Puumala and Tula Virus Differ in Replication Kinetics and Innate Immune Stimulation in Human Endothelial Cells and Macrophages


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Cultivation
2.2. Cell Lines
2.3. Infection
2.4. Focus Forming Unit Assay
2.5. Quantitative Reverse Transcription PCR
2.6. Immunoblotting
3. Results
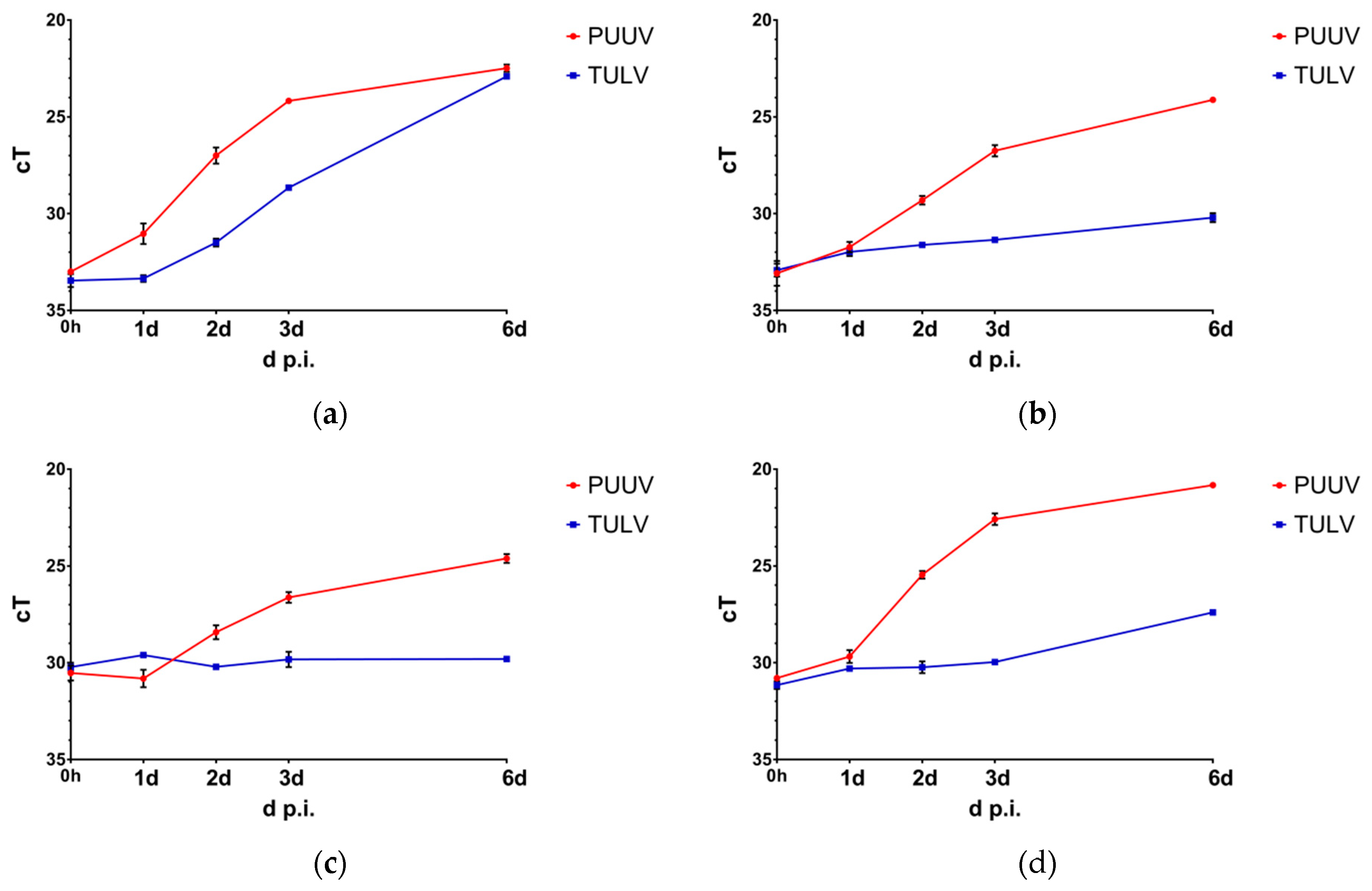
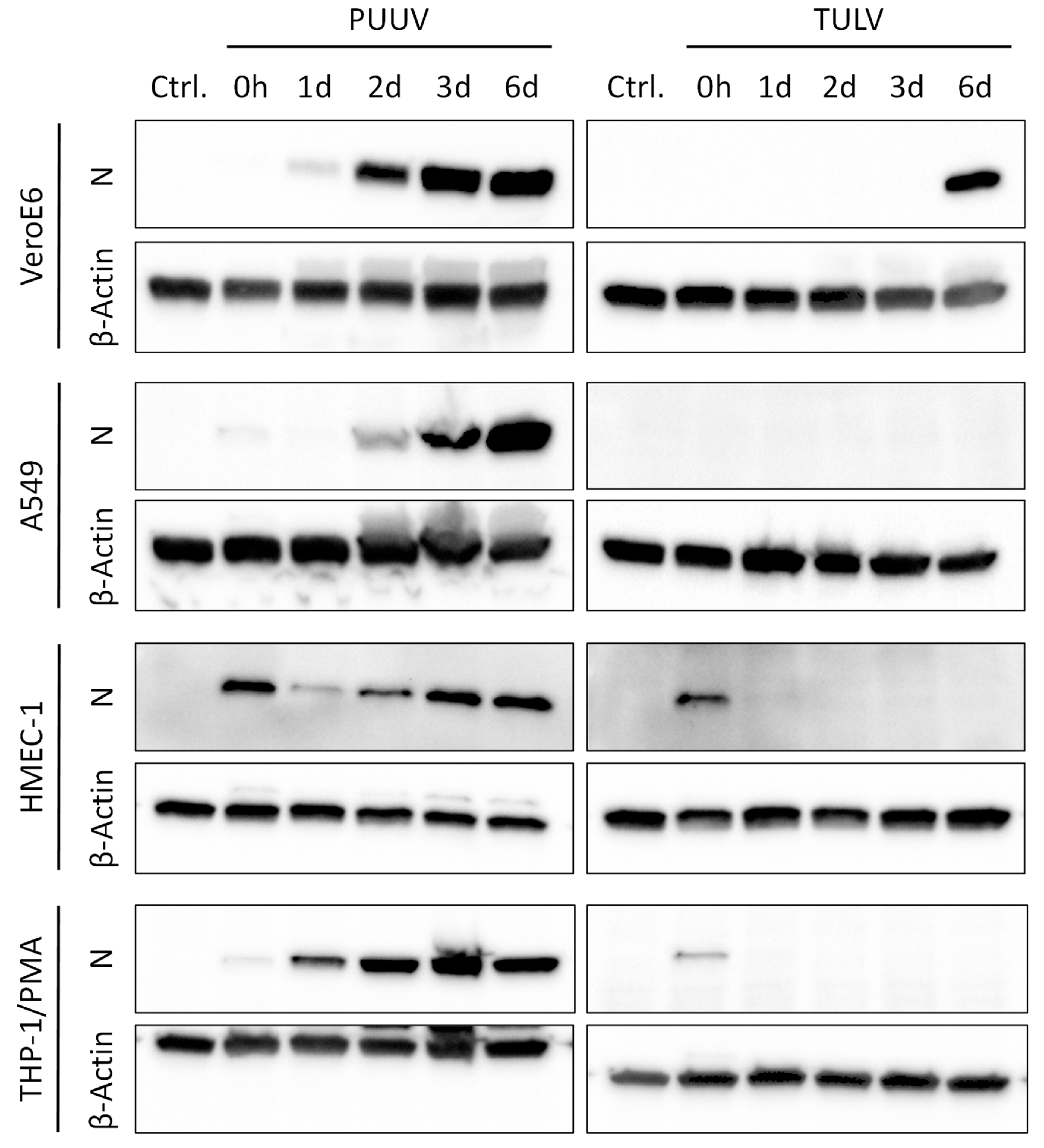
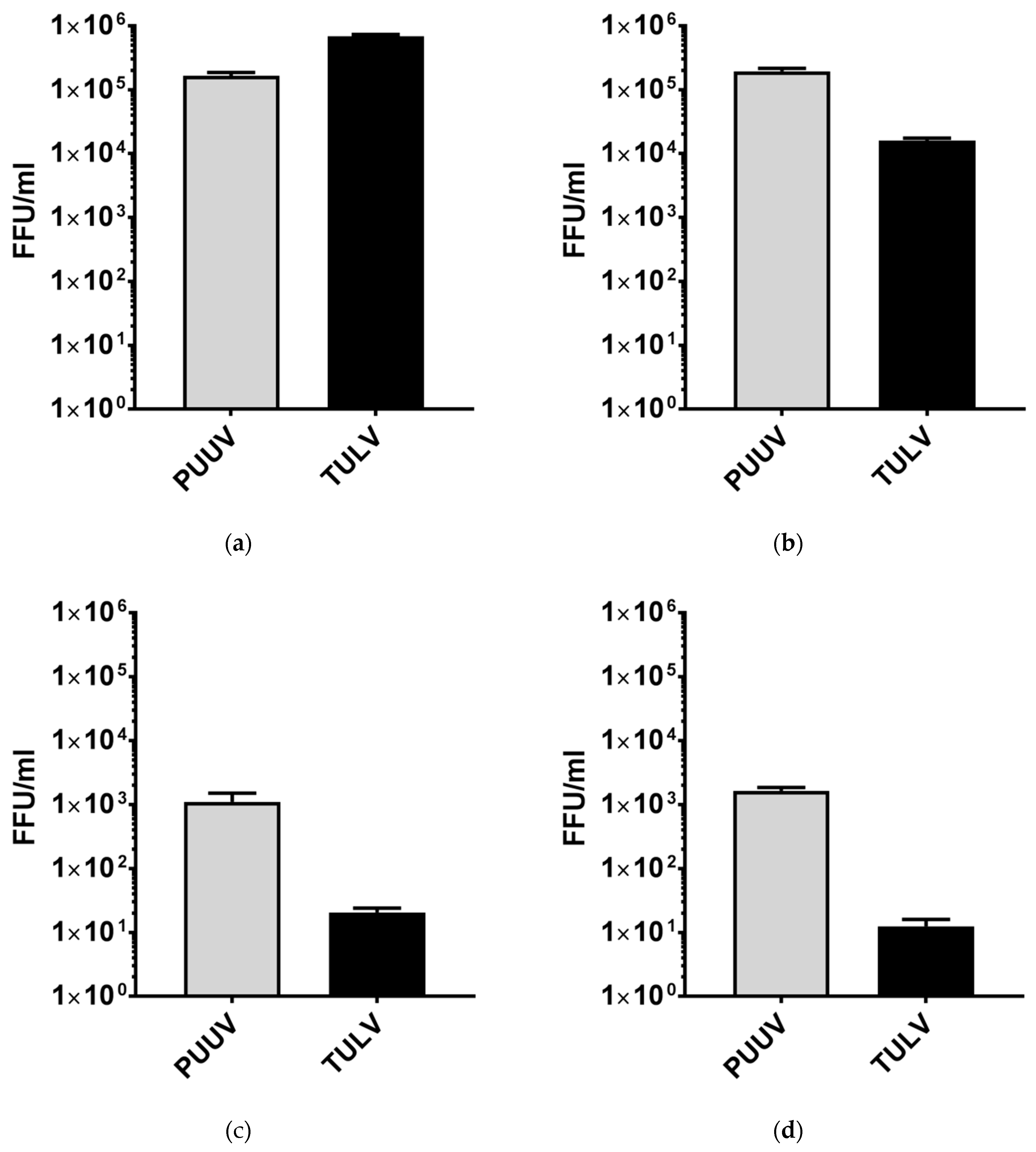
3.1. Puumala Virus Replicates More Efficiently Than Tula Virus in IFN-Competent Cell Types
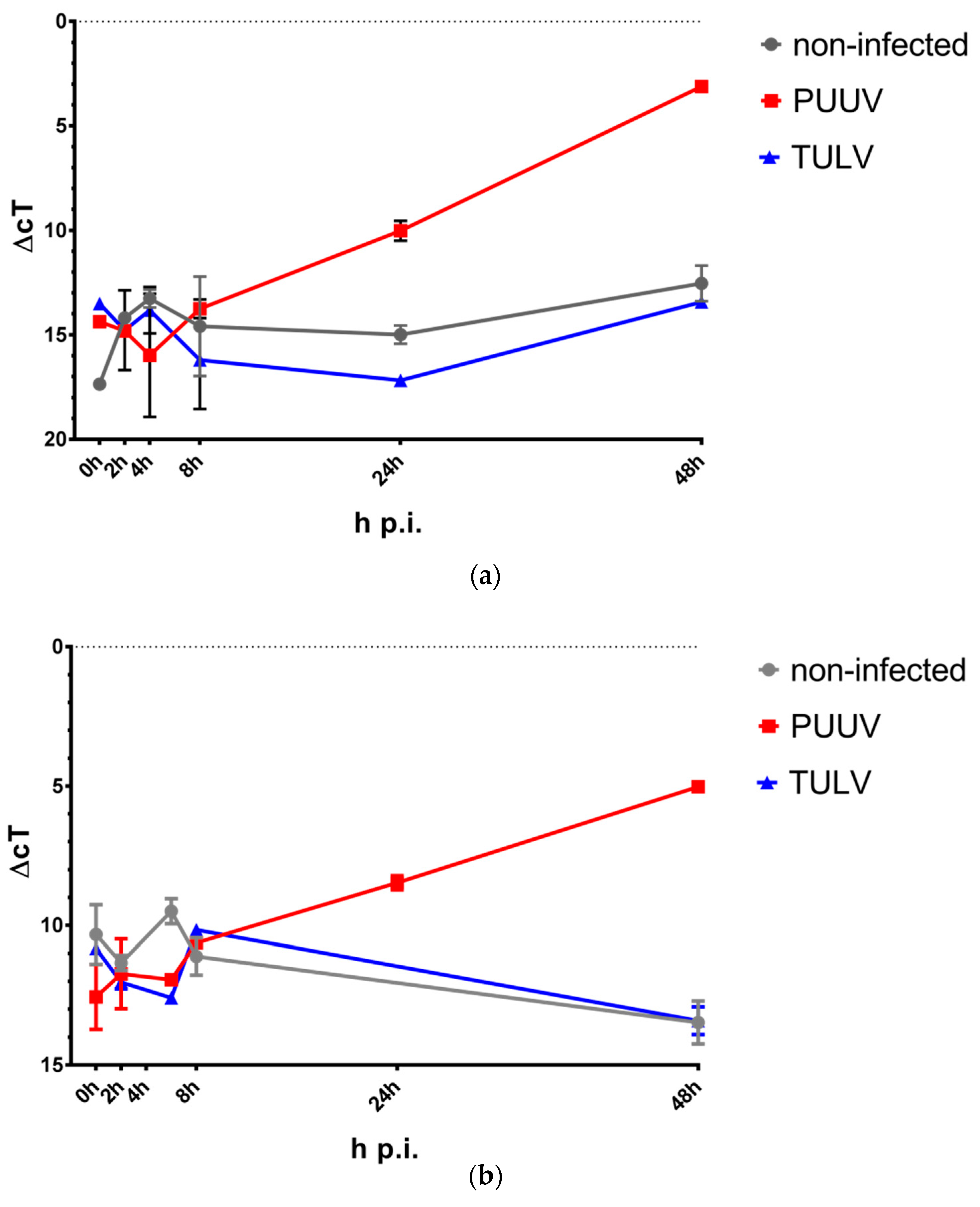
3.2. PUUV Infection Induces IFN-β and IFN-λ Gene Expression
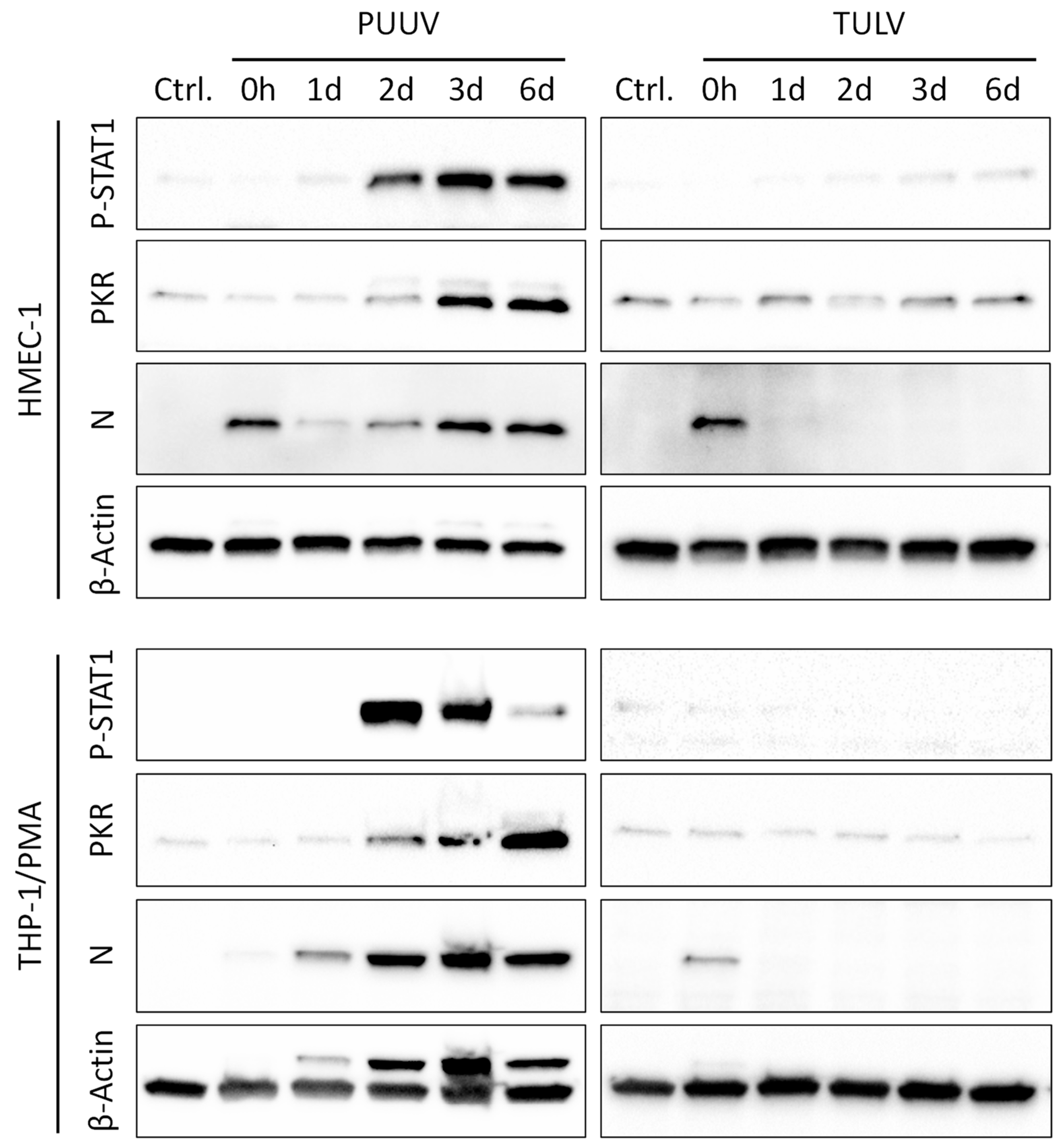
3.3. PUUV but Not TULV Infection Induces Phosphorylation of STAT1 and Interferon-stimulated Gene Expression in Endothelial Cells and Monocytes/Macrophages
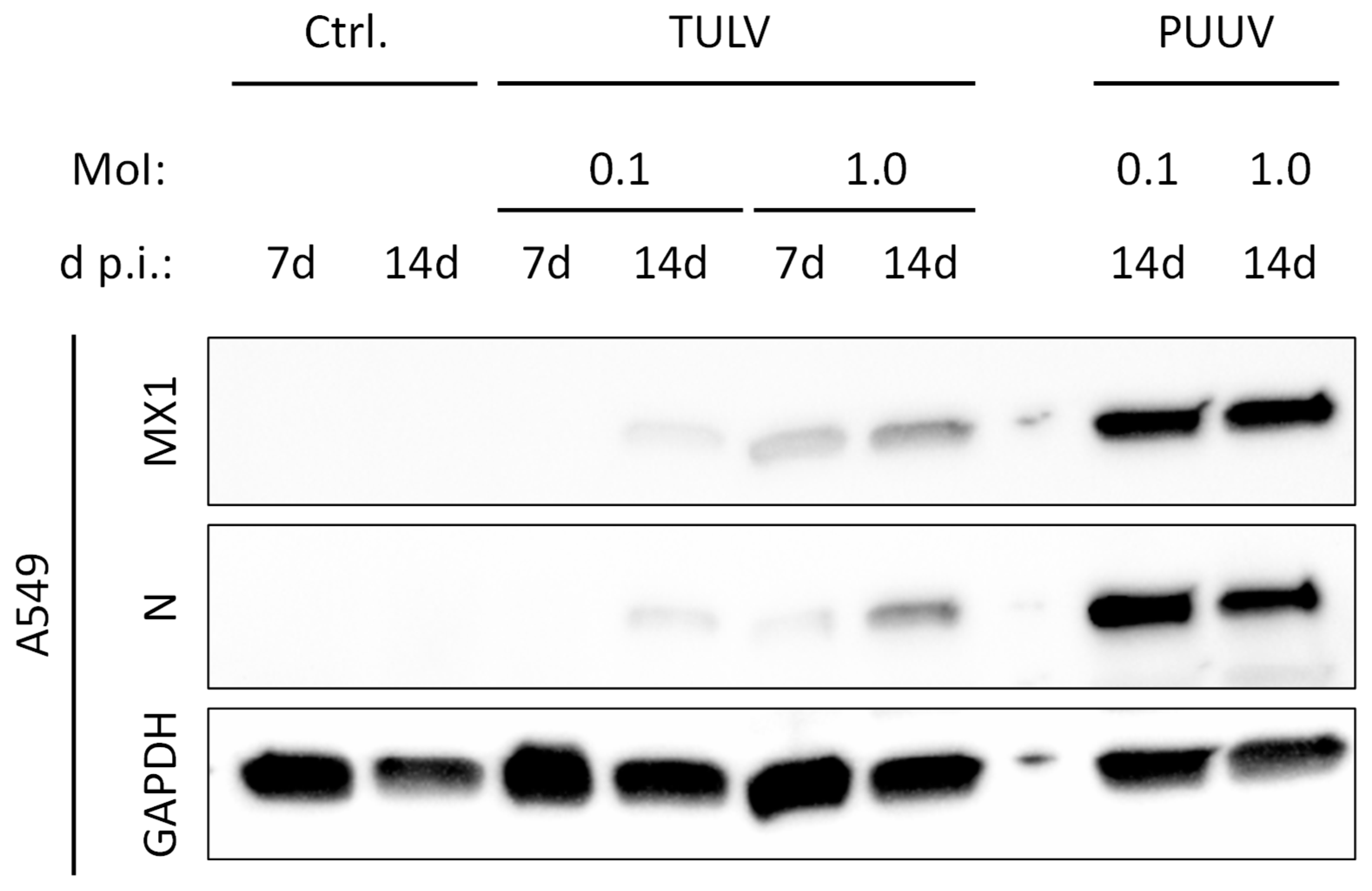
3.4. TULV Replication Induces Only a Weak IFN Response in Permissive A549 Cells in Comparison to PUUV
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kruger, D.H.; Figueiredo, L.T.; Song, J.W.; Klempa, B. Hantaviruses—Globally emerging pathogens. J. Clin. Virol. 2015, 64, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Vaheri, A.; Henttonen, H.; Voutilainen, L.; Mustonen, J.; Sironen, T.; Vapalahti, O. Hantavirus infections in Europe and their impact on public health. Rev. Med. Virol. 2013, 23, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Makary, P.; Kanerva, M.; Ollgren, J.; Virtanen, M.J.; Vapalahti, O.; Lyytikainen, O. Disease burden of Puumala virus infections, 1995–2008. Epidemiol. Infect. 2010, 138, 1484–1492. [Google Scholar] [CrossRef] [PubMed]
- Kruger, D.H.; Ulrich, R.G.; Hofmann, J. Hantaviruses as zoonotic pathogens in Germany. Dtsch. Arztebl. Int. 2013, 110, 461–467. [Google Scholar] [CrossRef]
- Maas, M.; de Vries, A.; van Roon, A.; Takumi, K.; van der Giessen, J.; Rockx, B. High Prevalence of Tula Hantavirus in Common Voles in The Netherlands. Vector Borne Zoonotic Dis. 2017, 17, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Vapalahti, O.; Mustonen, J.; Lundkvist, A.; Henttonen, H.; Plyusnin, A.; Vaheri, A. Hantavirus infections in Europe. Lancet Infect. Dis. 2003, 3, 653–661. [Google Scholar] [CrossRef]
