Updated Phylogeny of Chikungunya Virus Suggests Lineage-Specific RNA Architecture

 , , , ,
, , , , 
Abstract
:1. Introduction
1.1. Chikungunya Lineages
1.2. Conserved RNA Structure as Evolutionary Trait
2. Materials and Methods
2.1. Taxon Sampling
2.2. Phylogenetic Tree Search
2.3. Global Spread Of Virus
- Collapsing of branches with a bootstrap value under 80% [49].
- Replacing tree tip names with metadata information (i.e., geographic location).
2.4. Conserved RNA Structures in CHIKV 3’UTR
3. Results
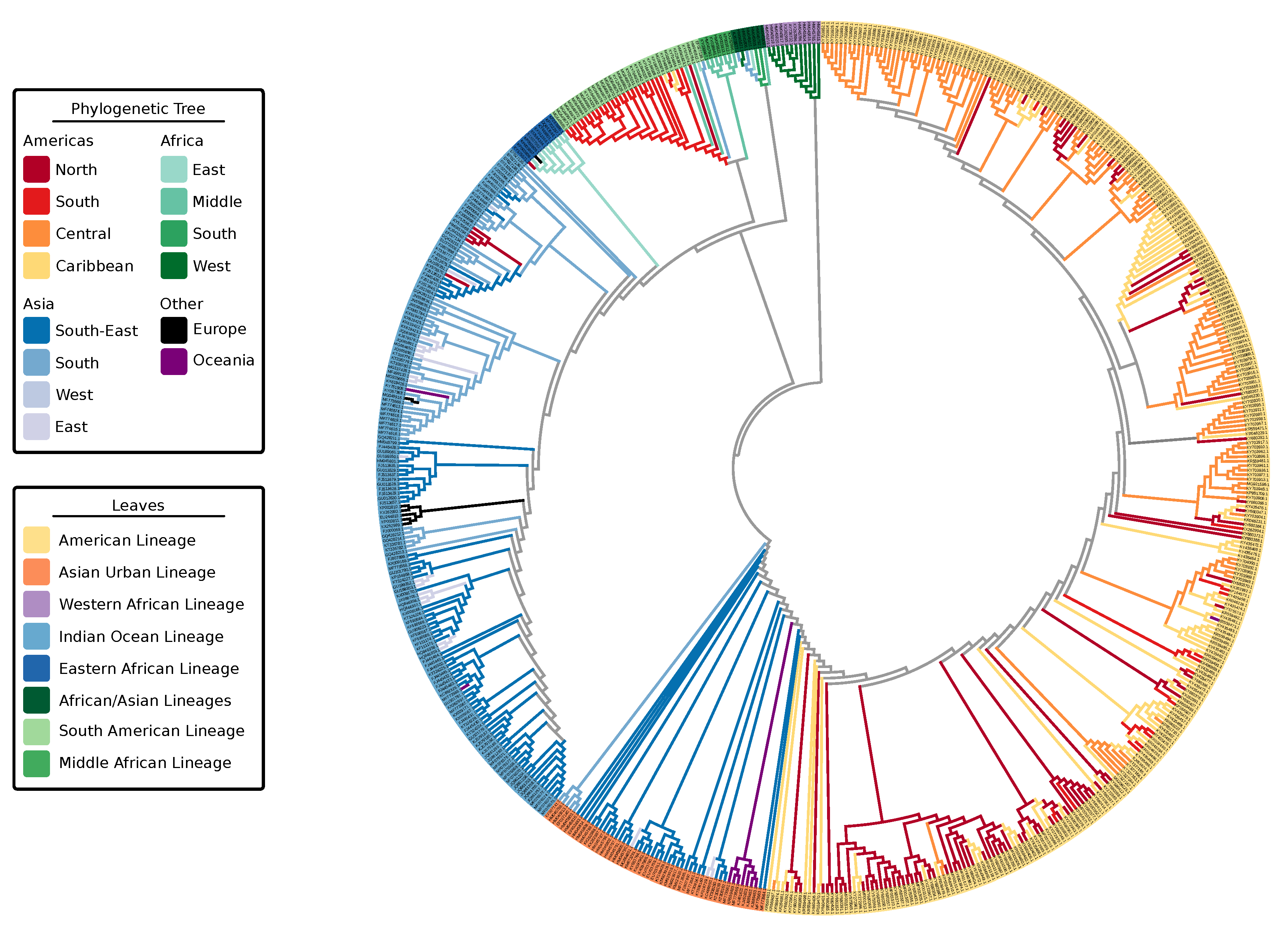
3.1. Phylogeny of Chikungunya Indicates Multiple Lineages In Africa
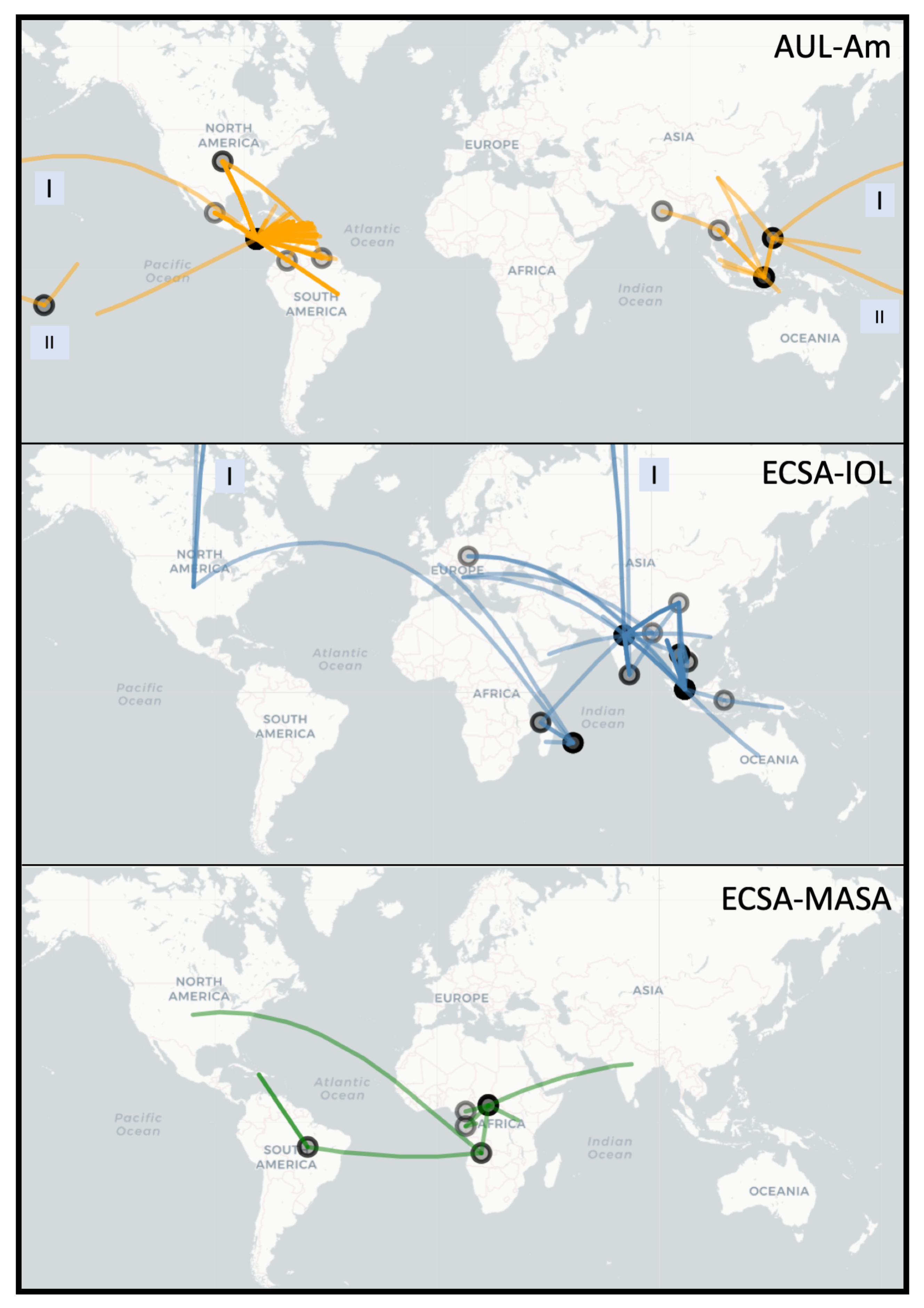
3.2. Spread of Major Chikungunya Epidemic Lineages
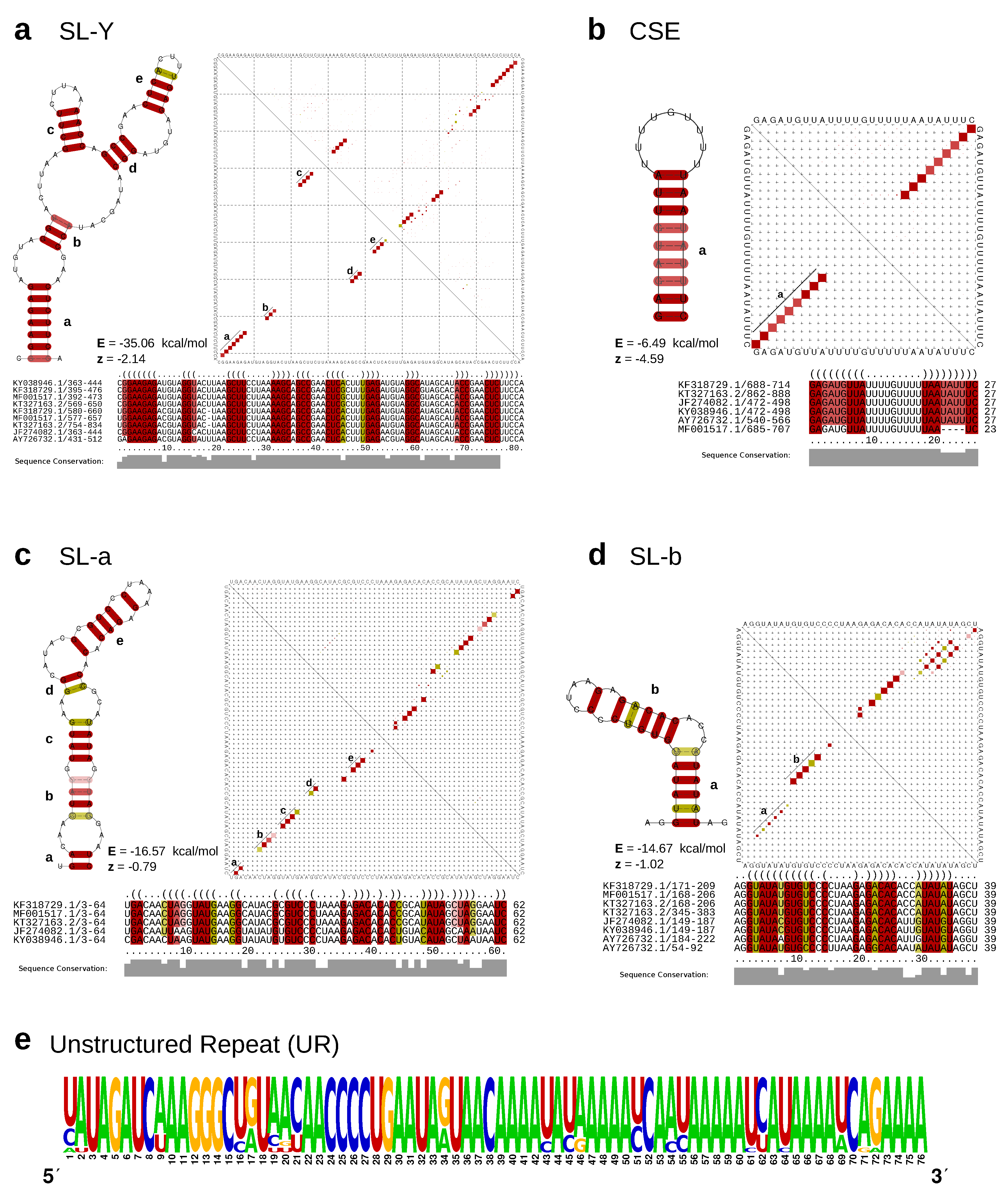
3.3. Lineage-Specific 3’UTR Organization
4. Discussion
4.1. Chikungunya Virus Nomenclature
4.2. Lineage Diversity in Chikungunya Virus
4.3. Lineage-Specific 3’UTR Architecture
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gould, E.; Pettersson, J.; Higgs, S.; Charrel, R.; de Lamballerie, X. Emerging arboviruses: Why today? ONE Health 2017, 4, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.; Kinney, R.M.; Kaaden, O.R. The Alphavirus 3’-Nontranslated Region: Size Heterogeneity and Arrangement of Repeated Sequence Elements. Virology 1998, 240, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Campbell, L.P.; Luther, C.; Moo-Llanes, D.; Ramsey, J.M.; Danis-Lozano, R.; Peterson, A.T. Climate change influences on global distributions of dengue and chikungunya virus vectors. Philos. Trans. R. Soc. B 2015, 370, 20140135. [Google Scholar] [CrossRef] [PubMed]
- Powers, A.M.; Brault, A.C.; Shirako, Y.; Strauss, E.G.; Kang, W.; Strauss, J.H.; Weaver, S.C. Evolutionary Relationships and Systematics of the Alphaviruses. J. Virol. 2001, 75, 10118–10131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrester, N.; Palacios, G.; Tesh, R.; Savji, N.; Guzman, H.; Sherman, M.; Weaver, S.; Lipkin, W. Genome-scale phylogeny of the alphavirus genus suggests a marine origin. J. Virol. 2012, 86, 2729–2738. [Google Scholar] [CrossRef] [PubMed]
- Zeller, H.; Van Bortel, W.; Sudre, B. Chikungunya: Its history in Africa and Asia and its spread to new regions in 2013–2014. J. Infect. Dis. 2016, 214, S436–S440. [Google Scholar] [CrossRef]
- Chen, R.; Puri, V.; Fedorova, N.; Lin, D.; Hari, K.L.; Jain, R.; Rodas, J.D.; Das, S.R.; Shabman, R.S.; Weaver, S.C. Comprehensive genome scale phylogenetic study provides new insights on the global expansion of Chikungunya virus. J. Virol. 2016, 90, 10600–10611. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. The Newala epidemic: III. The virus: Isolation, pathogenic properties and relationship to the epidemic. Epidemiol. Infect. 1956, 54, 177–191. [Google Scholar] [CrossRef]
