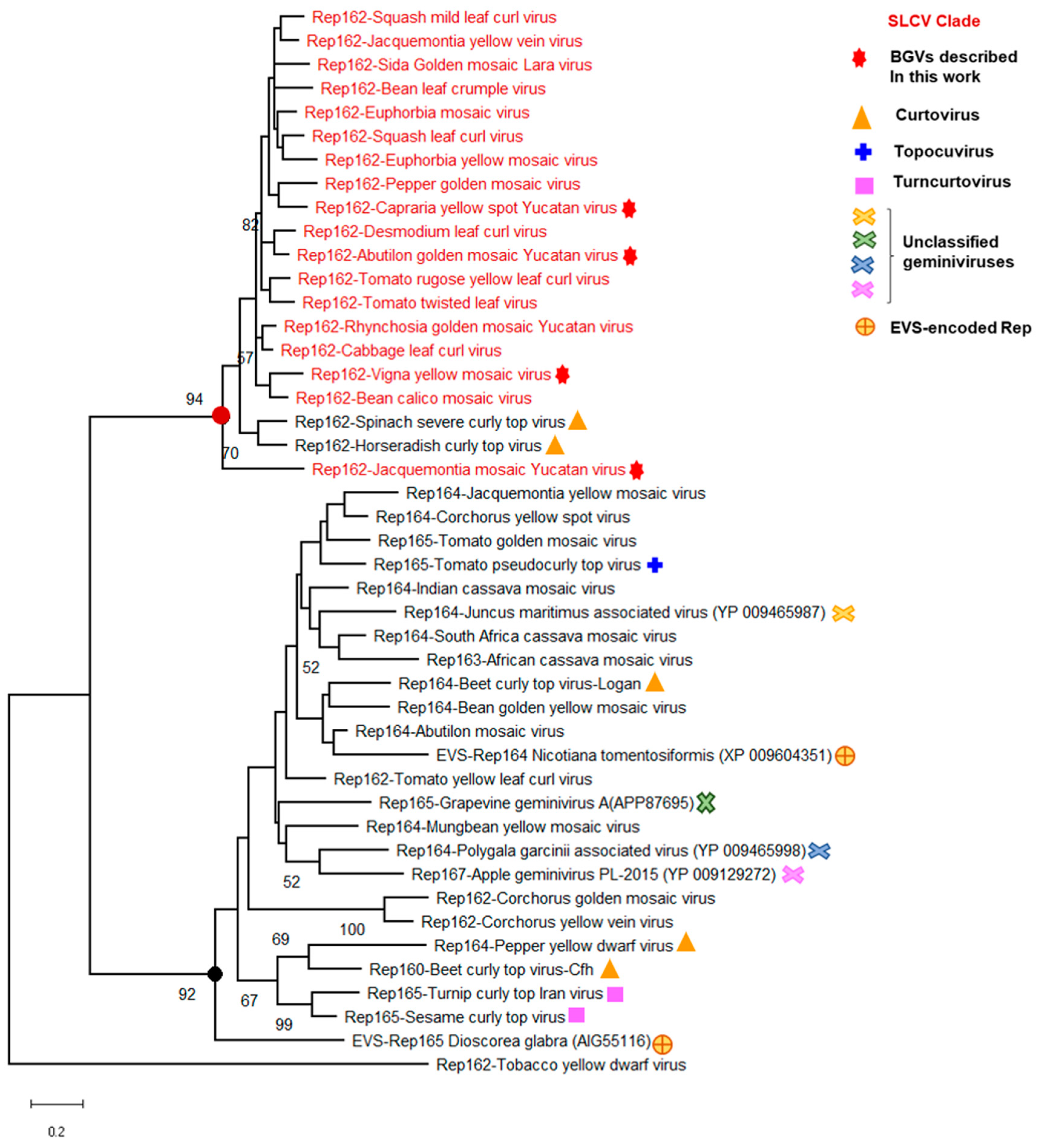
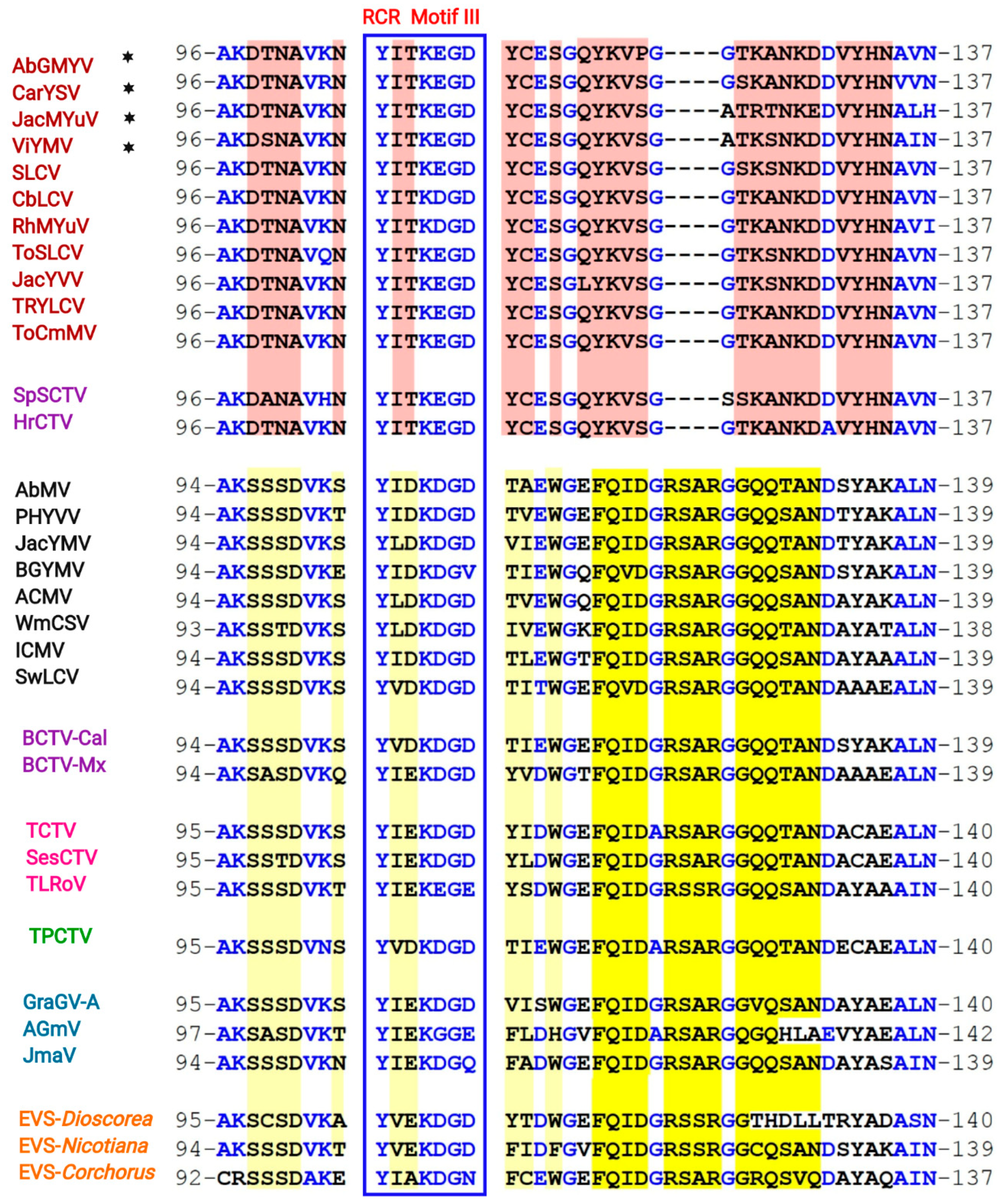
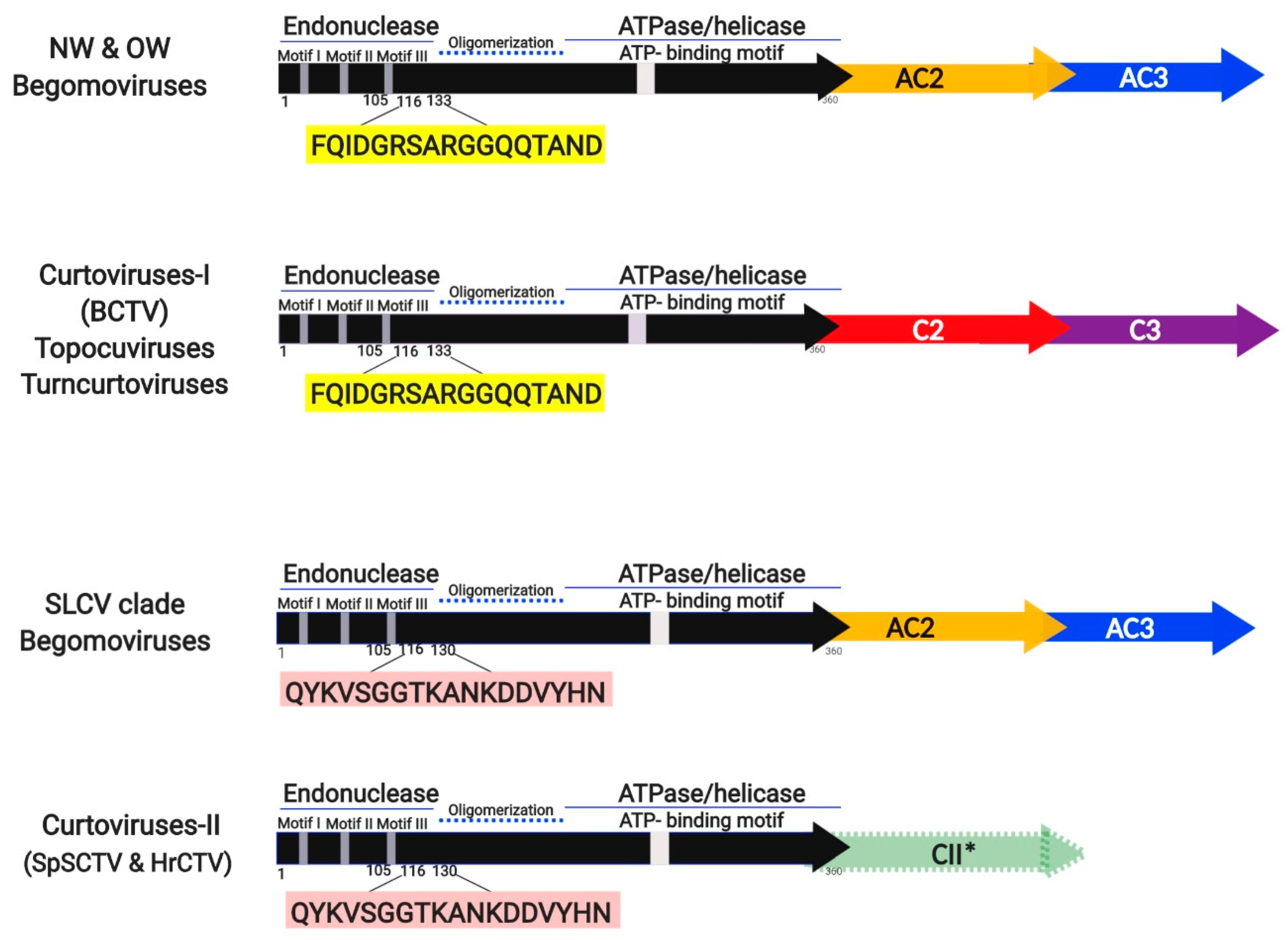
3.5. Identification of a Distinctive Rep Domain Encoded by SLCV-Lin BGVs
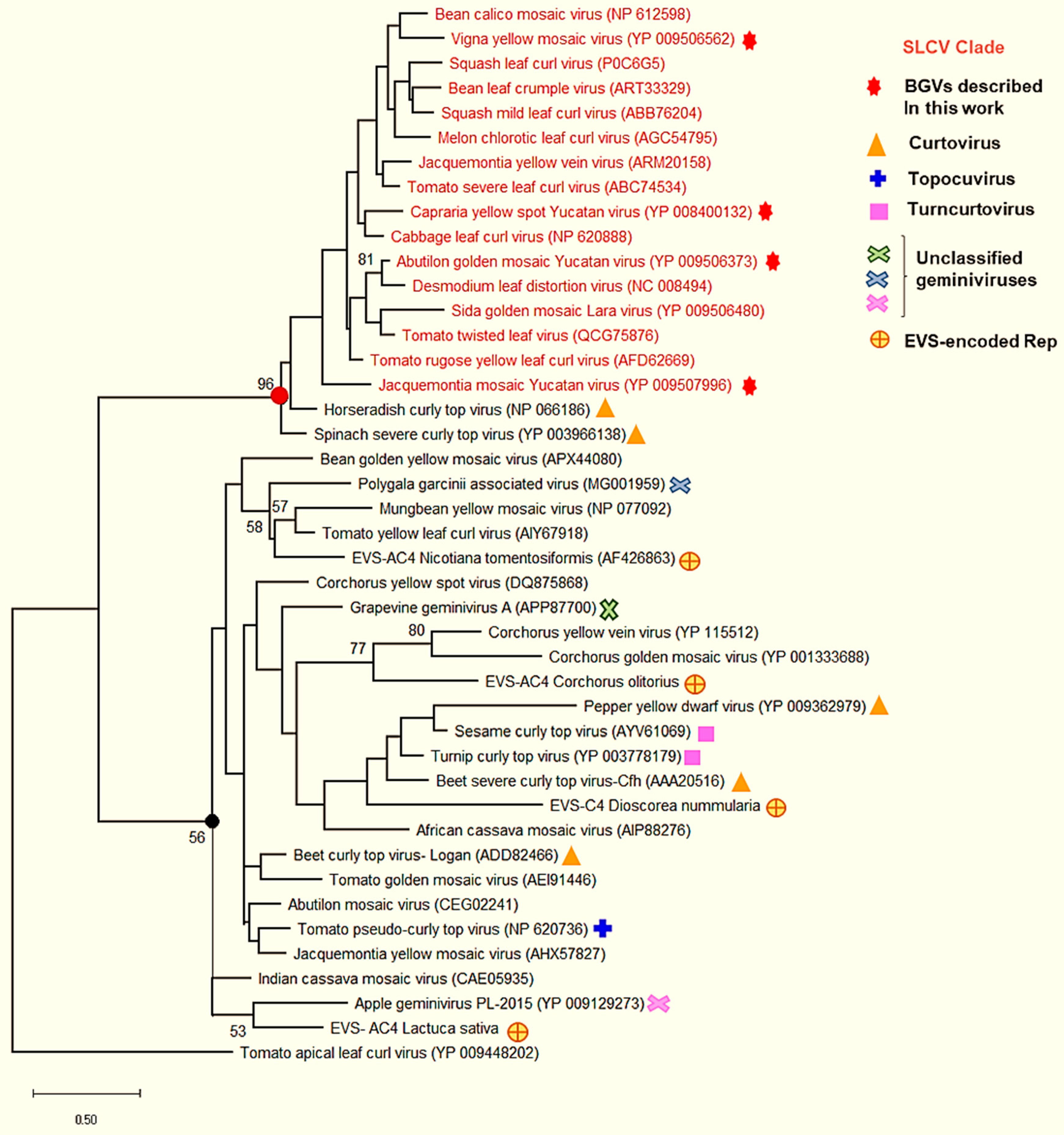
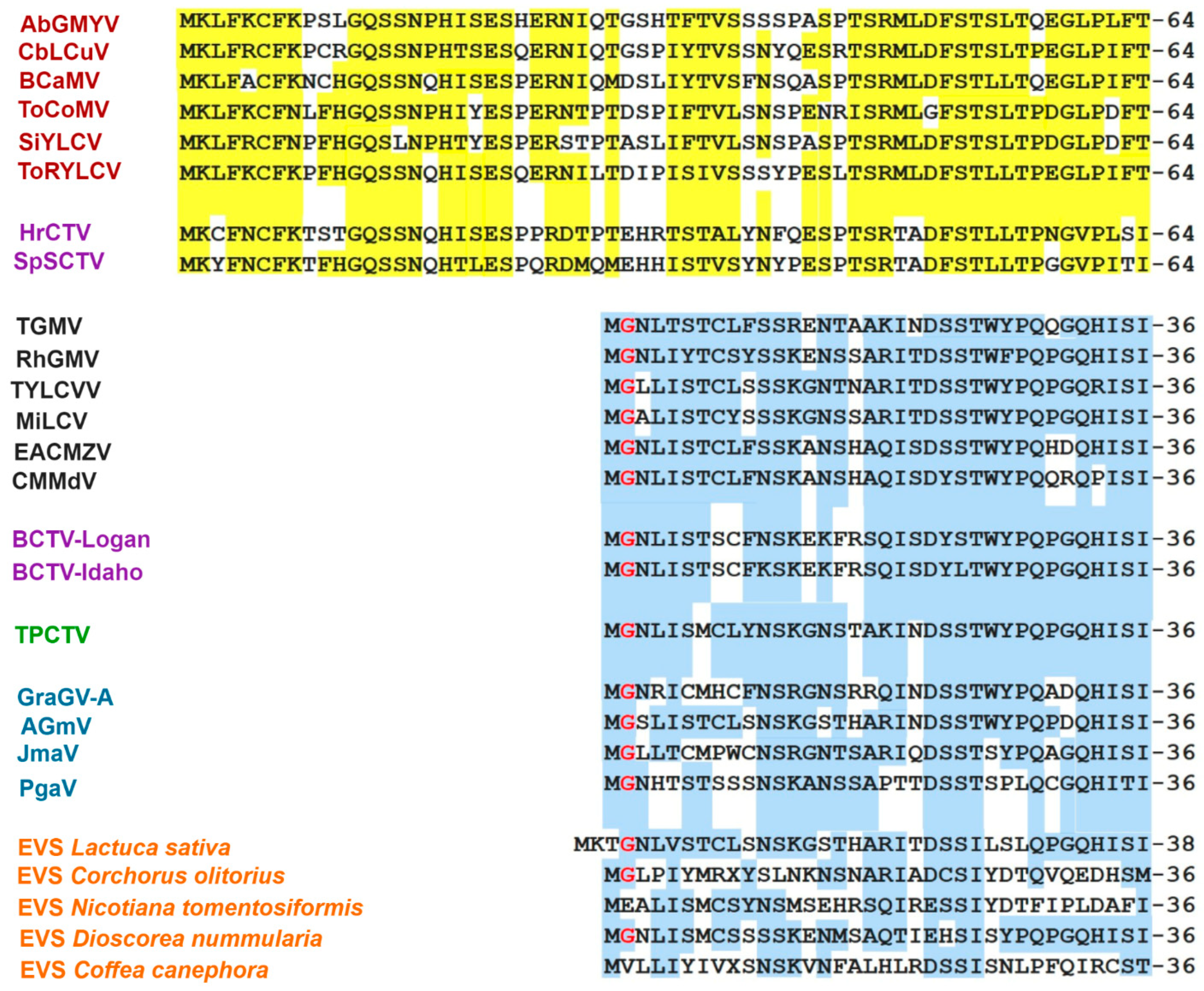
3.6. Comparative Analysis of the AC4 Protein of SLCV Lineage Viruses
3.7. Comparative Analysis of Curtovirus and S-Lin BGV Genomes
3.8. Searching for the Common Ancestor of the SLCV Clade Members
4. Discussion
Evolutionary Relationships of Curtoviruses with S-Lin Begomoviruses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanley-Bowdoin, L.; Bejarano, E.R.; Robertson, D.; Mansoor, S. Geminiviruses: Masters at redirecting and reprogramming plant processes. Nat. Rev. Microbiol. 2013, 11, 777. [Google Scholar] [CrossRef] [PubMed]
- Hesketh, E.L.; Saunders, K.; Fisher, C.; Potze, J.; Stanley, J.; Lomonossoff, G.P.; Ranson, N.A. The 3.3 A structure of a plant geminivirus using cryo -EM. Nat. Commun. 2018, 9, 2369. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, Q.; Hong, J.; Li, Z.; Zhang, X.; Zhou, X. Cryo-EM Structure of a Begomovirus Geminate Particle. Int. J. Mol. Sci. 2019, 20, 1738. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.R.; Macedo, M.A.; Maliano, M.R.; Soto-Aguilar, M.; Souza, J.O.; Briddon, R.W.; Kenyon, L.; Rivera-Bustamante, R.F.; Zerbini, F.M.; Adkins, S.; et al. World Management of Geminiviruses. Annu. Rev. Phytopathol. 2018, 56, 637–677. [Google Scholar] [CrossRef] [PubMed]
- Zerbini, F.M.; Briddon, R.W.; Idris, A.; Martin, D.P.; Moriones, E.; Navas-Castillo, J.; Rivera-Bustamante, R.; Rourmagnac, P.; Varsani, A. ICTV virus taxonomy profile: Geminiviridae. J. Gen. Virol. 2017, 98, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Varma, A.; Malathi, V.G. Emerging geminivirus problems: A serious threat to crop production. Ann. Appl. Biol. 2003, 142, 145–164. [Google Scholar] [CrossRef]