- Schmidt-Chanasit, J.; Essbauer, S.; Petraityte, R.; Yoshimatsu, K.; Tackmann, K.; Conraths, F.J.; Sasnauskas, K.; Arikawa, J.; Thomas, A.; Pfeffer, M.; et al. Extensive host sharing of central European Tula virus. J. Virol. 2010, 84, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Vapalahti, O.; Lundkvist, A.; Kukkonen, S.K.; Cheng, Y.; Gilljam, M.; Kanerva, M.; Manni, T.; Pejcoch, M.; Niemimaa, J.; Kaikusalo, A.; et al. Isolation and characterization of Tula virus, a distinct serotype in the genus Hantavirus, family Bunyaviridae. J. Gen. Virol. 1996, 77 Pt 12, 3063–3067. [Google Scholar] [CrossRef]
- Mertens, M.; Hofmann, J.; Petraityte-Burneikiene, R.; Ziller, M.; Sasnauskas, K.; Friedrich, R.; Niederstrasser, O.; Kruger, D.H.; Groschup, M.H.; Petri, E.; et al. Seroprevalence study in forestry workers of a non-endemic region in eastern Germany reveals infections by Tula and Dobrava-Belgrade hantaviruses. Med. Microbiol. Immunol. 2011, 200, 263–268. [Google Scholar] [CrossRef]
- Klempa, B.; Meisel, H.; Rath, S.; Bartel, J.; Ulrich, R.; Kruger, D.H. Occurrence of renal and pulmonary syndrome in a region of northeast Germany where Tula hantavirus circulates. J. Clin. Microbiol. 2003, 41, 4894–4897. [Google Scholar] [CrossRef]
- Zelena, H.; Mrazek, J.; Kuhn, T. Tula hantavirus infection in immunocompromised host, Czech Republic. Emerg. Infect. Dis. 2013, 19, 1873–1875. [Google Scholar] [CrossRef] [PubMed]
- Reynes, J.M.; Carli, D.; Boukezia, N.; Debruyne, M.; Herti, S. Tula hantavirus infection in a hospitalised patient, France, June 2015. Euro Surveill. 2015, 20. [Google Scholar] [CrossRef] [PubMed]
- Clement, J.; Van Ranst, M. Three vole species and one (?) novel arvicolid hantavirus pathogen: Tula virus revisited. Euro Surveill. 2016, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hepojoki, J.; Vaheri, A.; Strandin, T. The fundamental role of endothelial cells in hantavirus pathogenesis. Front. Microbiol. 2014, 5, 727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackow, E.R.; Gavrilovskaya, I.N. Hantavirus regulation of endothelial cell functions. Thromb. Haemost. 2009, 102, 1030–1041. [Google Scholar] [CrossRef] [PubMed]
