MiDRMpol: A High-Throughput Multiplexed Amplicon Sequencing Workflow to Quantify HIV-1 Drug Resistance Mutations against Protease, Reverse Transcriptase, and Integrase Inhibitors

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Specimens, Plasmids, and Control Virus
2.2. High-Throughput Sequencing
2.3. Bioinformatics Pipeline for Analysis
2.4. Standard Population Sequencing
2.5. HIV-1 Subtyping and Phylogenetic Analysis
2.6. Drug Resistance Mutations
2.7. Availability of Data
2.8. Ethics Approval and Consent of Participants
3. Results
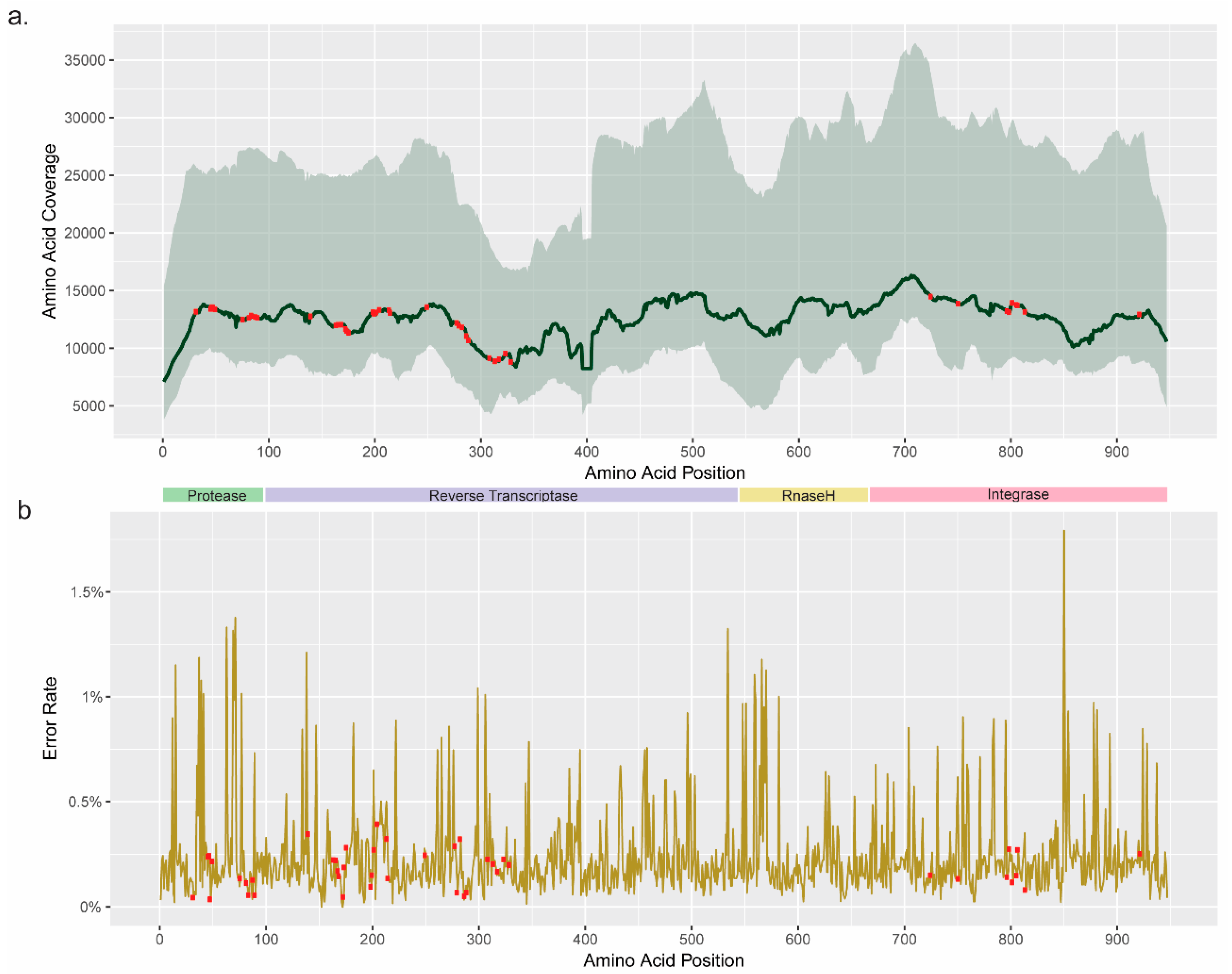
3.1. Coverage and Error Rate
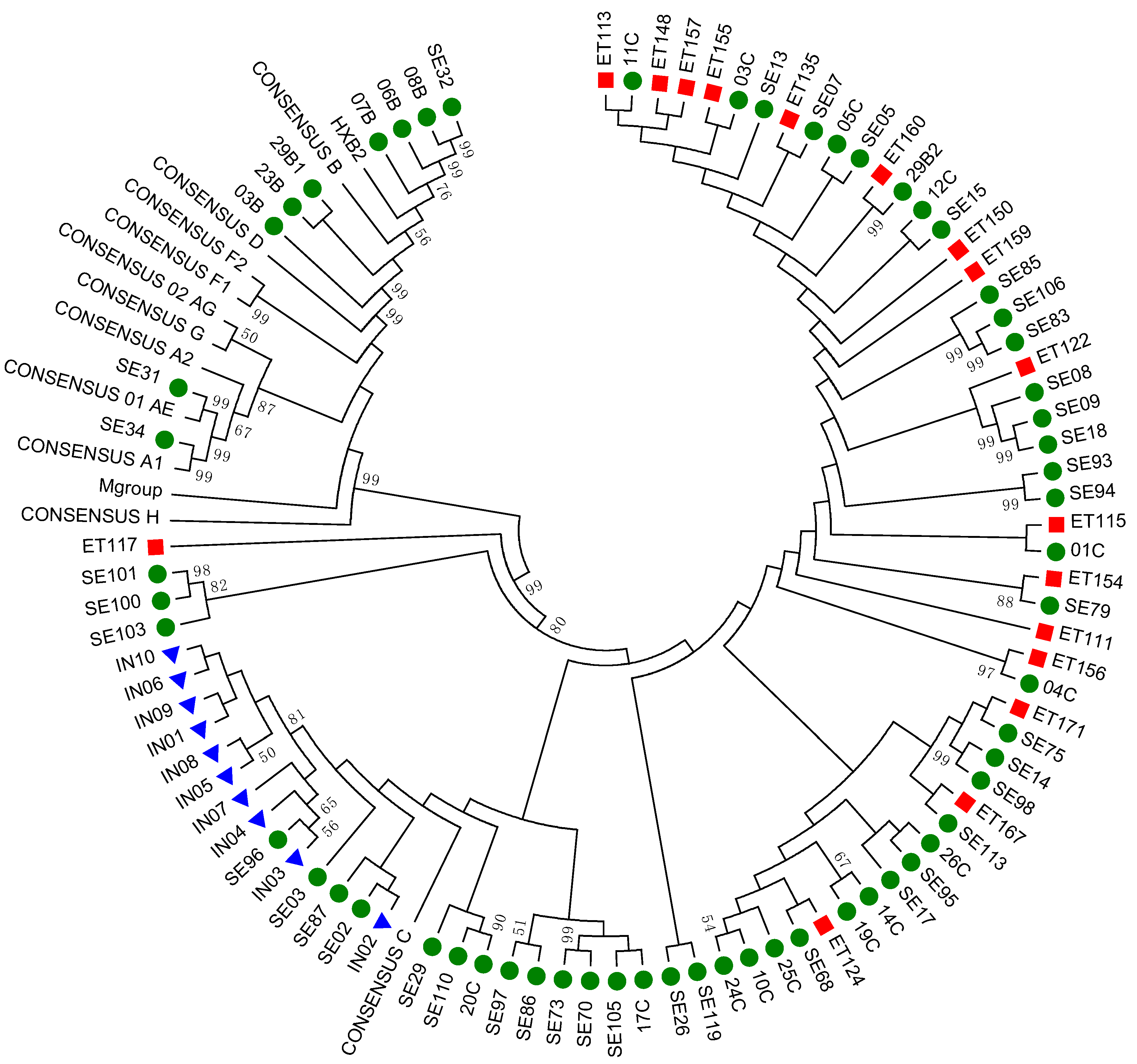
3.2. Subtyping and Phylogenetic Analysis
3.3. Identification of DRM by GRT-HTS
3.4. Comparison of DRM Detected by GRT-PS and GRT-HTS
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Inzaule, S.C.; Hamers, R.L.; Noguera-Julian, M.; Casadella, M.; Parera, M.; Kityo, C.; Steegen, K.; Naniche, D.; Clotet, B.; Rinke de Wit, T.F.; et al. Clinically relevant thresholds for ultrasensitive hiv drug resistance testing: A multi-country nested case-control study. Lancet HIV 2018, 5, e638–e646. [Google Scholar] [CrossRef]