- Varsani, A.; Navas-Castillo, J.; Moriones, E.; Hernández-Zepeda, C.; Idris, A.; Brown, J.K.; Murilo Zerbini, F.; Martin, D.P. Establishment of three new genera in the family Geminiviridae: Becurtovirus, Eragrovirus and Turncurtovirus. Arch. Virol. 2014, 159, 2193–2203. [Google Scholar] [CrossRef]
- Varsani, A.; Roumagnac, P.; Fuchs, M.; Navas-Castillo, J.; Moriones, E.; Idris, A.; Briddon, R.W.; Rivera-Bustamante, R.; Murilo Zerbini, F.; Martin, D.P. Capulavirus and Grablovirus: Two new genera in the family Geminiviridae. Arch. Virol. 2017, 162, 1819–1831. [Google Scholar] [CrossRef]
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org/taxonomy/ (accessed on 8 July 2019).
- De Barro, P.J.; Liu, S.S.; Boykin, L.M.; Dinsdale, A.B. Bemisia tabaci: A statement of species status. Ann. Rev. Entomol. 2011, 56, 1–19. [Google Scholar] [CrossRef]
- Waqar, I.; Akutse, K.S.; Qasim, M.; Khan, K.A.; Ghramh, H.A.; Idrees, A.; Latif, S. Bemisia tabaci-mediated facilitation in diversity of begomoviruses: Evidence from recent molecular studies. Microb. Pathog. 2018, 123, 162–168. [Google Scholar] [CrossRef]
- Briddon, R.W.; Patil, B.L.; Bagewadi, B.; Nawaz-ul-Rehman, M.S.; Fauquet, C.M. Distinct evolutionary histories of the DNA-A and DNA-B components of bipartite begomoviruses. BMC Evol. Biol. 2010, 10, 97. [Google Scholar] [CrossRef] [PubMed]
- Rybicki, E.P. A phylogenetic and evolutionary justification for three genera of Geminiviridae. Arch. Virol. 1994, 139, 49–77. [Google Scholar] [CrossRef] [PubMed]
- Macedo, M.A.; Albuquerque, L.C.; Maliano, M.R.; Souza, J.O.; Rojas, M.R.; Inoue-Nagata, A.K.; Gilbertson, R.L. Characterization of tomato leaf curl purple vein virus, a new monopartite New World begomovirus infecting tomato in Northeast Brazil. Arch. Virol. 2018, 163, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Melgarejo, T.A.; Kon, T.; Rojas, M.R.; Paz-Carrasco, L.; Zerbini, F.M.; Gilbertson, R.L. Characterization of a new world monopartite begomovirus causing leaf curl disease of tomato in Ecuador and Peru reveals a new direction in geminivirus evolution. J. Virol. 2013, 87, 5397–5413. [Google Scholar] [CrossRef] [PubMed]
