Immune-Escape Hepatitis B Virus Mutations Associated with Viral Reactivation upon Immunosuppression


Abstract
:1. Introduction
1.1. Natural History of HBV Infection and HBV Reactivation
1.2. Molecular Virology and Genetic Variability of HBV
1.3. Biology of HBV Surface Antigen and Immune-Escape Mutations
2. Mutations Associated with HBV Reactivation
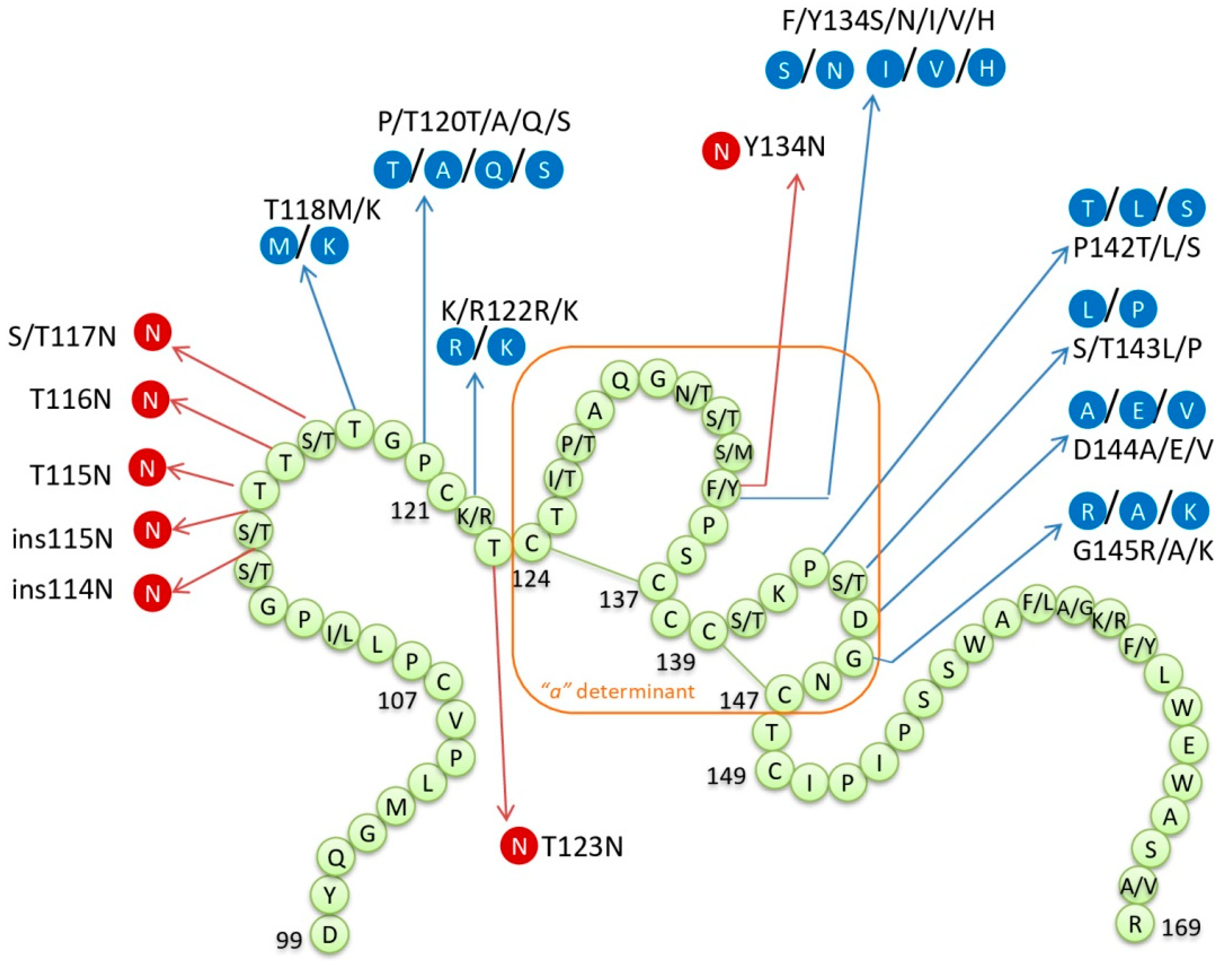
2.1. The Implications of HBsAg Variability
2.1.1. Mutations within “a” Determinant
2.1.2. Additional N-linked glycosylation Sites
2.1.3. MHR Mutations outside “a” Determinant
2.1.4. Mutations outside MHR
2.2. Mutations in the Overlapping Reverse Transcriptase Region
2.3. Mutations in Basal Core Promoter and Precore Regions
3. Summary and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lavanchy, D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J. Viral. Hepat. 2004, 11, 97–107. [Google Scholar] [CrossRef] [PubMed]
- WHO Guidelines on Hepatitis B and C Testing; World Health Organization: Geneva, Switzerland, 2017; Available online: https://apps.who.int/iris/rest/bitstreams/1080581/retrieve (accessed on 4 July 2019).
- Guidelines for the Prevention, Care and Treatment of Persons with Chronic Hepatitis B Infection; World Health Organization: Geneva, Switzerland, 2015; Available online: https://apps.who.int/iris/bitstream/handle/10665/154590/9789241549059_eng.pdf (accessed on 4 July 2019).