- Mason, P.; Haddow, A. An epidemic of virus disease in Southern Province, Tanganyika Territory, in 1952–1953: An additional note on Chikungunya virus isolations and serum antibodies. Trans. R. Soc. Trop. Med. H 1957, 51, 238–240. [Google Scholar] [CrossRef]
- Rezza, G.; Nicoletti, L.; Angelini, R.; Romi, R.; Finarelli, A.; Panning, M.; Cordioli, P.; Fortuna, C.; Boros, S.; Magurano, F.; et al. Infection with Chikungunya virus in Italy: An outbreak in a temperate region. Lancet 2007, 370, 1840–1846. [Google Scholar] [CrossRef]
- Van Bortel, W.; Dorleans, F.; Rosine, J.; Blateau, A.; Rousset, D.; Matheus, S.; Leparc-Goffart, I.; Flusin, O.; Prat, C.; Cesaire, R.; et al. Chikungunya outbreak in the Caribbean region, December 2013 to March 2014, and the significance for Europe. Eurosurveillance 2014, 19, 20759. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.G.; Andrade, A.M.; Maria da Conceição, N.C.; Castro, J.S.; Oliveira, F.L.; Goes, C.S.; Maia, M.; Santana, E.B.; Nunes, B.T.; Vasconcelos, P.F. East/Central/South African genotype Chikungunya virus, Brazil, 2014. Emerg. Infect. Dis. 2015, 21, 906. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Lecuit, M. Chikungunya virus and the global spread of a mosquito-borne disease. N. Engl. J. Med. 2015, 372, 1231–1239. [Google Scholar] [CrossRef]
- Rezza, G.; Weaver, S.C. Chikungunya as a paradigm for emerging viral diseases: Evaluating disease impact and hurdles to vaccine development. PLoS Negl. Trop. D 2019, 13, e0006919. [Google Scholar] [CrossRef] [PubMed]
- Borgherini, G.; Poubeau, P.; Staikowsky, F.; Lory, M.; Moullec, N.L.; Becquart, J.P.; Wengling, C.; Michault, A.; Paganin, F. Outbreak of Chikungunya on Reunion Island: Early clinical and laboratory features in 157 adult patients. Clin. Infect. Dis. 2007, 44, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Pulmanausahakul, R.; Roytrakul, S.; Auewarakul, P.; Smith, D.R. Chikungunya in Southeast Asia: Understanding the emergence and finding solutions. Int. J. Infect. Dis. 2011, 15, e671–e676. [Google Scholar] [CrossRef]
- Suhrbier, A.; La Linn, M. Clinical and pathologic aspects of arthritis due to Ross River virus and other alphaviruses. Curr. Opin. Rheumatol. 2004, 16, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Wahid, B.; Ali, A.; Rafique, S.; Idrees, M. Global expansion of chikungunya virus: Mapping the 64-year history. Int. J. Infect. Dis. 2017, 58, 69–76. [Google Scholar] [CrossRef]