- Romay, G.; Geraud-Pouey, F.; Chirinos, D.T.; Mahillon, M.; Gillis, A.; Mahillon, J.; Bragard, C. Tomato Twisted Leaf Virus: A Novel Indigenous New World Monopartite Begomovirus Infecting Tomato in Venezuela. Viruses 2019, 11, 327. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.K.; Zerbini, F.M.; Navas-Castillo, J.; Moriones, E.; Ramos-Sobrinho, R.; Silva, J.C.; Fiallo-Olivé, E.; Briddon, R.W.; Hernández-Zepeda, C.; Idris, A.; et al. Revision of Begomovirus taxonomy based on pairwise sequence comparisons. Arch. Virol. 2015, 160, 1593–1619. [Google Scholar] [CrossRef]
- Fondong, V.N. Geminivirus protein structure and function. Mol. Plant Patho. 2013, 14, 635–649. [Google Scholar] [CrossRef]
- Rojas, M.R.; Hagen, C.; Lucas, W.J.; Gilbertson, R.L. Exploiting chinks in the plant’s armor: Evolution and emergence of geminiviruses. Annu. Rev. Phytopathol. 2005, 43, 361–394. [Google Scholar] [CrossRef]
- Lazarowitz, S.G.; Beachy, R.N. Viral movement proteins asprobes for intracellular and intercellular trafficking in plants. Plant Cell 1999, 11, 535–548. [Google Scholar] [CrossRef]
- Laufs, J.; Traut, W.; Heyraud, F.; Matzeit, V.; Rogers, S.G.; Schell, J.; Gronenborn, B. In vitro cleavage and joining at the viral origin of replication by the replication initiator protein of tomato yellow leaf curl virus. Proc. Natl. Acad. Sci. USA 1995, 92, 3879–3883. [Google Scholar] [CrossRef]
- Ha, C.; Coombs, S.; Revill, P.; Harding, R.; Vu, M.; Dale, J. Corchorus yellow vein virus, a New World geminivirus from the Old World. J. Gen. Virol. 