- Chen, Y.M.; Yang, S.S.; Chen, D.Y. Risk-stratified management strategies for HBV reactivation in RA patients receiving biological and targeted therapy: A narrative review. J. Microbiol. Immunol. Infect. 2019, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mizukoshi, E.; Sidney, J.; Livingston, B.; Ghany, M.; Hoofnagle, J.H.; Sette, A.; Rehermann, B. Cellular immune responses to the hepatitis B virus polymerase. J. Immunol. 2004, 173, 5863–5871. [Google Scholar] [CrossRef]
- Jung, M.C.; Stemler, M.; Weimer, T.; Spengler, U.; Dohrmann, J.; Hoffmann, R.; Eichenlaub, D.; Eisenburg, J.; Paumgartner, G.; Riethmüller, G.; et al. Immune response of peripheral blood mononuclear cells to HBx-antigen of hepatitis B virus. Hepatology 1991, 13, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Penna, A.; Bertoletti, A.; Cavalli, A.; Valli, A.; Missale, G.; Pilli, M.; Marchelli, S.; Giuberti, T.; Fowler, P.; Chisari, F.V.; et al. Fine specificity of the human T cell response to hepatitis B virus core antigen. Arch. Virol. Suppl. 1992, 4, 23–28. [Google Scholar]
- Ferrari, C. HBV and the immune response. Liver Int. 2015, 35, 121–128. [Google Scholar] [CrossRef]
- Boeijen, L.L.; Hoogeveen, R.C.; Boonstra, A.; Lauer, G.M. Hepatitis B virus infection and the immune response: The big questions. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Bertoletti, A.; Ferrari, C. Adaptive immunity in HBV infection. J. Hepatol. 2016, 64, S71–S83. [Google Scholar] [CrossRef]
- Boni, C.; Laccabue, D.; Lampertico, P.; Giuberti, T.; Viganò, M.; Schivazappa, S.; Alfieri, A.; Pesci, M.; Gaeta, G.B.; Brancaccio, G.; et al. Restored function of HBV-specific T cells after long term effective therapy with nucleos(t)ide anlogues. Gastroenterology 2012, 143, 963–973. [Google Scholar] [CrossRef]
- Terrault, N.A.; Lok, A.S.F.; McMahon, B.J.; Chang, K.M.; Hwang, J.P.; Jonas, M.M.; Brown, R.S., Jr.; Bzowej, N.H.; Wong, J.B. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology 2018, 67, 1560–1599. [Google Scholar] [CrossRef]
- Pattullo, V. Prevention of Hepatitis B reactivation in the setting of immunosuppression. Clin. Mol. Hepatol. 2016, 22, 219–237. [Google Scholar] [CrossRef] [Green Version]
- Elefsiniotis, I.; Vezali, E.; Vrachatis, D.; Hatzianastasiou, S.; Pappas, S.; Farmakidis, G.; Vrioni, G.; Tsakris, A. Post-partum reactivation of chronic hepatitis B virus infection among hepatitis B e-antigen-negative women. World J. Gastroenterol. 2015, 21, 1261–1267. [Google Scholar] [CrossRef]
- Kamitsukasa, H.; Iri, M.; Tanaka, A.; Nagashima, S.; Takahashi, M.; Nishizawa, T.; Okamoto, H. Spontaneous reactivation of hepatitis B virus (HBV) infection in patients with resolved or occult HBV infection. J. Med. Virol. 2015, 87, 589–600. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398. [Google Scholar] [CrossRef] [Green Version]
- Yeo, W.; Zee, B.; Zhong, S.; Chan, P.K.; Wong, W.L.; Ho, W.M.; Lam, K.C.; Johnson, P.J. Comprehensive analysis of risk factors associating with Hepatitis B virus (HBV) reactivation in cancer patients undergoing cytotoxic chemotherapy. Br. J. Cancer 2004, 90, 1306–1311. [Google Scholar] [CrossRef]
- Shouval, D.; Shibolet, O. Immunosuppression and HBV reactivation. Semin. Liver Dis. 2013, 33, 167–177. [Google Scholar] [CrossRef]
- Macera, M.; Stanzione, M.; Messina, V.; D’Adamo, G.; Sangiovanni, V.; Mioglioresi, L.; Fontanella, L.; De Pascalis, S.; Stornaiuolo, G.; Galeota Lanza, A.; et al. Interferon-Free Regimens in Hepatitis B Surface Antigen/Anti-Hepatitis C Virus Positive Patients: The Need to Control Hepatitis B Virus Replication to Avoid Hepatitis B Virus Reactivation. Clin. Gastroenterol. Hepatol. 2017, 15, 1800–1802. [Google Scholar] [CrossRef]
- Sagnelli, E.; Coppola, N.; Scolastico, C.; Filippini, P.; Santantonio, T.; Stroffolini, T.; Piccinino, F. Virologic and clinical expressions of reciprocal inhibitory effect of hepatitis B, C, and delta viruses in patients with chronic hepatitis. Hepatology 2000, 32, 1106–1110. [Google Scholar] [CrossRef]