- Krautkramer, E.; Zeier, M. Old World hantaviruses: Aspects of pathogenesis and clinical course of acute renal failure. Virus Res. 2014, 187, 59–64. [Google Scholar] [CrossRef]
- Mackow, E.R.; Dalrymple, N.A.; Cimica, V.; Matthys, V.; Gorbunova, E.; Gavrilovskaya, I. Hantavirus interferon regulation and virulence determinants. Virus Res. 2014, 187, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Matthys, V.; Mackow, E.R. Hantavirus regulation of type I interferon responses. Adv. Virol. 2012, 2012, 524024. [Google Scholar] [CrossRef]
- Rang, A. Modulation of innate immune responses by hantaviruses. Crit. Rev. Immunol. 2010, 30, 515–527. [Google Scholar] [CrossRef]
- Ermonval, M.; Baychelier, F.; Tordo, N. What Do We Know about How Hantaviruses Interact with Their Different Hosts? Viruses 2016, 8. [Google Scholar] [CrossRef]
- Alff, P.J.; Gavrilovskaya, I.N.; Gorbunova, E.; Endriss, K.; Chong, Y.; Geimonen, E.; Sen, N.; Reich, N.C.; Mackow, E.R. The pathogenic NY-1 hantavirus G1 cytoplasmic tail inhibits RIG-I- and TBK-1-directed interferon responses. J. Virol. 2006, 80, 9676–9686. [Google Scholar] [CrossRef] [PubMed]
- Geimonen, E.; Neff, S.; Raymond, T.; Kocer, S.S.; Gavrilovskaya, I.N.; Mackow, E.R. Pathogenic and nonpathogenic hantaviruses differentially regulate endothelial cell responses. Proc. Natl. Acad. Sci. USA 2002, 99, 13837–13842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brocato, R.L.; Wahl, V.; Hammerbeck, C.D.; Josleyn, M.D.; McElroy, A.K.; Smith, J.M.; Hooper, J.W. Innate immune responses elicited by Sin Nombre virus or type I IFN agonists protect hamsters from lethal Andes virus infections. J. Gen. Virol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Safronetz, D.; Zivcec, M.; Lacasse, R.; Feldmann, F.; Rosenke, R.; Long, D.; Haddock, E.; Brining, D.; Gardner, D.; Feldmann, H.; et al. Pathogenesis and host response in Syrian hamsters following intranasal infection with Andes virus. PLoS Pathog. 2011, 7, e1002426. [Google Scholar] [CrossRef] [PubMed]
- Schonrich, G.; Kruger, D.H.; Raftery, M.J. Hantavirus-induced disruption of the endothelial barrier: Neutrophils are on the payroll. Front. Microbiol. 2015, 6, 222. [Google Scholar] [CrossRef]