2006, 87, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Argüello-Astorga, G.R.; Guevara-Gonzalez, R.G.; Herrera-Estrella, L.R.; Rivera-Bustamante, R.F. Geminivirus replication origins have a group-specific organization of iterative elements: A model for replication. Virology 1994, 203, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Argüello-Astorga, G.R.; Ruiz-Medrano, R. An iteron-related domain is associated to Motif 1 in the replication proteins of geminiviruses: Identification of potential interacting amino acid–base pairs by a comparative approach. Arch. Virol. 2001, 146, 465–485. [Google Scholar] [CrossRef]
- Ha, C.; Coombs, S.; Revill, P.; Harding, R.; Vu, M.; Dale, J. Molecular characterization of begomoviruses and DNA satellites from Vietnam: Additional evidence that the New World geminiviruses were present in the Old World prior to continental separation. J. Gen. Virol. 2008, 89, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Mauricio-Castillo, J.A.; Torres-Herrera, S.I.; Cardenas-Conejo, Y.; Pastor-Palacios, G.; Mendez-Lozano, J.; Arguello-Astorga, G.R. A novel begomovirus isolated from sida contains putative cisand trans-acting replication specificity determinants that have evolved independently in several geographical lineages. Arch. Virol. 2014, 159, 2283–2294. [Google Scholar] [CrossRef]
- Nawaz-ul-Rehman, M.S.; Fauquet, C.M. Evolution of geminiviruses and their satellites. FEBS Lett. 2009, 583, 1825–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefeuvre, P.; Harkins, G.W.; Lett, J.M.; Briddon, R.W.; Chase, M.W.; Moury, B.; Martin, D.P. Evolutionary timescale of the begomoviruses: Evidence from integrated sequences in the Nicotiana genome. PLoS ONE 2011, 6, e19193. [Google Scholar] [CrossRef]
- Brown, J.K.; Idris, A.M.; Alteri, C.; Stenger, D.C. Emergence of a New Cucurbit-Infecting Begomovirus Species Capable of Forming Viable Reassortants with Related Viruses in the Squash leaf curl virus Cluster. Phytopathology 2002, 92, 734–742. [Google Scholar] [CrossRef]