- Diallo, M.; Thonnon, J.; Traore-Lamizana, M.; Fontenille, D. Vectors of Chikungunya virus in Senegal: Current data and transmission cycles. Am. J. Trop. Med. Hyg. 1999, 60, 281–286. [Google Scholar] [CrossRef]
- Weaver, S.C.; Forrester, N.L. Chikungunya: Evolutionary history and recent epidemic spread. Antivir. Res. 2015, 120, 32–39. [Google Scholar] [CrossRef]
- Langsjoen, R.M.; Haller, S.L.; Roy, C.J.; Vinet-Oliphant, H.; Bergren, N.A.; Erasmus, J.H.; Livengood, J.A.; Powell, T.D.; Weaver, S.C.; Rossi, S.L. Chikungunya virus strains show lineage-specific variations in virulence and cross-protective ability in murine and nonhuman primate models. mBio 2018, 9, e02449-17. [Google Scholar] [CrossRef] [PubMed]
- Volk, S.M.; Chen, R.; Tsetsarkin, K.A.; Adams, A.P.; Garcia, T.I.; Sall, A.A.; Nasar, F.; Schuh, A.J.; Holmes, E.C.; Higgs, S.; et al. Genome-Scale Phylogenetic Analyses of Chikungunya Virus Reveal Independent Emergences of Recent Epidemics and Various Evolutionary Rates. J. Virol. 2010, 84, 6497–6504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, S.K.; Mavian, C.; Salemi, M.; Morris, J.G., Jr.; Elbadry, M.A.; Okech, B.A.; Lednicky, J.A.; Dunford, J.C. A new “American” subgroup of African-lineage Chikungunya virus detected in and isolated from mosquitoes collected in Haiti, 2016. PLoS ONE 2018, 13, e0196857. [Google Scholar] [CrossRef] [PubMed]
- Li, X.F.; Jiang, T.; Deng, Y.Q.; Zhao, H.; Yu, X.D.; Ye, Q.; Wang, H.J.; Zhu, S.Y.; Zhang, F.C.; Qin, E.D.; et al. Complete genome sequence of a Chikungunya virus isolated in Guangdong, China. J. Virol. 2012, 86, 8904–8905. [Google Scholar] [CrossRef] [PubMed]
- Hyde, J.L.; Chen, R.; Trobaugh, D.W.; Diamond, M.S.; Weaver, S.C.; Klimstra, W.B.; Wilusz, J. The 5’ and 3’ ends of alphavirus RNAs–non-coding is not non-functional. Virus Res. 2015, 206, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Casal, P.E.; Chouhy, D.; Bolatti, E.M.; Perez, G.R.; Stella, E.J.; Giri, A.A. Evidence for homologous recombination in Chikungunya Virus. Mol. Phylogenet. Evol. 2015, 85, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Roby, J.A.; Pijlman, G.P.; Wilusz, J.; Khromykh, A.A. Noncoding subgenomic flavivirus RNA: Multiple functions in West Nile virus pathogenesis and modulation of host responses. Viruses 2014, 6, 404–427. [Google Scholar] [CrossRef] [PubMed]