- Yu, G.; Chi, X.; Wu, R.; Wang, X.; Gao, X.; Kong, F.; Feng, X.; Gao, Y.; Huang, X.; Jin, J.; et al. Replication inhibition of hepatitis B virus and hepatitis C virus in co-infected patients in Chinese population. PLoS ONE 2015, 10, e0139015. [Google Scholar] [CrossRef]
- Stroffolini, T.; Sagnelli, E.; Sagnelli, C.; Smedile, A.; Furlan, C.; Morisco, F.; Coppola, N.; Andriulli, A.; Almasio, P.L. The burden of HBV infection in HCV patients in Italy and the risk of reactivation under DAA therapy. Dig. Liver Dis. 2019, 51, 434–437. [Google Scholar] [CrossRef]
- Valaydon, Z.S.; Locarnini, S.A. The virological aspects of hepatitis B. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 257–264. [Google Scholar] [CrossRef]
- Leistner, C.M.; Gruen-Bernhard, S.; Glebe, D. Role of glycosaminoglycans for binding and infection of hepatitis B virus. Cell Microbiol. 2008, 10, 122–133. [Google Scholar] [CrossRef]
- Ni, Y.; Lempp, F.A.; Mehrle, S.; Nkongolo, S.; Kaufman, C.; Falth, M.; Stindt, J.; Koniger, C.; Nassal, M.; Kubitz, R.; et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 2014, 146, 1070–1083. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1, e00049. [Google Scholar] [CrossRef]
- Rabe, B.; Vlachou, A.; Pante, N.; Helenius, A.; Kann, M. Nuclear import of hepatitis B virus capsids and release of the viral genome. Proc. Natl. Acad. Sci. USA 2003, 100, e54. [Google Scholar] [CrossRef]
- Belloni, L.; Pollicino, T.; de Nicola, F.; Guerrieri, F.; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979. [Google Scholar] [CrossRef] [Green Version]
- Milich, D.; Liang, T.J. Exploring the biological basis of hepatitis B e antigen in hepatitis B virus infection. Hepatology 2003, 38, 1075–1086. [Google Scholar] [CrossRef]
- Summers, J.; Mason, W.S. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 1982, 29, 403–415. [Google Scholar] [CrossRef]
- Caballero, A.; Tabernero, D.; Buti, M.; Rodriguez-Frias, F. Hepatitis B virus: The challenge of an ancient virus with multiple faces and a remarkable replication strategy. Antivir. Res. 2018, 158, 34–44. [Google Scholar] [CrossRef]
- Tuttleman, J.S.; Pourcel, C.; Summers, J. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell 1986, 47, 451–460. [Google Scholar] [CrossRef]
- Vierling, J.M. The immunology of hepatitis B. Clin. Liver Dis. 2007, 11, 727–759. [Google Scholar] [CrossRef]
- Summers, J.; Jilbert, A.R.; Yang, W.; Aldrich, C.E.; Saputelli, J.; Litwin, S.; Toll, E.; Mason, W.S. Hepatocyte turnover during resolution of a transient hepadnaviral infection. Proc. Natl. Acad. Sci. USA 2003, 100, 11652–11659. [Google Scholar] [CrossRef] [Green Version]
- Kay, A.; Zoulim, F. Hepatitis B virus genetic variability and evolution. Virus Res. 2007, 127, 164–176. [Google Scholar] [CrossRef]
- Tong, S.; Revill, P. Overview of hepatitis B viral replication and genetic variability. J. Hepatol. 2016, 64, S4–S16. [Google Scholar] [CrossRef] [Green Version]
- Rajoriya, N.; Combet, C.; Zoulim, F.; Janssen, H.L.A. How viral genetic variants and genotypes influence disease and treatment outcome of chronic hepatitis B. Time for an individualised approach? J. Hepatol. 2017, 67, 1281–1297. [Google Scholar] [CrossRef] [Green Version]
- Nowak, M.A.; Bonhoeffer, S.; Hill, A.M.; Boehme, R.; Thomas, H.C.; McDade, H. Viral dynamics in hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 1996, 93, 4398–4402. [Google Scholar] [CrossRef]
- Okamoto, H.; Imai, M.; Kametani, M.; Nakamura, T.; Mayumi, M. Genomic heterogeneity of hepatitis B virus in a 54-yearold woman who contracted the infection through maternofetal transmission. Jpn. J. Exp. Med. 1987, 57, 231–236. [Google Scholar]
- Heermann, K.H.; Goldmann, U.; Schwartz, W.; Seyffarth, T.; Baumgarten, H.; Gerlich, W.H. Large surface proteins of hepatitis B virus containing the pre-s sequence. J. Virol. 1984, 52, 396–402. [Google Scholar] [Green Version]
- Glebe, D.; Bremer, C.M. The molecular virology of hepatitis B virus. Semin. Liver Dis. 2013, 33, 103–112. [Google Scholar] [CrossRef]