- Gregorio-Jorge, J.; Bernal-Alcocer, A.; Bañuelos-Hernández, B.; Alpuche-Solís, A.G.; Hernández-Zepeda, C.; Moreno-Valenzuela, O.; Frías-Treviño, G.; Argüello-Astorga, G.R. Analysis of a new strain of Euphorbia mosaic virus with distinct replication specificity unveils a lineage of begomoviruses with short Rep sequences in the DNA-B intergenic region. Virol. J. 2010, 7, 275. [Google Scholar] [CrossRef]
- Márquez-Martín, B.; Maeso, D.; Martínez-Ayala, A.; Bernal, R.; Teresa-Federici, M.; Vincelli, P.; Navas-Castillo, J.; Moriones, E. Diverse population of a new bipartite begomovirus infecting tomato crops in Uruguay. Arch. Virol. 2012, 157, 1137–1142. [Google Scholar] [CrossRef]
- Arguello-Astorga, G.; Herrera-Estrella, L.; Rivera-Bustamante, R. Experimental and theoretical definition of geminivirus origin of replication. Plant Mol. Biol. 1994, 26, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Dellaporta, S.L.; Wood, J.; Hicks, J.B. A plant DNA minipreparation: Version II. Plant Mol. Biol. Rep. 1983, 1, 19–21. [Google Scholar] [CrossRef]
- Mauricio-Castillo, J.A.; Argüello-Astorga, G.R.; Ambriz-Granados, S.; Alpuche-Solís, A.G.; Monreal-Vargas, C.T. First Report of Tomato golden mottle virus on Lycopersicon esculentum and Solanum rostratum in Mexico. Plant Dis. 2007, 91, 1513. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evolu. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Lemey, P.; Lott, M.; Moulton, V.; Posada, D.; Lefeuvre, P. RDP3: A flexible and fast computer program for analyzing recombination. Bioinformatics 2010, 26, 2462–2463. [Google Scholar] [CrossRef]
- Hernandez-Zepeda, C.; Idris, A.M.; Carnevali, G.; Brown, J.K.; Moreno-Valenzuela, O.A. Molecular characterization and phylogenetic relationships of two new bipartite begomovirus infecting malvaceous plants in Yucatan, Mexico. Virus Genes 2007, 35, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Zepeda, C.; Argüello-Astorga, G.R.; Idris, A.M.; Carnevali, G.; Brown, J.K.; Moreno-Valenzuela, O.A. Molecular characterization and phylogenetic relationships of Desmodium leaf distortion virus (DeLDV): A new begomovirus infecting Desmodium glabrum in Yucatan, Mexico. Virus Genes 2009, 39, 371–374. [Google Scholar] [CrossRef]