- Villordo, S.M.; Carballeda, J.M.; Filomatori, C.V.; Gamarnik, A.V. RNA Structure Duplications and Flavivirus Host Adaptation. Trends Microbiol. 2016, 24, 270–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, E.G.; Moon, S.L.; Wilusz, J.; Kieft, J.S. RNA structures that resist degradation by Xrn1 produce a pathogenic Dengue virus RNA. eLife 2014, 3, e01892. [Google Scholar] [CrossRef]
- MacFadden, A.; O’Donoghue, Z.; Silva, P.A.; Chapman, E.G.; Olsthoorn, R.C.; Sterken, M.G.; Pijlman, G.P.; Bredenbeek, P.J.; Kieft, J.S. Mechanism and structural diversity of exoribonuclease-resistant RNA structures in flaviviral RNAs. Nat. Commun. 2018, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- De Bernardi Schneider, A.; Wolfinger, M.T. Musashi binding elements in Zika and related Flavivirus 3’ UTRs: A comparative study in silico. Sci. Rep. 2019, 9, 6911. [Google Scholar] [CrossRef] [PubMed]
- Filomatori, C.V.; Bardossy, E.S.; Merwaiss, F.; Suzuki, Y.; Henrion, A.; Saleh, M.C.; Alvarez, D.E. RNA recombination at Chikungunya virus 3’UTR as an evolutionary mechanism that provides adaptability. PLoS Pathog. 2019, 15, e1007706. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.H.; Trent, D.W.; Strauss, J.H. The 3’-non-coding regions of alpharivus RNAs contain repeating sequences. J. Mol. Biol. 1982, 156, 719–730. [Google Scholar] [CrossRef]
- Chen, R.; Wang, E.; Tsetsarkin, K.A.; Weaver, S.C. Chikungunya virus 3’ untranslated region: Adaptation to mosquitoes and a population bottleneck as major evolutionary forces. PLoS Pathog. 2013, 9, e1003591. [Google Scholar] [CrossRef] [PubMed]
- Ochsenreiter, R.; Hofacker, I.L.; Wolfinger, M.T. Functional RNA Structures in the 3’UTR of Tick-Borne, Insect-Specific and No-Known-Vector Flaviviruses. Viruses 2019, 11, 298. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.; Jayaraman, B.; Frankel, A. The HIV-1 Rev response element: An RNA scaffold that directs the cooperative assembly of a homo-oligomeric ribonucleoprotein complex. RNA Biol. 2012, 9, 6–11. [Google Scholar] [CrossRef]
- Kutchko, K.M.; Madden, E.A.; Morrison, C.; Plante, K.S.; Sanders, W.; Vincent, H.A.; Cruz Cisneros, M.C.; Long, K.M.; Moorman, N.J.; Heise, M.T.; et al. Structural divergence creates new functional features in alphavirus genomes. Nucleic Acids Res. 2018, 46, 3657–3670. [Google Scholar] [CrossRef] [Green Version]
- Benson, D.A.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 42, D32–D37. [Google Scholar] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- De Bernardi Schneider, A. Arboviruses: The Hidden Path of an Imminent Threat. Ph.D. Thesis, The University of North Carolina at Charlotte, Charlotte, NC, USA, 2018. [Google Scholar]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Schliep, K.P. phangorn: Phylogenetic analysis in R. Bioinformatics 2010, 27, 592–593. [Google Scholar] [CrossRef] [PubMed]