- Wu, C.C.; Chen, Y.S.; Cao, L.; Chen, X.W.; Lu, M.J. Hepatitis B virus infection: Defective surface antigen expression and pathogenesis. World J. Gastroenterol. 2018, 24, 3488–3499. [Google Scholar] [CrossRef]
- Ostapchuk, P.; Hearing, P.; Ganem, D. A dramatic shift in the transmembrane topology of a viral envelope glycoprotein accompanies hepatitis B viral morphogenesis. EMBO J. 1994, 13, 1048–1057. [Google Scholar] [CrossRef]
- Bruss, V.; Lu, X.; Thomssen, R.; Gerlich, W.H. Posttranslational alterations in transmembrane topology of the hepatitis B virus large envelope protein. EMBO J. 1994, 13, 2273–2279. [Google Scholar] [CrossRef]
- Zhao, K.; Wu, C.; Yao, Y.; Cao, L.; Zhang, Z.; Yuan, Y.; Wang, Y.; Pei, R.; Chen, J.; Hu, X.; et al. Ceruloplasmin inhibits the production of extracellular hepatitis B virions by targeting its middle surface protein. J. Gen. Virol. 2017, 98, 1410–1421. [Google Scholar] [CrossRef]
- Kekule, A.S.; Lauer, U.; Meyer, M.; Caselmann, W.H.; Hofschneider, P.H.; Koshy, R. The preS2/S region of integrated hepatitis B virus DNA encodes a transcriptional transactivator. Nature 1990, 343, 457–461. [Google Scholar] [CrossRef]
- Peterson, D.L.; Nath, N.; Gavilanes, F. Structure of hepatitis B surface antigen. Correlation of subtype with amino acid sequence and location of the carbohydrate moiety. J. Biol. Chem. 1982, 257, 10414–10420. [Google Scholar]
- Stibbe, W.; Gerlich, W.H. Structural relationships between minor and major proteins of hepatitis B surface antigen. J. Virol. 1983, 46, 626–628. [Google Scholar]
- Persing, D.H.; Varmus, H.E.; Ganem, D. The preS1 protein of hepatitis B virus is acylated at its amino terminus with myristic acid. J. Virol. 1987, 61, 1672–1677. [Google Scholar] [Green Version]
- Bruss, V.; Hagelstein, J.; Gerhardt, E.; Galle, P.R. Myristylation of the large surface protein is required for hepatitis B virus in vitro infectivity. Virology 1996, 218, 396–399. [Google Scholar] [CrossRef]
- Lentz, T.B.; Loeb, D.D. Roles of the envelope proteins in the amplification of covalently closed circular DNA and completion of synthesis of the plus-strand DNA in hepatitis B virus. J. Virol. 2011, 85, 11916–11927. [Google Scholar] [CrossRef]
- Summers, J.; Smith, P.M.; Horwich, A.L. Hepadnavirus envelope proteins regulate covalently closed circular DNA amplification. J. Virol. 1990, 64, 2819–2824. [Google Scholar] [Green Version]
- Pollicino, T.; Amaddeo, G.; Restuccia, A.; Raffa, G.; Alibrandi, A.; Cutroneo, G.; Favaloro, A.; Maimone, S.; Squadrito, G.; Raimondo, G. Impact of hepatitis B virus (HBV) preS/S genomic variability on HBV surface antigen and HBV DNA serum levels. Hepatology 2012, 56, 434–443. [Google Scholar] [CrossRef]
- Lada, O.; Benhamou, Y.; Poynard, T.; Thibault, V. Coexistence of hepatitis B surface antigen (HBs Ag) and anti-HBs antibodies in chronic hepatitis B virus carriers: Influence of “a” determinant variants. J. Virol. 2006, 80, 2968–2975. [Google Scholar] [CrossRef]
- Cremer, J.; Hofstraat, S.H.I.; van Heiningen, F.; Veldhuijzen, I.K.; van Benthem, B.H.B.; Benschop, K.S.M. Genetic variation of hepatitis B surface antigen among acute and chronic hepatitis B virus infections in The Netherlands. J. Med. Virol. 2018, 90, 1576–1585. [Google Scholar] [CrossRef]
- Zanetti, A.R.; Tanzi, E.; Manzillo, G.; Maio, G.; Sbreglia, C.; Caporaso, N.; Thomas, H.; Zuckerman, A.J. Hepatitis B variant in Europe. Lancet 1988, 2, 1132–1133. [Google Scholar] [CrossRef]
- Carman, W.F.; Zanetti, A.R.; Karayiannis, P.; Waters, J.; Manzillo, G.; Tanzi, E.; Zuckerman, A.J.; Thomas, H.C. Vaccine-induced escape mutant of hepatitis B virus. Lancet 1990, 336, 325–329. [Google Scholar] [CrossRef]
- Echevarría, J.M.; Avellón, A. Hepatitis B virus genetic diversity. J. Med. Virol. 2006, 78, S36–S42. [Google Scholar] [CrossRef]
- Lazarevic, I. Clinical implications of hepatitis B virus mutations: Recent advances. World J. Gastroenterol. 2014, 28, 7653–7664. [Google Scholar] [CrossRef]
- Allain, J.P.; Cox, L. Challenges in hepatitis B detection among blood donors. Curr. Opin. Hematol. 2011, 18, 461–466. [Google Scholar] [CrossRef]