- Arguello-Astorga, G.R.; Department of Molecular Biology, IPICYT, San Luis Potosí, S.L.P., Mexico. Begomovirirus phylogeny based based on the alignment of the full-length DNA-A. Unpublished work. 2019. [Google Scholar]
- Qazi, J.; Ilyas, M.; Mansoor, S.; Briddon, R.W. Legume yellow mosaic viruses: Genetically isolated begomoviruses. Mol. Plant Pathol. 2007, 8, 343–438. [Google Scholar] [CrossRef]
- Klute, K.A.; Nadler, S.A.; Stenger, D.C. Horseradish curly top virus is a distinct subgroup II geminivirus species with rep and C4 genes derived from a subgroup III ancestor. J. Gen. Virol. 1996, 77, 1369–1378. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Zepeda, C.; Brown, J.K. First report of a new curtovirus species, Spinach severe curly top virus, in commercial spinach plants (Spinacia oleracea) from south-central Arizona. Plant Dis. 2010, 94, 917. [Google Scholar] [CrossRef] [PubMed]
- Fondong, V.N.; Reddy, R.C.; Lu, C.; Hankoua, B.; Felton, C.; Czymmek, K.; Achenjang, F. The consensus N-myristoylation motif of a geminivirus AC4 protein is required for membrane binding and pathogenicity. Mol. Plant Microbe Interact. 2007, 20, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Hipp, K.; Rau, P.; Schäfer, B.; Pfannstiel, J.; Jeske, H. Translation, modification and cellular distribution of two AC4 variants of African cassava mosaic virus in yeast and their pathogenic potential in plants. Virology 2016, 498, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zeng, R.; Chen, Z.; Liu, X.; Cao, Z.; Xie, Q.; Yang, C.; Lai, J. S-acylation of a geminivirus C4 protein is essential for regulating the CLAVATA pathway in symptom determination. J. Exp. Bot. 2018, 69, 4459–4468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejarano, E.R.; Khashoggi, A.; Witty, M.; Lichtenstein, C. Integration of multiple repeats of geminiviral DNA into the nuclear genome of tobacco during evolution. Proc. Natl. Acad. Sci. USA 1996, 23, 759–764. [Google Scholar] [CrossRef]