- Charif, D.; Lobry, J.R. SeqinR 1.0-2: A contributed package to the R project for statistical computing devoted to biological sequences retrieval and analysis. In Structural Approaches to Sequence Evolution; Springer: Berlin/Heidelberg, Germany, 2007; pp. 207–232. [Google Scholar]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef]
- De Bernardi Schneider, A.; Ford, C.T.; Hostager, R.; Williams, J.; Cioce, M.; Çatalyürek, Ü.V.; Wertheim, J.O.; Janies, D. StrainHub: A phylogenetic tool to construct pathogen transmission networks. bioRxiv 2019. [Google Scholar] [CrossRef]
- Louca, S.; Doebeli, M. Efficient comparative phylogenetics on large trees. Bioinformatics 2017, 34, 1053–1055. [Google Scholar] [CrossRef]
- Graul, C. LeafletR: Interactive Web-Maps Based on the Leaflet JavaScript Library. R-Package Version 0.4-0. 2016. Available online: https://github.com/chgrl/leafletR (accessed on 28 August 2019).
- Hijmans, R.J. Geosphere: Spherical Trigonometry. R Package Version 1.5-5. 2016. Available online: https://cran.r-project.org/package=geosphere (accessed on 28 August 2019).
- Eddy, S.R.; Durbin, R. RNA sequence analysis using covariance models. Nucleic Acids Res. 1994, 22, 2079–2088. [Google Scholar] [CrossRef] [Green Version]
- Wolfinger, M.T.; Fallmann, J.; Eggenhofer, F.; Amman, F. ViennaNGS: A toolbox for building efficient next-generation sequencing analysis pipelines. F1000Research 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Will, S.; Reiche, K.; Hofacker, I.L.; Stadler, P.F.; Backofen, R. Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput. Biol. 2007, 3, e65. [Google Scholar] [CrossRef] [PubMed]
- Bernhart, S.H.; Hofacker, I.L.; Will, S.; Gruber, A.R.; Stadler, P.F. RNAalifold: Improved consensus structure prediction for RNA alignments. BMC Bioinform. 2008, 9, 474. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bernhart, S.H.; Zu Siederdissen, C.H.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithm. Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold Faster RNA Homology Searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef]
- Lindsey, N.P.; Staples, J.E.; Fischer, M. Chikungunya virus disease among travelers—United States, 2014–2016. Am. J. Trop. Med. Hyg. 2018, 98, 192–197. [Google Scholar] [CrossRef]
- Hapuarachchi, H.; Bandara, K.; Sumanadasa, S.; Hapugoda, M.; Lai, Y.L.; Lee, K.S.; Tan, L.K.; Lin, R.T.; Ng, L.F.; Bucht, G.; et al. Re-emergence of Chikungunya virus in South-east Asia: virological evidence from Sri Lanka and Singapore. J. Gen. Virol. 2010, 91, 1067–1076. [Google Scholar] [CrossRef]