- Hollinger, F.B. Hepatitis B virus infection and transfusion medicine: Science and the occult. Transfusion 2008, 48, 1001–1026. [Google Scholar] [CrossRef]
- Svicher, V.; Cento, V.; Bernassola, M.; Neumann-Fraune, M.; van Hemert, F.; Chen, M.; Salpini, R.; Liu, C.; Longo, R.; Visca, M.; et al. Novel HBsAg markers tightly correlate with occult HBV infection and strongly affect HBsAg detection. Antivir. Res. 2012, 93, 86–93. [Google Scholar] [CrossRef]
- Huang, C.H.; Yuan, Q.; Chen, P.J.; Zhang, Y.L.; Chen, C.R.; Zheng, Q.B.; Yeh, S.H.; Yu, H.; Xue, Y.; Chen, Y.X.; et al. Influence of mutations in hepatitis B virus surface protein on viral antigenicity and phenotype in occult HBV strains from blood donors. J. Hepatol. 2012, 57, 720–729. [Google Scholar] [CrossRef]
- Zhu, H.L.; Li, X.; Li, J.; Zhang, Z.H. Genetic variation of occult hepatitis B virus infection. World J. Gastroenterol. 2016, 22, 3531–3546. [Google Scholar] [CrossRef]
- Chen, S.J.; Zhao, Y.X.; Fang, Y.; Xu, W.Z.; Ma, Y.X.; Song, Z.W.; Teng, X.; Gu, H.X. Viral deletions among healthy young Chinese adults with occult hepatitis B virus infection. Virus Res. 2012, 163, 197–201. [Google Scholar] [CrossRef]
- Seto, W.K.; Chan, T.S.Y.; Hwang, Y.Y.; Wong, D.K.H.; Fung, J.; Liu, K.S.H.; Gill, H.; Lam, Y.F.; Lie, A.K.W.; Lai, C.L.; et al. Hepatitis B reactivation in patients with previous hepatitis B virus exposure undergoing rituximab-containing chemotherapy for lymphoma: A prospective study. J. Clin. Oncol. 2014, 32, 3736–3743. [Google Scholar] [CrossRef]
- Salpini, R.; Colagrossi, L.; Bellocchi, M.C.; Surdo, M.; Becker, C.; Alteri, C.; Aragri, M.; Ricciardi, A.; Armenia, D.; Pollicita, M.; et al. Hepatitis B surface antigen genetic elements critical for immune escape correlate with hepatitis B virus reactivation upon immunosuppression. Hepatology 2015, 61, 823–833. [Google Scholar] [CrossRef]
- Colson, P.; Borentain, P.; Coso, D.; Motte, A.; Aurran-Schleinitz, T.; Charbonnier, A.; Stoppa, A.M.; Chabannon, C.; Serrero, M.; Bertrand, J.; et al. Hepatitis B virus reactivation in HBsAg-negative patients is associated with emergence of viral strains with mutated HBsAg and reverse transcriptase. Virology 2015, 484, 354–463. [Google Scholar] [CrossRef]
- Inoue, J.; Kondo, Y.; Wakui, Y.; Kogure, T.; Morosawa, T.; Fujisaka, Y.; Umetsu, T.; Takai, S.; Nakamura, T.; Shimosegawa, T. Reactivation of resolved hepatitis B virus infection with immune escape mutations after long-term corticosteroid therapy. Clin. J. Gastroenterol. 2016, 9, 93–98. [Google Scholar] [CrossRef]
- Martel, N.; Cotte, L.; Trabaud, M.A.; Trepo, C.; Zoulim, F.; Gomes, S.A.; Kay, A. Probable Corticosteroid-Induced Reactivation of Latent Hepatitis B Virus Infection in an HIV-Positive Patient Involving Immune Escape. J. Infect. Dis. 2012, 205, 1757–1761. [Google Scholar] [CrossRef] [Green Version]
- Ceccarelli, L.; Salpini, R.; Sarmati, L.; Svicher, V.; Bertoli, A.; Sordillo, P.; Ricciardi, A.; Perno, C.F.; Andreoni, M.; Sarrecchia, C. Late hepatitis B virus reactivation after lamivudine prophylaxis interruption in an anti-HBs-positive and anti-HBc-negative patient treated with rituximab-containing therapy. J. Infect. 2012, 65, 180–183. [Google Scholar] [CrossRef]
- Anastasiou, O.E.; Almpani, F.; Herrmann, A.; Gerken, G.; Ditschkowski, M.; Ciesek, S. HBV reactivation in allogeneic stem cell transplant recipients: Risk factors, outcome, and role of hepatitis B virus mutations. Hepatol. Commun. 2017, 1, 1014–1023. [Google Scholar] [CrossRef]
- Ando, T.; Kojima, K.; Isoda, H.; Eguchi, Y.; Honda, T.; Ishigami, M.; Kimura, S. Reactivation of resolved infection with the hepatitis B virus immune escape mutant G145R during dasatinib treatment for chronic myeloid leukemia. Int. J. Hematol. 2015, 102, 379–382. [Google Scholar] [CrossRef]
- Pei, R.; Grund, S.; Verheyen, J.; Esser, S.; Chen, X.; Lu, M. Spontaneous reactivation of hepatitis B virus replication in an HIV coinfected patient with isolated anti-Hepatitis B core antibodies. Virol. J. 2014, 11, 9. [Google Scholar] [CrossRef]