- Sundstrom, K.B.; Nguyen Hoang, A.T.; Gupta, S.; Ahlm, C.; Svensson, M.; Klingstrom, J. Andes Hantavirus-Infection of a 3D Human Lung Tissue Model Reveals a Late Peak in Progeny Virus Production Followed by Increased Levels of Proinflammatory Cytokines and VEGF-A. PLoS ONE 2016, 11, e0149354. [Google Scholar] [CrossRef]
- Ades, E.W.; Candal, F.J.; Swerlick, R.A.; George, V.G.; Summers, S.; Bosse, D.C.; Lawley, T.J. HMEC-1: Establishment of an immortalized human microvascular endothelial cell line. J. Investig. Dermatol. 1992, 99, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Vega, M.; Masso, F.; Paez, A.; Carreon-Torres, E.; Cabrera-Fuentes, H.A.; Fragoso, J.M.; Perez-Hernandez, N.; Martinez, L.O.; Najib, S.; Vargas-Alarcon, G.; et al. Characterization of immortalized human dermal microvascular endothelial cells (HMEC-1) for the study of HDL functionality. Lipids Health Dis. 2018, 17, 44. [Google Scholar] [CrossRef]
- Xu, Y.; Swerlick, R.A.; Sepp, N.; Bosse, D.; Ades, E.W.; Lawley, T.J. Characterization of expression and modulation of cell adhesion molecules on an immortalized human dermal microvascular endothelial cell line (HMEC-1). J. Investig. Dermatol. 1994, 102, 833–837. [Google Scholar] [CrossRef]
- Lee, M.H.; Lalwani, P.; Raftery, M.J.; Matthaei, M.; Lutteke, N.; Kirsanovs, S.; Binder, M.; Ulrich, R.G.; Giese, T.; Wolff, T.; et al. RNA helicase retinoic acid-inducible gene I as a sensor of Hantaan virus replication. J. Gen. Virol. 2011, 92, 2191–2200. [Google Scholar] [CrossRef]
- Oelschlegel, R.; Kruger, D.H.; Rang, A. MxA-independent inhibition of Hantaan virus replication induced by type I and type II interferon in vitro. Virus Res. 2007, 127, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Popugaeva, E.; Witkowski, P.T.; Schlegel, M.; Ulrich, R.G.; Auste, B.; Rang, A.; Kruger, D.H.; Klempa, B. Dobrava-Belgrade hantavirus from Germany shows receptor usage and innate immunity induction consistent with the pathogenicity of the virus in humans. PLoS ONE 2012, 7, e35587. [Google Scholar] [CrossRef]