- Da Costa, A.C.; Thézé, J.; Komninakis, S.C.V.; Sanz-Duro, R.L.; Felinto, M.R.L.; Moura, L.C.C.; de Oliveira Barroso, I.M.; Santos, L.E.C.; de Lemos Nunes, M.A.; Moura, A.A.; et al. Spread of Chikungunya virus east/central/south African genotype in northeast Brazil. Emerg. Infect. Dis. 2017, 23, 1742. [Google Scholar] [CrossRef]
- Souza, T.M.A.; Azeredo, E.L.; Badolato-Corrêa, J.; Damasco, P.V.; Santos, C.; Petitinga-Paiva, F.; Nunes, P.C.G.; Barbosa, L.S.; Cipitelli, M.C.; Chouin-Carneiro, T.; et al. First report of the East-Central South African genotype of Chikungunya virus in Rio de Janeiro, Brazil. PLoS Curr. 2017, 9. [Google Scholar] [CrossRef]
- Hardy, R.W.; Rice, C.M. Requirements at the 3’ end of the Sindbis Virus Genome Efficient Synthesis Minus-Strand RNA. J. Virol. 2005, 79, 4630–4639. [Google Scholar] [CrossRef]
- Gruber, A.R.; Findeiß, S.; Washietl, S.; Hofacker, I.L.; Stadler, P.F. RNAz 2.0: Improved noncoding RNA detection. In Biocomp 2010; World Scientific: London, UK, 2010; pp. 69–79. [Google Scholar]
- Stapleford, K.A.; Moratorio, G.; Henningsson, R.; Chen, R.; Matheus, S.; Enfissi, A.; Weissglas-Volkov, D.; Isakov, O.; Blanc, H.; Mounce, B.C.; et al. Whole-Genome Sequencing Analysis from the Chikungunya Virus Caribbean Outbreak Reveals Novel Evolutionary Genomic Elements. PLoS Negl. Trop. D 2016, 10, e0004402. [Google Scholar] [CrossRef] [PubMed]
- Wiley, E.O. The evolutionary species concept reconsidered. Syst. Zool 1978, 27, 17–26. [Google Scholar] [CrossRef]
- Peterson, A.T. Defining viral species: Making taxonomy useful. Virol. J. 2014, 11, 131. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.F.; Su, C.L.; Hsu, T.C.; Chang, S.F.; Lin, C.C.; Huang, J.C.; Shu, P.Y. Imported Chikungunya Virus Strains, Taiwan, 2006–2014. Emerg. Infect. Dis. 2016, 22, 1981. [Google Scholar] [CrossRef] [PubMed]
- Maha, M.S.; Susilarini, N.K.; Nur Ika Hariastuti, S. Chikungunya virus mutation, Indonesia, 2011. Emerg. Infect. Dis. 2015, 21, 379. [Google Scholar] [CrossRef] [PubMed]
- Nunes, M.R.T.; Faria, N.R.; de Vasconcelos, J.M.; Golding, N.; Kraemer, M.U.; de Oliveira, L.F.; da Silva Azevedo, R.d.S.; da Silva, D.E.A.; da Silva, E.V.P.; da Silva, S.P.; et al. Emergence and potential for spread of Chikungunya virus in Brazil. BMC Med. 2015, 13, 102. [Google Scholar] [CrossRef]
- Yactayo, S.; Staples, J.E.; Millot, V.; Cibrelus, L.; Ramon-Pardo, P. Epidemiology of Chikungunya in the Americas. J. Infect. Dis. 2016, 214, S441–S445. [Google Scholar] [CrossRef]