- Murad, L.; Bielawski, J.P.; Matyasek, R.; Kovarıík, A.; Nichols, R.A.; Leitch, A.R.; Lichtenstein, C.P. The origin and evolution of geminivirus-related DNA sequences in Nicotiana. Heredity 2004, 92, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Filloux, D.; Murrell, S.; Koohapitagtam, M.; Golden, M.; Julian, C.; Galzi, S.; Uzest, M.; Rodier-Goud, M.; D’Hont, A.; Vernerey, M.S.; et al. The genomes of many yam species contain transcriptionally active endogenous geminiviral sequences that may be functionally expressed. Virus Evol. 2015, 1, vev002. [Google Scholar] [CrossRef]
- Chandler, M.; de la Cruz, F.; Dyda, F.; Hickman, A.B.; Moncalian, G.; Ton-Hoang, B. Breaking and joining single-stranded DNA: The HUH endonuclease superfamily. Nat. Rev. Microbiol. 2013, 11, 525–538. [Google Scholar] [CrossRef]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M. Origins and evolution of viruses of eukaryotes: The ultimate modularity. Virology 2015, 479–480, 2–25. [Google Scholar] [CrossRef]
- Cardenas-Conejo, Y. Reconstruction of Genome Evolution of Begomoviruses through an Integrated Multidisciplinary Approach. Ph.D. Thesis, Instituto Potosino de Investigación Científica y Tecnológica (IPICYT), México, 2012. (In Spanish). [Google Scholar]
- Fauquet, C.M.; Briddon, R.W.; Brown, J.K.; Moriones, E.; Stanley, J.; Zerbini, M.; Zhou, X. Geminivirus strain demarcation and nomenclature. Arch. Virol. 2008, 153, 783–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orozco, B.M.; Gladfelter, H.J.; Settlage, S.B.; Eagle, P.A.; Gentry, R.N.; Hanley-Bowdoin, L. Multiple cis elements contribute to geminivirus origin function. Virology 1998, 242, 346–356. [Google Scholar] [CrossRef] [PubMed]
- SoftBerry. Available online: http://www.softberry.com/ (accessed on 18 June 2019).