- Shim, S.H.; Park, M.S.; Moon, S.; Park, K.S.; Song, J.W.; Song, K.J.; Baek, L.J. Comparison of innate immune responses to pathogenic and putative non-pathogenic hantaviruses in vitro. Virus Res. 2011, 160, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Stoltz, M.; Klingstrom, J. Alpha/beta interferon (IFN-alpha/beta)-independent induction of IFN-lambda1 (interleukin-29) in response to Hantaan virus infection. J. Virol. 2010, 84, 9140–9148. [Google Scholar] [CrossRef]
- Witkowski, P.T.; Bourquain, D.; Bankov, K.; Auste, B.; Dabrowski, P.W.; Nitsche, A.; Kruger, D.H.; Schaade, L. Infection of human airway epithelial cells by different subtypes of Dobrava-Belgrade virus reveals gene expression patterns corresponding to their virulence potential. Virology 2016, 493, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Gavrilovskaya, I.N.; Chumakov, M.P.; Apekina, N.S.; Ryltseva, E.V.; Martiyanova, L.I.; Gorbachkova, E.A.; Bernshtein, A.D.; Zakharova, M.A.; Boiko, V.A. Adaptation to laboratory and wild animals of the haemorrhagic fever with renal syndrome virus present in the foci of European U.S.S.R. Brief report. Arch. Virol. 1983, 77, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Kramski, M.; Meisel, H.; Klempa, B.; Kruger, D.H.; Pauli, G.; Nitsche, A. Detection and typing of human pathogenic hantaviruses by real-time reverse transcription-PCR and pyrosequencing. Clin. Chem. 2007, 53, 1899–1905. [Google Scholar] [CrossRef] [PubMed]
- Kramski, M.; Matz-Rensing, K.; Stahl-Hennig, C.; Kaup, F.J.; Nitsche, A.; Pauli, G.; Ellerbrok, H. A novel highly reproducible and lethal nonhuman primate model for orthopox virus infection. PLoS ONE 2010, 5, e10412. [Google Scholar] [CrossRef] [PubMed]
- Schonrich, G.; Rang, A.; Lutteke, N.; Raftery, M.J.; Charbonnel, N.; Ulrich, R.G. Hantavirus-induced immunity in rodent reservoirs and humans. Immunol. Rev. 2008, 225, 163–189. [Google Scholar] [CrossRef]
- Handke, W.; Oelschlegel, R.; Franke, R.; Kruger, D.H.; Rang, A. Hantaan virus triggers TLR3-dependent innate immune responses. J. Immunol. 2009, 182, 2849–2858. [Google Scholar] [CrossRef]
- Xi, Y.; Day, S.L.; Jackson, R.J.; Ranasinghe, C. Role of novel type I interferon epsilon in viral infection and mucosal immunity. Mucosal Immunol. 2012, 5, 610–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemann, E.A.; Gale, M., Jr.; Savan, R. Interferon Lambda Genetics and Biology in Regulation of Viral Control. Front. Immunol. 2017, 8, 1707. [Google Scholar] [CrossRef] [PubMed]