- Sato, A.; Ishii, T.; Sano, F.; Yamada, T.; Takahashi, H.; Matsumoto, N. Severe de novo Hepatitis B Recovered from Late-Onset Liver Insufficiency with Prolonged Ascites and Hypoalbuminemia due to Hepatitis B Virus Genotype Bj with Precore Mutation. Case Rep. Gastroenterol. 2016, 10, 553–559. [Google Scholar] [CrossRef] [Green Version]
- Cerva, C.; Maffongelli, G.; Svicher, V.; Salpini, R.; Colagrossi, L.; Battisti, A.; Mariotti, B.; Cerretti, R.; Cudillo, L.; Sarmati, L. Hepatitis B reactivation characterized by HBsAg negativity and anti-HbsAg antibodies persistence in haematopoietic stem cell transplanted patient after lamivudine withdrawal. BMC Infect. Dis. 2017, 17, 566. [Google Scholar] [CrossRef]
- Saffioti, F.; Raimondo, G. What do we know about occult hepatitis B virus infection? Med. Biol. Sci. 2017, 105. [Google Scholar] [CrossRef]
- Samal, J.; Kandpal, M.; Vivekanandan, P. Molecular mechanisms underlying occult hepatitis B virus infection. Clin. Microbiol. Rev. 2012, 25, 142–163. [Google Scholar] [CrossRef]
- Johnstone, R.W.; Licht, J.D. Histone deacetylase inhibitors in cancer therapy: Is transcription the primary target? Cancer Cell 2003, 4, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Shi, H.; Wang, Y.; Lu, M.; Xu, Y.; Chen, X. A case of hepatitis B reactivation due to the hepatitis B virus escape mutant in a patient undergoing chemotherapy. Virol. Sin. 2012, 27, 369–372. [Google Scholar] [CrossRef]
- Cerva, C.; Colagrossi, L.; Maffongelli, G.; Salpini, R.; di Carlo, D.; Malagnino, V.; Battisti, A.; Ricciardi, A.; Pollicita, M.; Bianchi, A.; et al. Persistent risk of HBV reactivation despite extensive lamivudine prophylaxis in haematopoietic stem cell transplant recipients who are anti-HBc-positive or HBV-negative recipients with an anti-HBc-positive donor. Clin. Microbiol. Infect. 2016, 22, 946.e1–946.e8. [Google Scholar] [CrossRef] [Green Version]
- Westhoff, T.H.; Jochimsen, F.; Schmittel, A.; Stoffler-Meilicke, M.; Schafer, J.H.; Zidek, W.; Gerlich, W.H.; Thiel, E. Fatal hepatitis B virus reactivation by an escape mutant following rituximab therapy. Blood 2003, 102, 1930. [Google Scholar] [CrossRef]
- Ciardi, M.R.; Iannetta, M.; Zingaropoli, M.A.; Salpini, R.; Aragri, M.; Annecca, R.; Pontecorvo, S.; Altieri, M.; Russo, G.; Svicher, V.; et al. Reactivation of Hepatitis B Virus With Immune-Escape Mutations After Ocrelizumab Treatment for Multiple Sclerosis. Open Forum Infect. Dis. 2018, 6, ofy356. [Google Scholar] [CrossRef]
- Fusco, D.N.; Ganova-Raeva, L.; Khudyakov, Y.; Punkova, L.; Mohamed, A.; Cheon, S.S.Y.; Koirala, P.; Andersson, K.L.; Jourdain, G.; Sureau, C.; et al. Reactivation of a Vaccine Escape Hepatitis B Virus Mutant in a Cambodian Patient During Anti-Hepatitis C Virus Therapy. Front. Med. 2018, 5, 97. [Google Scholar] [CrossRef] [Green Version]
- Schlabe, S.; van Bremen, K.; Aldabbagh, S.; Glebe, D.; Bremer, C.M.; Marsen, T.; Mellin, W.; Cristanziano, V.D.; Eis-Hübinger, A.M.; Spengler, U. Hepatitis B virus subgenotype F3 reactivation with vaccine escape mutations: A case report and review of the literature. World J. Hepatol. 2018, 10, 509–516. [Google Scholar] [CrossRef]
- Elkady, A.; Aboulfotuh, S.; Ali, E.M.; Sayed, D.; Abdel-Aziz, N.M.; Ali, A.M.; Murakami, S.; Iijima, S.; Tanaka, Y. Incidence and characteristics of HBV reactivation in hematological malignant patients in south Egypt. World J. Gastroenterol. 2013, 19, 6214–6220. [Google Scholar] [CrossRef]
- Blaich, A.; Manz, M.; Dumoulin, A.; Schüttler, C.G.; Hirsch, H.H.; Gerlich, W.H.; Frei, R. Reactivation of hepatitis B virus with mutated hepatitis B surface antigen in a liver transplant recipient receiving a graft from an antibody to hepatitis B surface antigen- and antibody to hepatitis B core antigen-positive donor. Transfusion 2012, 52, 1999–2006. [Google Scholar] [CrossRef]
- Silva-Pinto, A.; Andrade, J.; Araújo, F.; Santos, L.; Sarmento, A. Reactivation of hepatitis B virus without core antibody. J. Clin. Microbiol. 2015, 53, 1434–1435. [Google Scholar] [CrossRef]