- Posada-Cespedes, S.; Seifert, D.; Beerenwinkel, N. Recent advances in inferring viral diversity from high-throughput sequencing data. Virus Res. 2017, 239, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Enns, E.; Brumme, C.J.; Parkin, N.; Howison, M.; Lee, E.R.; Capina, R.; Marinier, E.; Avila-Rios, S.; Sandstrom, P.; et al. Bioinformatic data processing pipelines in support of next-generation sequencing-based hiv drug resistance testing: The winnipeg consensus. J. Int. AIDS Soc. 2018, 21, e25193. [Google Scholar] [CrossRef] [PubMed]
- Ndung’u, T.; Renjifo, B.; Essex, M. Construction and analysis of an infectious human immunodeficiency virus type 1 subtype c molecular clone. J. Virol. 2001, 75, 4964–4972. [Google Scholar] [CrossRef] [PubMed]
- Aralaguppe, S.G.; Siddik, A.B.; Manickam, A.; Ambikan, A.T.; Kumar, M.M.; Fernandes, S.J.; Amogne, W.; Bangaruswamy, D.K.; Hanna, L.E.; Sonnerborg, A.; et al. Multiplexed next-generation sequencing and de novo assembly to obtain near full-length hiv-1 genome from plasma virus. J. Virol. Methods 2016, 236, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Joshi, N.; Fass, J. Sickle: A sliding-window, adaptive, quality-based trimming tool for fastq files (version 1.33) [software]. 2011. Available online: https://github.com/najoshi/sickle (accessed on 2 January 2019).
- Xu, H.; Luo, X.; Qian, J.; Pang, X.; Song, J.; Qian, G.; Chen, J.; Chen, S. Fastuniq: A fast de novo duplicates removal tool for paired short reads. PLoS ONE 2012, 7, e52249. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with bowtie 2. Nat Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Amogne, W.; Bontell, I.; Grossmann, S.; Aderaye, G.; Lindquist, L.; Sonnerborg, A.; Neogi, U. Phylogenetic analysis of ethiopian hiv-1 subtype c near full-length genomes reveals high intrasubtype diversity and a strong geographical cluster. AIDS Res. Hum. Retroviruses 2016, 32, 471–474. [Google Scholar] [CrossRef]
- Regav3. Available online: http://dbpartners.stanford.edu:8080/RegaSubtyping/stanford-hiv/typingtool/ (accessed on 30 March 2016).
- Comet-HIV. Available online: https://comet.lih.lu/ (accessed on 30 March 2016).