- Strandin, T.; Hepojoki, J.; Laine, O.; Makela, S.; Klingstrom, J.; Lundkvist, A.; Julkunen, I.; Mustonen, J.; Vaheri, A. Interferons Induce STAT1-Dependent Expression of Tissue Plasminogen Activator, a Pathogenicity Factor in Puumala Hantavirus Disease. J. Infect. Dis. 2016, 213, 1632–1641. [Google Scholar] [CrossRef] [PubMed]
- Raftery, M.J.; Lalwani, P.; Krautkrmer, E.; Peters, T.; Scharffetter-Kochanek, K.; Kruger, R.; Hofmann, J.; Seeger, K.; Kruger, D.H.; Schonrich, G. beta2 integrin mediates hantavirus-induced release of neutrophil extracellular traps. J. Exp. Med. 2014, 211, 1485–1497. [Google Scholar] [CrossRef] [PubMed]
- Strandin, T.; Makela, S.; Mustonen, J.; Vaheri, A. Neutrophil Activation in Acute Hemorrhagic Fever With Renal Syndrome Is Mediated by Hantavirus-Infected Microvascular Endothelial Cells. Front. Immunol. 2018, 9, 2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagihara, R.; Silverman, D.J. Experimental infection of human vascular endothelial cells by pathogenic and nonpathogenic hantaviruses. Arch. Virol. 1990, 111, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Goeijenbier, M.; Meijers, J.C.; Anfasa, F.; Roose, J.M.; van de Weg, C.A.; Bakhtiari, K.; Henttonen, H.; Vaheri, A.; Osterhaus, A.D.; van Gorp, E.C.; et al. Effect of Puumala hantavirus infection on human umbilical vein endothelial cell hemostatic function: Platelet interactions, increased tissue factor expression and fibrinolysis regulator release. Front. Microbiol. 2015, 6, 220. [Google Scholar] [CrossRef] [PubMed]
- Temonen, M.; Lankinen, H.; Vapalahti, O.; Ronni, T.; Julkunen, I.; Vaheri, A. Effect of interferon-alpha and cell differentiation on Puumala virus infection in human monocyte/macrophages. Virology 1995, 206, 8–15. [Google Scholar] [CrossRef]
- Krautkramer, E.; Grouls, S.; Stein, N.; Reiser, J.; Zeier, M. Pathogenic old world hantaviruses infect renal glomerular and tubular cells and induce disassembling of cell-to-cell contacts. J. Virol. 2011, 85, 9811–9823. [Google Scholar] [CrossRef]