- Fylaktou, A.; Daoudaki, M.; Dimou, V.; Sianou, E.; Papaventsis, D.; Mavrovouniotis, I.; Fouzas, I.; Papanikolaou, V. Hepatitis B reactivation in a renal transplant patient due to a surface antigen mutant strain: A case report. Transplant. Proc. 2012, 44, 2773–2775. [Google Scholar] [CrossRef]
- Kusumoto, S.; Tanaka, Y.; Suzuki, R.; Watanabe, T.; Nakata, M.; Takasaki, H.; Fukushima, N.; Fukushima, T.; Moriuchi, Y.; Itoh, K.; et al. Monitoring of Hepatitis B Virus (HBV) DNA and Risk of HBV Reactivation in B-Cell Lymphoma: A Prospective Observational Study. Clin. Infect. Dis. 2015, 61, 719–729. [Google Scholar] [CrossRef] [Green Version]
- Sureau, C.; Salisse, J. A conformational heparan sulfate binding site essential to infectivity overlaps with the conserved hepatitis B virus a-determinant. Hepatology 2013, 57, 985–994. [Google Scholar] [CrossRef]
- Rezaee, R.; Poorebrahim, M.; Najafi, S.; Sadeghi, S.; Pourdast, A.; Alavian, S.M.; Alavian, S.E.; Poortahmasebi, V. Impacts of the G145R Mutation on the Structure and Immunogenic Activity of the Hepatitis B Surface Antigen: A Computational Analysis. Hepat. Mon. 2016, 16, e39097. [Google Scholar] [CrossRef]
- Kreutz, C. Molecular, immunological and clinical properties of mutated hepatitis B viruses. J. Cell Mol. Med. 2002, 6, 113–143. [Google Scholar] [CrossRef]
- Ma, Q.; Wang, Y. Comprehensive analysis of the prevalence of hepatitis B virus escape mutations in the major hydrophilic region of surface antigen. J. Med. Virol. 2012, 84, 198–206. [Google Scholar] [CrossRef]
- Vigerust, D.J.; Shepherd, V.L. Virus glycosylation: Role in virulence and immune interactions. Trends Microbiol. 2007, 15, 211–218. [Google Scholar] [CrossRef]
- Kwei, K.; Tang, X.; Lok, A.S.; Sureau, C.; Garcia, T.; Li, J.; Wands, J.; Tong, S. Impaired virion secretion by hepatitis B virus immune escape mutants and its rescue by wild-type envelope proteins or a secondsite mutation. J. Virol. 2013, 87, 2352–2357. [Google Scholar] [CrossRef]
- Yu, D.M.; Li, X.H.; Mom, V.; Lu, Z.H.; Liao, X.W.; Han, Y.; Pichoud, C.; Gong, Q.M.; Zhang, D.H.; Zhang, Y.; et al. N-glycosylation mutations within hepatitis B virus surface major hydrophilic region contribute mostly to immune-escape. J. Hepatol. 2014, 60, 515–522. [Google Scholar] [CrossRef]
- Oon, C.J.; Chen, W.N. Current aspects of hepatitis B surface antigen mutants in Singapore. J. Viral. Hepat. 1998, 5, 17–23. [Google Scholar] [CrossRef]
- Norder, H.; Courouce, A.M.; Coursaget, P.; Echevarria, J.M.; Lee, S.D.; Mushahwar, I.K.; Robertson, B.H.; Locarnini, S.; Magniusm, L.O. Genetic diversity of hepatitis B virus strains derived worldwide: Genotypes, subgenotypes, and HBsAg subtypes. Intervirology 2004, 47, 289–309. [Google Scholar] [CrossRef]
- Le Bouvier, G.L.; Capper, R.A.; Williams, A.E.; Pelletier, M.; Katz, A.J. Concurrently circulating hepatitis B surface antigen and heterotypic anti-HBs antibody. J. Immunol. 1976, 117, 2262–2264. [Google Scholar]
- Desmond, C.P.; Bartholomeusz, A.; Gaudieri, S.; Revill, P.A.; Lewin, S.R. A systematic review of T-cell epitopes in hepatitis B virus: Identification, genotypic variation and relevance to antiviral therapeutics. Antivir. Ther. 2008, 13, 161–175. [Google Scholar]
- Ando, K.; Moriyama, T.; Guidotti, L.G.; Wirth, S.; Schreiber, R.D.; Schlicht, H.J.; Huang, S.N.; Chisari, F.V. Mechanisms of class I restricted immunopathology. A transgenic mouse model of fulminant hepatitis. J. Exp. Med. 1993, 178, 1541–1554. [Google Scholar] [CrossRef]
- Churin, Y.; Roderfeld, M.; Roeb, E. Hepatitis B virus large surface protein: Function and fame. HepatoBiliary Surg. Nutr. 2015, 4, 1–10. [Google Scholar] [CrossRef]
- Lai, M.W.; Yeh, C.T. The oncogenic potential of hepatitis B virus rtA181T/ surface truncation mutant. Antivir. Ther. 2008, 13, 875–879. [Google Scholar]
- Yeh, C.T. Development of HBV S gene mutants in chronic hepatitis B patients receiving nucleotide/nucleoside analogue therapy. Antivir. Ther. 2010, 15, 471–475. [Google Scholar] [CrossRef] [Green Version]