- Higo, K.; Ugawa, Y.; Iwamoto, M.; Korenaga, T. Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res. 1999, 27, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Chabouté, M.E.; Clément, B.; Sekine, M.; Philipps, G.; Chaubet-Gigota, N. Cell Cycle Regulation of the Tobacco Ribonucleotide Reductase Small Subunit Gene Is Mediated by E2F-like Elements. Plant Cell. 2000, 12, 1987–1999. [Google Scholar] [CrossRef] [PubMed]
- Vandepoele, K.; Vlieghe, K.; Florquin, K.; Hennig, L.; Beemster, G.T.; Gruissem, W.; van de Peer, Y.; Inzé, D.; de Veylder, L. Genome-wide Identification of potential plant E2F target genes. Plant Physiol. 2005, 139, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Fenoll, C.; Schwarz, J.J.; Black, D.M.; Schneider, M.; Howell, S.H. The intergenic region of maize streak virus contains a GC-rich element that activates rightward transcription and binds maize nuclear factors. Plant Mol. Biol. 1990, 15, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Claverie, S.; Bernardo, P.; Kraberger, S.; Hartnadyk, P.; Lefeuvre, P.; Lett, J.M.; Galzi, S.; Filloux, D.; Harkins, G.W.; Varsani, A.; et al. From Spatial Metagenomics to Molecular Characterization of Plant Viruses: A Geminivirus Case Study. Adv. Virus Res. 2018, 101, 55–83. [Google Scholar] [PubMed]
- Varsani, A.; Martin, D.P.; Navas-Castillo, J.; Moriones, E.; Hernández-Zepeda, C.; Idris, A.; Murilo Zerbini, F.; Brown, J.K. Revisiting the classificationof curtoviruses based on genome-wide pairwise identity. Arch. Virol. 2014, 159, 1873–1882. [Google Scholar] [CrossRef]
- Briddon, R.W.; Bedford, I.D.; Tsai, J.H.; Markham, P.G. Analysis of the nucleotide sequence of the treehopper-transmitted geminivirus, tomato pseudo-curly top virus, suggests a recombinant origin. Virology 1996, 219, 387–394. [Google Scholar] [CrossRef]
- Orozco, B.M.; Kong, L.-J.; Batts, L.A.; Elledge, S.; Hanley-Bowdoin, L. The multifunctional character of a geminivirus replication protein is reflected by its complex oligomerization properties. J. Biol. Chem. 2000, 275, 6114–6122. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres-Herrera, S.I.; Romero-Osorio, A.; Moreno-Valenzuela, O.; Pastor-Palacios, G.; Cardenas-Conejo, Y.; Ramírez-Prado, J.H.; Riego-Ruiz, L.; Minero-García, Y.; Ambriz-Granados, S.; Argüello-Astorga, G.R. A Lineage of Begomoviruses Encode Rep and AC4 Proteins of Enigmatic Ancestry: Hints on the Evolution of Geminiviruses in the New World. Viruses 2019, 11, 644. https://doi.org/10.3390/v11070644
Torres-Herrera SI, Romero-Osorio A, Moreno-Valenzuela O, Pastor-Palacios G, Cardenas-Conejo Y, Ramírez-Prado JH, Riego-Ruiz L, Minero-García Y, Ambriz-Granados S, Argüello-Astorga GR. A Lineage of Begomoviruses Encode Rep and AC4 Proteins of Enigmatic Ancestry: Hints on the Evolution of Geminiviruses in the New World. Viruses. 2019; 11(7):644. https://doi.org/10.3390/v11070644
Chicago/Turabian StyleTorres-Herrera, Sandra Iliana, Angélica Romero-Osorio, Oscar Moreno-Valenzuela, Guillermo Pastor-Palacios, Yair Cardenas-Conejo, Jorge H. Ramírez-Prado, Lina Riego-Ruiz, Yereni Minero-García, Salvador Ambriz-Granados, and Gerardo R. Argüello-Astorga. 2019. "A Lineage of Begomoviruses Encode Rep and AC4 Proteins of Enigmatic Ancestry: Hints on the Evolution of Geminiviruses in the New World" Viruses 11, no. 7: 644. https://doi.org/10.3390/v11070644
APA StyleTorres-Herrera, S. I., Romero-Osorio, A., Moreno-Valenzuela, O., Pastor-Palacios, G., Cardenas-Conejo, Y., Ramírez-Prado, J. H., Riego-Ruiz, L., Minero-García, Y., Ambriz-Granados, S., & Argüello-Astorga, G. R. (2019). A Lineage of Begomoviruses Encode Rep and AC4 Proteins of Enigmatic Ancestry: Hints on the Evolution of Geminiviruses in the New World. Viruses, 11(7), 644. https://doi.org/10.3390/v11070644