- Temonen, M.; Vapalahti, O.; Holthofer, H.; Brummer-Korvenkontio, M.; Vaheri, A.; Lankinen, H. Susceptibility of human cells to Puumala virus infection. J. Gen. Virol. 1993, 74 Pt 3, 515–518. [Google Scholar] [CrossRef]
- Kanerva, M.; Melen, K.; Vaheri, A.; Julkunen, I. Inhibition of puumala and tula hantaviruses in Vero cells by MxA protein. Virology 1996, 224, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Kukkonen, S.; Vapalahti, O.; Plyusnin, A.; Lankinen, H.; Vaheri, A. Tula hantavirus infection of Vero E6 cells induces apoptosis involving caspase 8 activation. J. Gen. Virol. 2004, 85, 3261–3268. [Google Scholar] [CrossRef] [PubMed]
- Lutteke, N.; Raftery, M.J.; Lalwani, P.; Lee, M.H.; Giese, T.; Voigt, S.; Bannert, N.; Schulze, H.; Kruger, D.H.; Schonrich, G. Switch to high-level virus replication and HLA class I upregulation in differentiating megakaryocytic cells after infection with pathogenic hantavirus. Virology 2010, 405, 70–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, A.A.; Raftery, M.J.; Giese, T.; Ulrich, R.; Zawatzky, R.; Hippenstiel, S.; Suttorp, N.; Kruger, D.H.; Schonrich, G. Differential antiviral response of endothelial cells after infection with pathogenic and nonpathogenic hantaviruses. J. Virol. 2004, 78, 6143–6150. [Google Scholar] [CrossRef] [PubMed]
- Matthys, V.; Gorbunova, E.E.; Gavrilovskaya, I.N.; Pepini, T.; Mackow, E.R. The C-terminal 42 residues of the Tula virus Gn protein regulate interferon induction. J. Virol. 2011, 85, 4752–4760. [Google Scholar] [CrossRef] [PubMed]
- Resman Rus, K.; Korva, M.; Bogovic, P.; Pal, E.; Strle, F.; Avsic-Zupanc, T. Delayed Interferon Type 1-Induced Antiviral State Is a Potential Factor for Hemorrhagic Fever With Renal Syndrome Severity. J. Infect. Dis. 2018, 217, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef]
- Kraus, A.A.; Priemer, C.; Heider, H.; Kruger, D.H.; Ulrich, R. Inactivation of Hantaan virus-containing samples for subsequent investigations outside biosafety level 3 facilities. Intervirology 2005, 48, 255–261. [Google Scholar] [CrossRef]
- Jaaskelainen, K.M.; Kaukinen, P.; Minskaya, E.S.; Plyusnina, A.; Vapalahti, O.; Elliott, R.M.; Weber, F.; Vaheri, A.; Plyusnin, A. Tula and Puumala hantavirus NSs ORFs are functional and the products inhibit activation of the interferon-beta promoter. J. Med. Virol. 2007, 79, 1527–1536. [Google Scholar] [CrossRef]