- Torresi, J.; Earnest-Silveira, L.; Civitico, G.; Walters, T.E.; Lewin, S.R.; Fyfe, J.; Locarnini, S.A.; Manns, M.; Trautwein, C.; Bock, T.C. Restoration of replication phenotype of lamivudine-resistant hepatitis B virus mutants by compensatory changes in the “fingers” subdomain of the viral polymerase selected as a consequence of mutations in the overlapping S gene. Virology 2002, 299, 88–99. [Google Scholar] [CrossRef]
- Villet, S.; Pichoud, C.; Villeneuve, J.P.; Trépo, C.; Zoulim, F. Selection of a multiple drug-resistant hepatitis B virus strain in a liver-transplanted patient. Gastroenterology 2006, 131, 1253–1261. [Google Scholar] [CrossRef]
- Lee, S.A.; Kim, K.; Kim, H.; Kim, B.J. Nucleotide change of codon 182 in the surface gene of hepatitis B virus genotype C leading to truncated surface protein is associated with progression of liver diseases. J. Hepatol. 2012, 56, 63–69. [Google Scholar] [CrossRef]
- Funk, M.L.; Rosenberg, D.M.; Lok, A.S. World-wide epidemiology of HBeAg-negative chronic hepatitis B and associated precore and core promoter variants. J. Viral Hepat. 2002, 9, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Parekh, S.; Zoulim, F.; Ahn, S.H.; Tsai, A.; Li, J.; Kawai, S.; Khan, N.; Trepo, C.; Wands, J.; Tong, S. Genome replication, virion secretion, and e antigen expression of naturally occurring hepatitis B virus core promoter mutants. J. Virol. 2003, 77, 6601–6612. [Google Scholar] [CrossRef]
- Li, J.; Buckwold, V.E.; Hon, M.W.; Ou, J.H. Mechanism of suppression of hepatitis B virus precore RNA transcription by a frequent double mutation. J. Virol. 1999, 73, 1239–1244. [Google Scholar]
- Guidotti, L.G.; Matzke, B.; Pasquinelli, C.; Shoenberger, J.M.; Rogler, C.E.; Chisari, F.V. The hepatitis B virus (HBV) precore protein inhibits HBV replication in transgenic mice. J. Virol. 1996, 70, 7056–7061. [Google Scholar] [Green Version]
- Lamberts, C.; Nassal, M.; Velhagen, I.; Zentgraf, H.; Schroder, C.H. Precore-mediated inhibition of hepatitis B virus progeny DNA synthesis. J. Virol. 1993, 67, 3756–3762. [Google Scholar] [Green Version]
- Chen, M.T.; Billaud, J.N.; Sallberg, M.; Guidotti, L.G.; Chisari, F.V.; Jones, J.; Hughes, J.; Milich, D.R. A function of the hepatitis B virus pre-core protein is to regulate the immune response to the core antigen. Proc. Natl. Acad. Sci. USA 2004, 101, 14913–14918. [Google Scholar] [CrossRef]
- Chen, M.; Sallberg, M.; Hughes, J.; Jones, J.; Guidotti, L.G.; Chisari, F.V.; Billaud, J.N.; Milich, D.R. Immune tolerance split between hepatitis B virus pre-core and core proteins. J. Virol. 2005, 79, 3016–3027. [Google Scholar] [CrossRef]
- Inoue, J.; Ueno, Y.; Nagasaki, F.; Wakui, Y.; Kondo, Y.; Fukushima, K.; Niitsuma, H.; Shimosegawa, T. Enhanced intracellular retention of a hepatitis B virus strain associated with fulminant hepatitis. Virology 2009, 395, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Ning, B.; Shih, C. Nucleolar localization of human hepatitis B virus capsid protein. J. Virol. 2004, 78, 13653–13668. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazarevic, I.; Banko, A.; Miljanovic, D.; Cupic, M. Immune-Escape Hepatitis B Virus Mutations Associated with Viral Reactivation upon Immunosuppression. Viruses 2019, 11, 778. https://doi.org/10.3390/v11090778
Lazarevic I, Banko A, Miljanovic D, Cupic M. Immune-Escape Hepatitis B Virus Mutations Associated with Viral Reactivation upon Immunosuppression. Viruses. 2019; 11(9):778. https://doi.org/10.3390/v11090778
Chicago/Turabian StyleLazarevic, Ivana, Ana Banko, Danijela Miljanovic, and Maja Cupic. 2019. "Immune-Escape Hepatitis B Virus Mutations Associated with Viral Reactivation upon Immunosuppression" Viruses 11, no. 9: 778. https://doi.org/10.3390/v11090778
APA StyleLazarevic, I., Banko, A., Miljanovic, D., & Cupic, M. (2019). Immune-Escape Hepatitis B Virus Mutations Associated with Viral Reactivation upon Immunosuppression. Viruses, 11(9), 778. https://doi.org/10.3390/v11090778