- Bennett, D.E.; Camacho, R.J.; Otelea, D.; Kuritzkes, D.R.; Fleury, H.; Kiuchi, M.; Heneine, W.; Kantor, R.; Jordan, M.R.; Schapiro, J.M.; et al. Drug resistance mutations for surveillance of transmitted hiv-1 drug-resistance: 2009 update. PLoS ONE 2009, 4, e4724. [Google Scholar] [CrossRef] [PubMed]
- Stanford hiv db. Available online: https://hivdb.stanford.edu/assets/media/resistance-mutation-handout-Feb2017.516aee6f.pdf (accessed on 2 January 2018).
- Rhee, S.Y.; Gonzales, M.J.; Kantor, R.; Betts, B.J.; Ravela, J.; Shafer, R.W. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 2003, 31, 298–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DNA sequencing costs: Data from the nhgri genome sequencing program (gsp). Available online: https://www.genome.gov/about-genomics/fact-sheets/DNA-Sequencing-Costs-Data (accessed on 13 August 2019).
- Casadella, M.; Paredes, R. Deep sequencing for hiv-1 clinical management. Virus Res. 2017, 239, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Ekici, H.; Rao, S.D.; Sonnerborg, A.; Ramprasad, V.L.; Gupta, R.; Neogi, U. Cost-efficient hiv-1 drug resistance surveillance using multiplexed high-throughput amplicon sequencing: Implications for use in low- and middle-income countries. J. Antimicrob. Chemother. 2014, 69, 3349–3355. [Google Scholar] [CrossRef] [PubMed]
- Arias, A.; López, P.; Sánchez, R.; Yamamura, Y.; Rivera-Amill, V. Sanger and next generation sequencing approaches to evaluate hiv-1 virus in blood compartments. Int. J. Environ. Res. Public Health 2018, 15, 1697. [Google Scholar] [CrossRef]
- Dalmat, R.R.; Makhsous, N.; Pepper, G.G.; Magaret, A.; Jerome, K.R.; Wald, A.; Greninger, A.L. Limited marginal utility of deep sequencing for hiv drug resistance testing in the age of integrase inhibitors. J. Clin. Microbiol. 2018, 56. [Google Scholar] [CrossRef]
- Telele, N.F.; Kalu, A.W.; Gebre-Selassie, S.; Fekade, D.; Abdurahman, S.; Marrone, G.; Neogi, U.; Tegbaru, B.; Sonnerborg, A. Pretreatment drug resistance in a large countrywide ethiopian hiv-1c cohort: A comparison of sanger and high-throughput sequencing. Sci. Rep. 2018, 8, 7556. [Google Scholar] [CrossRef]
- Gianella, S.; Richman, D.D. Minority variants of drug-resistant hiv. J. Infect. Dis. 2010, 202, 657–666. [Google Scholar] [CrossRef]
- Kozal, M.J.; Chiarella, J.; St John, E.P.; Moreno, E.A.; Simen, B.B.; Arnold, T.E.; Lataillade, M. Prevalence of low-level hiv-1 variants with reverse transcriptase mutation k65r and the effect of antiretroviral drug exposure on variant levels. Antivir. Ther. 2011, 16, 925–929. [Google Scholar] [CrossRef]
- Lataillade, M.; Chiarella, J.; Yang, R.; DeGrosky, M.; Uy, J.; Seekins, D.; Simen, B.; St John, E.; Moreno, E.; Kozal, M. Virologic failures on initial boosted-pi regimen infrequently possess low-level variants with major pi resistance mutations by ultra-deep sequencing. PLoS ONE 2012, 7, e30118. [Google Scholar] [CrossRef]
- Li, J.Z.; Paredes, R.; Ribaudo, H.J.; Svarovskaia, E.S.; Metzner, K.J.; Kozal, M.J.; Hullsiek, K.H.; Balduin, M.; Jakobsen, M.R.; Geretti, A.M.; et al. Low-frequency hiv-1 drug resistance mutations and risk of nnrti-based antiretroviral treatment failure: A systematic review and pooled analysis. JAMA J. Am. Med Assoc. 2011, 305, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Z.; Paredes, R.; Ribaudo, H.J.; Kozal, M.J.; Svarovskaia, E.S.; Johnson, J.A.; Geretti, A.M.; Metzner, K.J.; Jakobsen, M.R.; Hullsiek, K.H.; et al. Impact of minority nonnucleoside reverse transcriptase inhibitor resistance mutations on resistance genotype after virologic failure. J. Infect. Dis. 2013, 207, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.G.; Smith, D.M.; Murrell, B.; Slabbert, R.; Kirby, B.M.; Edson, C.; Cotton, M.F.; Haubrich, R.H.; Kosakovsky Pond, S.L.; Van Zyl, G.U. Next generation sequencing improves detection of drug resistance mutations in infants after pmtct failure. J. Clin. Virol. 2015, 62, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.paseq.org/ (accessed on 2 January 2019).