- Strauss, J.H.; Strauss, E.G. The Alphaviruses: Gene Expression, Replication, and Evolution. Microbiol. Mol. Biol. R 1994, 58, 491–562. [Google Scholar]
- Morley, V.J.; Noval, M.G.; Chen, R.; Weaver, S.C.; Vignuzzi, M.; Stapleford, K.A.; Turner, P.E. Chikungunya virus evolution following a large 3’ UTR deletion results in host-specific molecular changes in protein-coding regions. Virus Evol. 2018, 4, vey012. [Google Scholar] [CrossRef]
- Charley, P.A.; Wilusz, J. Sponging of cellular proteins by viral RNAs. Curr. Opin. Virol. 2014, 9, 14–18. [Google Scholar] [CrossRef] [Green Version]
- Pardigon, N.; Strauss, J.H. Mosquito homolog of the La autoantigen binds to Sindbis virus RNA. J. Virol. 1996, 70, 1173–1181. [Google Scholar] [Green Version]
- Sokoloski, K.J.; Dickson, A.M.; Chaskey, E.L.; Garneau, N.L.; Wilusz, C.J.; Wilusz, J. Sindbis Virus Usurps the Cellular HuR Protein to Stabilize its Transcripts and Promote Productive Infections in Mmammalian and Mmosquito Cells. Cell Host Microbe 2010, 8, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Dickson, A.M.; Anderson, J.R.; Barnhart, M.D.; Sokoloski, K.J.; Oko, L.; Opyrchal, M.; Galanis, E.; Wilusz, C.J.; Morrison, T.E.; Wilusz, J. Dephosphorylation of HuR protein during alphavirus infection is Associated with HuR relocalization to the cytoplasm. J. Biol. Chem. 2012, 287, 36229–36238. [Google Scholar] [CrossRef] [PubMed]
- Chavali, P.L.; Stojic, L.; Meredith, L.W.; Joseph, N.; Nahorski, M.S.; Sanford, T.J.; Sweeney, T.R.; Krishna, B.A.; Hosmillo, M.; Firth, A.E.; et al. Neurodevelopmental protein Musashi 1 interacts with the Zika genome and promotes viral replication. Science 2017, 357, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Trobaugh, D.W.; Gardner, C.L.; Sun, C.; Haddow, A.D.; Wang, E.; Chapnik, E.; Mildner, A.; Weaver, S.C.; Ryman, K.D.; Klimstra, W.B. RNA viruses can hijack vertebrate microRNAs to suppress innate immunity. Nature 2014, 506, 245. [Google Scholar] [CrossRef] [PubMed]
- Dubey, S.K.; Shrinet, J.; Sunil, S. Aedes aegypti microRNA, miR-2944b-5p interacts with 3’UTR of Chikungunya virus and cellular target vps-13 to regulate viral replication. PLoS Negl. Trop. D 2019, 13, e0007429. [Google Scholar] [CrossRef]
- Yen, P.S.; Chen, C.H.; Sreenu, V.; Kohl, A.; Failloux, A.B. Assessing the Potential Interactions between Cellular miRNA and Arboviral Genomic RNA in the Yellow Fever Mosquito, Aedes Aegypti. Viruses 2019, 11, 540. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Torres, S.; Schnettler, E.; Funk, A.; Grundhoff, A.; Pijlman, G.P.; Khromykh, A.A.; Asgari, S. West Nile virus encodes a microRNA-like small RNA in the 3’ untranslated region which up-regulates GATA4 mRNA and facilitates virus replication in mosquito cells. Nucleic Acids Res. 2012, 40, 2210–2223. [Google Scholar] [CrossRef]