- Jaaskelainen, K.M.; Plyusnina, A.; Lundkvist, A.; Vaheri, A.; Plyusnin, A. Tula hantavirus isolate with the full-length ORF for nonstructural protein NSs survives for more consequent passages in interferon-competent cells than the isolate having truncated NSs ORF. Virol. J. 2008, 5, 3. [Google Scholar] [CrossRef]
- Osada, N.; Kohara, A.; Yamaji, T.; Hirayama, N.; Kasai, F.; Sekizuka, T.; Kuroda, M.; Hanada, K. The genome landscape of the african green monkey kidney-derived vero cell line. DNA Res. 2014, 21, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Prescott, J.; Hall, P.; Acuna-Retamar, M.; Ye, C.; Wathelet, M.G.; Ebihara, H.; Feldmann, H.; Hjelle, B. New World hantaviruses activate IFNlambda production in type I IFN-deficient vero E6 cells. PLoS ONE 2010, 5, e11159. [Google Scholar] [CrossRef] [PubMed]
- Sundstrom, K.B.; Stoltz, M.; Lagerqvist, N.; Lundkvist, A.; Nemirov, K.; Klingstrom, J. Characterization of two substrains of Puumala virus that show phenotypes that are different from each other and from the original strain. J. Virol. 2011, 85, 1747–1756. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bourquain, D.; Bodenstein, C.; Schürer, S.; Schaade, L. Puumala and Tula Virus Differ in Replication Kinetics and Innate Immune Stimulation in Human Endothelial Cells and Macrophages. Viruses 2019, 11, 855. https://doi.org/10.3390/v11090855
Bourquain D, Bodenstein C, Schürer S, Schaade L. Puumala and Tula Virus Differ in Replication Kinetics and Innate Immune Stimulation in Human Endothelial Cells and Macrophages. Viruses. 2019; 11(9):855. https://doi.org/10.3390/v11090855
Chicago/Turabian StyleBourquain, Daniel, Clemens Bodenstein, Stefanie Schürer, and Lars Schaade. 2019. "Puumala and Tula Virus Differ in Replication Kinetics and Innate Immune Stimulation in Human Endothelial Cells and Macrophages" Viruses 11, no. 9: 855. https://doi.org/10.3390/v11090855
APA StyleBourquain, D., Bodenstein, C., Schürer, S., & Schaade, L. (2019). Puumala and Tula Virus Differ in Replication Kinetics and Innate Immune Stimulation in Human Endothelial Cells and Macrophages. Viruses, 11(9), 855. https://doi.org/10.3390/v11090855