- Routh, A.; Chang, M.W.; Okulicz, J.F.; Johnson, J.E.; Torbett, B.E. Covama: Co-variation mapper for disequilibrium analysis of mutant loci in viral populations using next-generation sequence data. Methods 2015, 91, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Noguera-Julian, M.; Edgil, D.; Harrigan, P.R.; Sandstrom, P.; Godfrey, C.; Paredes, R. Next-generation human immunodeficiency virus sequencing for patient management and drug resistance surveillance. J. Infect. Dis. 2017, 216, S829–S833. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Cohort | PID | Treatment | MiDRMpol | PASeq | GRT-PS | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PI | NRTI | NNRTI | INI | PI | NRTI | NNRTI | INI | NRTI | NNRTI | |||
| Swedish | SE03 | Experienced | None | D67G (2.0%) | None | None | None | None | K103R (16.9%) | None | None | None |
| L74I (1.9%) | ||||||||||||
| SE70 | Experienced | None | D67N (99.7%) | K103N (99.8%) | None | None | D67N (99.9%) | K103N (99.9%) | None | D67N | K103N | |
| L210W (99.5%) | G190A (99.6%) | L210W (99.8%) | G190A (99.7%) | L210W | G190A | |||||||
| T215Y (99.4%) | T215Y (99.8%) | T215Y | ||||||||||
| SE73 | Experienced | None | D67N (44.2%) | K103N (95.9%) | None | None | D67N (49.2%) | K103N (96.1%) | None | None | K103N | |
| L210W (23.4%) | G190A (35.2%) | L210W (46.1%) | G190A (49.6%) | Y188C | ||||||||
| T215Y (19.7%) | T215Y (45.9%) | |||||||||||
| 19C | Naïve | M46I (4.6%) | None | None | None | Failed | None | None | ||||
| 20C | Naïve | None | T215S (2.1%) | None | None | Failed | None | None | ||||
| 26C | Naïve | None | M184I (1.1%) | None | None | Failed | None | None | ||||
| 05C | Naïve | None | K219R (2.3%) | None | None | Failed | None | None | ||||
| SE07 | Naïve | None | None | None | Q148H (1.1%) | None | None | None | None | None | None | |
| SE14 | Naïve | None | None | None | E138K (5.5%) | None | None | None | E138K (1.6%) | None | None | |
| SE31 | Naïve | M46I (30.2%) | None | None | None | M46I (28.9%) | None | None | None | None | None | |
| SE97 | Naïve | D30N (13.7%) | None | None | None | D30N (12.2%) | None | None | None | None | None | |
| 24C | Naïve | None | T69D (99.7%) | M230L (99.7%) | None | None | D67E (77.2%) | M230L (99.8%) | None | T69D | M230L | |
| T69D (98.1%) | ||||||||||||
| T215A (99.4%) | ||||||||||||
| Indian | IN04 | Naïve | None | None | K101E (99.7%) | None | None | None | K101E (99.9%) | None | NA | NA |
| IN05 | Naïve | None | V75M (7.8%) | None | None | None | V75M (8.6%) | None | None | NA | NA | |
| IN06 | Naïve | None | None | V106M (98.1%) | None | None | None | V106M (100.0%) | None | NA | NA | |