- Kulasegaran-Shylini, R.; Atasheva, S.; Gorenstein, D.G.; Frolov, I. Structural and Functional Elements of the Promoter Encoded by the 5’ Untranslated Region of the Venezuelan Equine Encephalitis Virus Genome. J. Virol. 2009, 83, 8327–8339. [Google Scholar] [CrossRef]
- Fayzulin, R.; Frolov, I. Changes of the Ssecondary Sstructure of the 5’ end of the Sindbis Virus Genome Inhibit Virus Growth in Mosquito Cells and Lead to Accumulation of Adaptive Mutations. J. Virol. 2004, 78, 4953–4964. [Google Scholar] [CrossRef]
- Hyde, J.L.; Gardner, C.L.; Kimura, T.; White, J.P.; Liu, G.; Trobaugh, D.W.; Huang, C.; Tonelli, M.; Paessler, S.; Takeda, K.; et al. A viral RNA structural element alters host recognition of nonself RNA. Science 2014, 343, 783–787. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Previous Work | This Study | |
|---|---|---|
| Lineages | Lineages | Epidemic Clades |
| Indian Ocean (IOL) | Indian Ocean | ECSA-IOL |
| East Central and South African (ECSA) | Eastern African | |
| South American | ECSA-MASA | |
| Middle African | ||
| African/Asian | N/A | |
| Asian Urban (AUL) | Asian Urban | Asian Urban American (AUL-Am) |
| American | ||
| West African (WA) | Western African | Western African (WA) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Bernardi Schneider, A.; Ochsenreiter, R.; Hostager, R.; Hofacker, I.L.; Janies, D.; Wolfinger, M.T. Updated Phylogeny of Chikungunya Virus Suggests Lineage-Specific RNA Architecture. Viruses 2019, 11, 798. https://doi.org/10.3390/v11090798
de Bernardi Schneider A, Ochsenreiter R, Hostager R, Hofacker IL, Janies D, Wolfinger MT. Updated Phylogeny of Chikungunya Virus Suggests Lineage-Specific RNA Architecture. Viruses. 2019; 11(9):798. https://doi.org/10.3390/v11090798
Chicago/Turabian Stylede Bernardi Schneider, Adriano, Roman Ochsenreiter, Reilly Hostager, Ivo L. Hofacker, Daniel Janies, and Michael T. Wolfinger. 2019. "Updated Phylogeny of Chikungunya Virus Suggests Lineage-Specific RNA Architecture" Viruses 11, no. 9: 798. https://doi.org/10.3390/v11090798
APA Stylede Bernardi Schneider, A., Ochsenreiter, R., Hostager, R., Hofacker, I. L., Janies, D., & Wolfinger, M. T. (2019). Updated Phylogeny of Chikungunya Virus Suggests Lineage-Specific RNA Architecture. Viruses, 11(9), 798. https://doi.org/10.3390/v11090798