| Y181C (22.8%) | Y181C (23.6%) | |||||||||||
| IN07 | Naïve | None | K65R (3.0%) | V106M (3.9%) | None | None | K65R (2.8%) | V106M (3.6%) | None | NA | NA | |
| K70E (3.4%) | G190S (95.6%) | K70E (3.2%) | G190S (96.1%) | |||||||||
| L74V (66.1%) | L74V (67.6%) | |||||||||||
| M184V (7.5%) | M184V (8.4%) | |||||||||||
| M184I (3.7%) | M184I (3.2%) | |||||||||||
| T215I (3.4%) | T215I (3.3%) | |||||||||||
| IN08 | Naïve | None | None | Y181C (99.5%) | None | None | None | Y181C (99.9%) | None | NA | NA | |
| Ethiopian | ET160 | Naïve | L76V (4.5%) | None | None | None | L76V (4.4%) | None | None | None | None | None |
| ET115 | Naïve | None | M184I (1.1%) | None | None | None | None | None | None | None | None | |
| ET122 | Naïve | None | None | M230L (4.8%) | None | None | None | None | None | None | None | |
| ET155 | Naïve | None | None | None | T66I (3.3%) | None | None | None | T66I (5.8%) | None | None | |
| ET159 | Naïve | None | M184I (1.0%) | None | None | None | None | None | None | None | None | |
| ET171 | Naïve | None | M184I (1.5%) | None | Q148H (1.2%) | None | None | None | None | None | None | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aralaguppe, S.G.; Ambikan, A.T.; Ashokkumar, M.; Kumar, M.M.; Hanna, L.E.; Amogne, W.; Sönnerborg, A.; Neogi, U. MiDRMpol: A High-Throughput Multiplexed Amplicon Sequencing Workflow to Quantify HIV-1 Drug Resistance Mutations against Protease, Reverse Transcriptase, and Integrase Inhibitors. Viruses 2019, 11, 806. https://doi.org/10.3390/v11090806
Aralaguppe SG, Ambikan AT, Ashokkumar M, Kumar MM, Hanna LE, Amogne W, Sönnerborg A, Neogi U. MiDRMpol: A High-Throughput Multiplexed Amplicon Sequencing Workflow to Quantify HIV-1 Drug Resistance Mutations against Protease, Reverse Transcriptase, and Integrase Inhibitors. Viruses. 2019; 11(9):806. https://doi.org/10.3390/v11090806
Chicago/Turabian StyleAralaguppe, Shambhu G., Anoop T. Ambikan, Manickam Ashokkumar, Milner M. Kumar, Luke Elizabeth Hanna, Wondwossen Amogne, Anders Sönnerborg, and Ujjwal Neogi. 2019. "MiDRMpol: A High-Throughput Multiplexed Amplicon Sequencing Workflow to Quantify HIV-1 Drug Resistance Mutations against Protease, Reverse Transcriptase, and Integrase Inhibitors" Viruses 11, no. 9: 806. https://doi.org/10.3390/v11090806
APA StyleAralaguppe, S. G., Ambikan, A. T., Ashokkumar, M., Kumar, M. M., Hanna, L. E., Amogne, W., Sönnerborg, A., & Neogi, U. (2019). MiDRMpol: A High-Throughput Multiplexed Amplicon Sequencing Workflow to Quantify HIV-1 Drug Resistance Mutations against Protease, Reverse Transcriptase, and Integrase Inhibitors. Viruses, 11(9), 806. https://doi.org/10.3390/v11090806