Caspase-Mediated Cleavage of Human Cortactin during Influenza A Virus Infection Occurs in Its Actin-Binding Domains and Is Associated with Released Virus Titres


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Virus, and Plasmid
2.2. Infection
2.3. Western Blotting
2.4. Quantitative Real-Time RT-PCR
2.5. RNA Interference
2.6. Site-Directed Mutagenesis
2.7. Ectopic Expression
2.8. Microplaque Assay
2.9. Statistical Analysis
3. Results
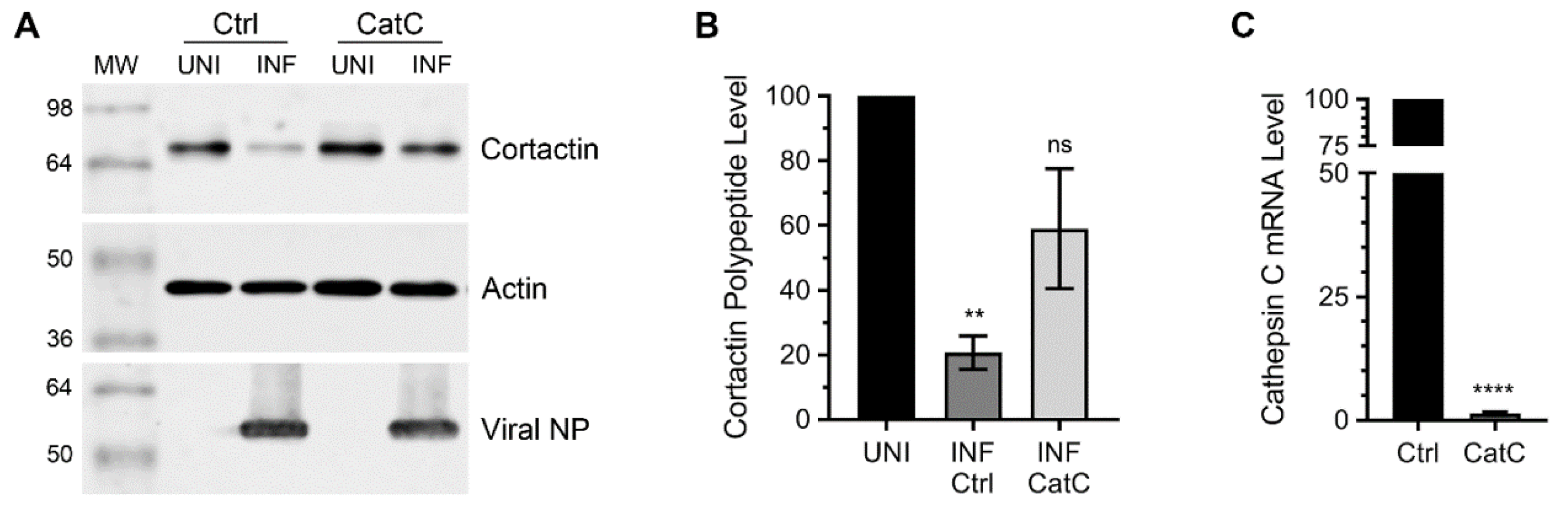
3.1. Lysosomal Protease, Cathepsin C is Involved in Cortactin Degradation during IAV Infection
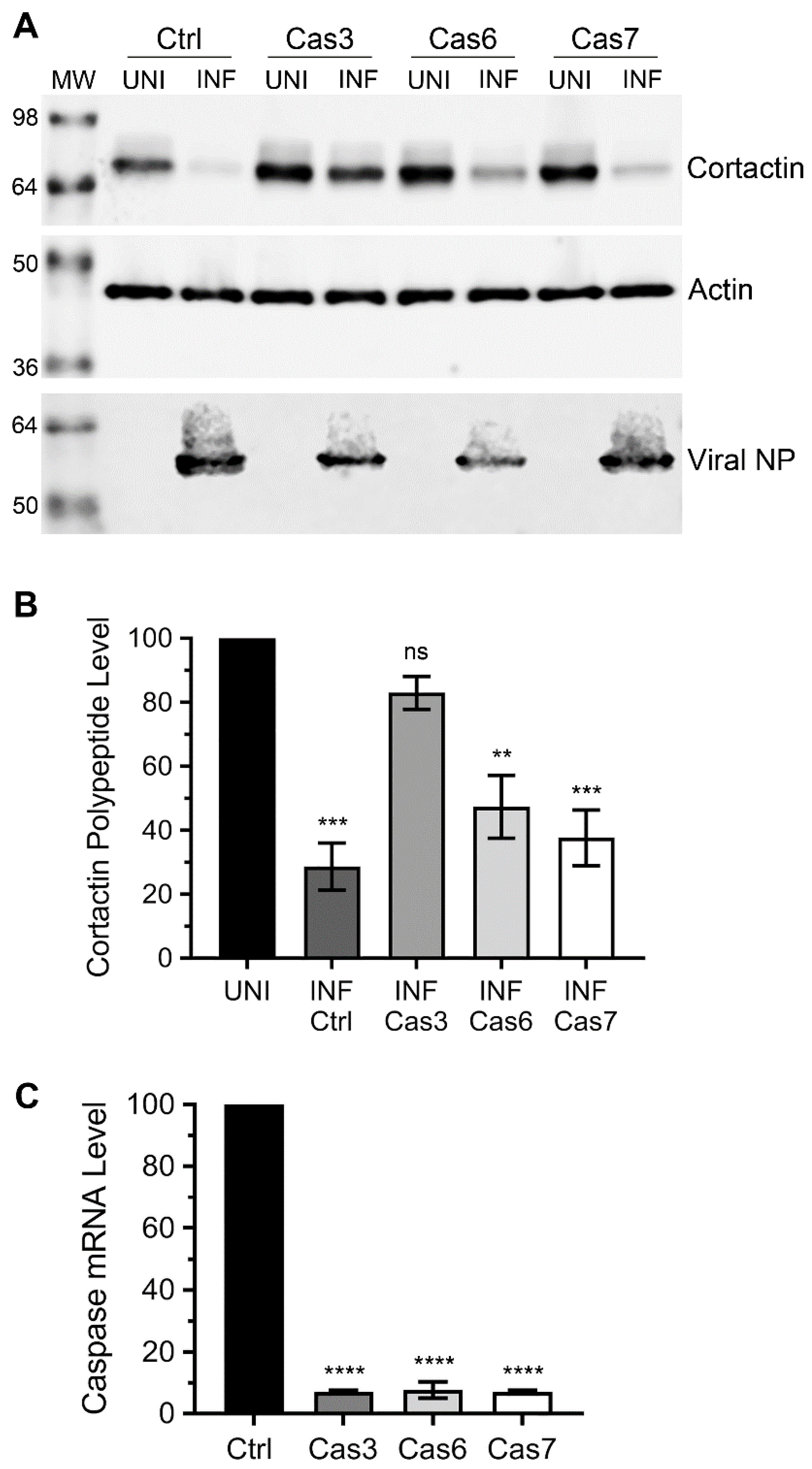
3.2. Caspase-3, not Caspase-6 or Caspase-7 is Involved in Cortactin Degradation during IAV Infection
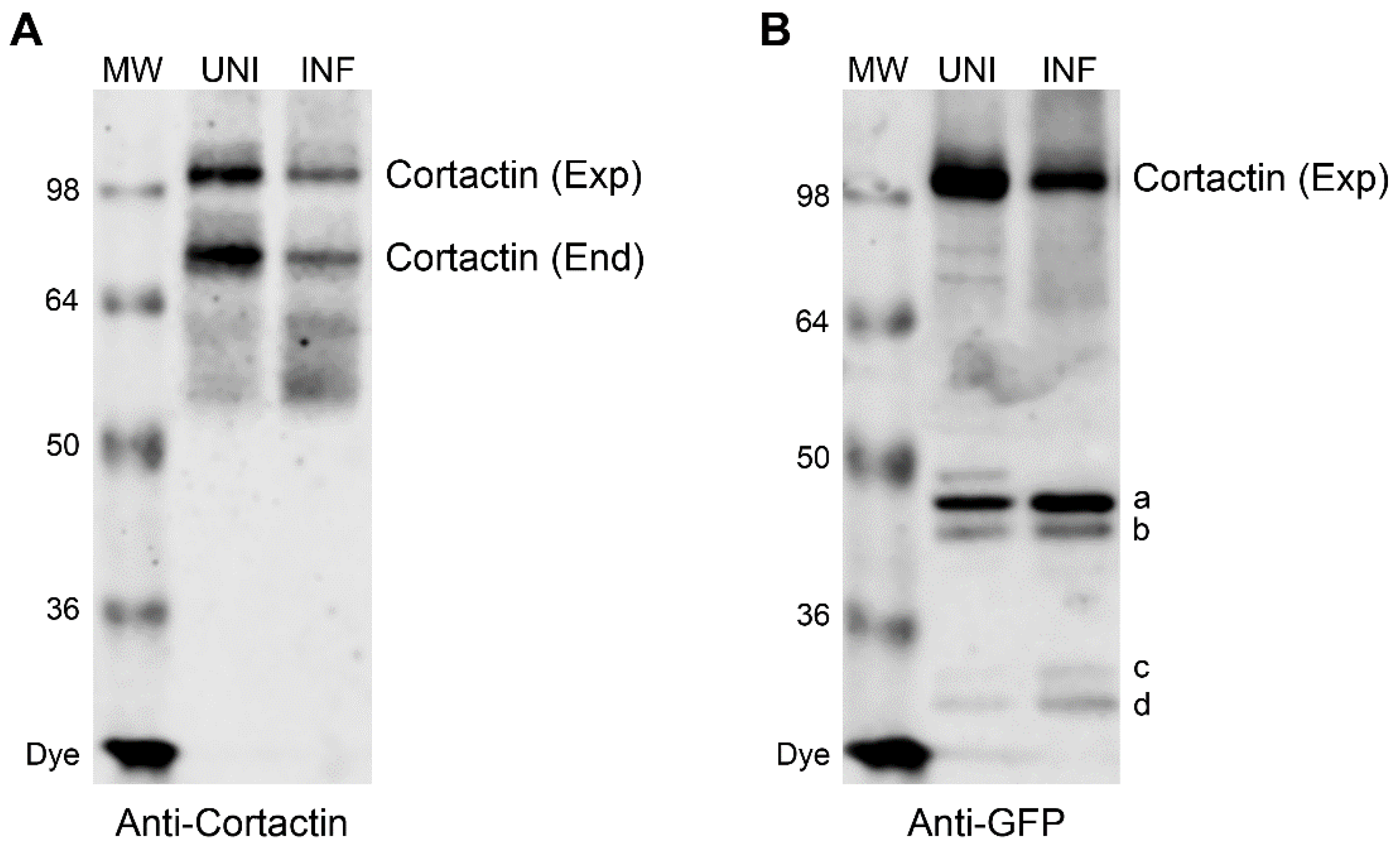
3.3. Caspase Cleavage Sites are Located within Actin-Binding Repeat 1 of Cortactin Polypeptide
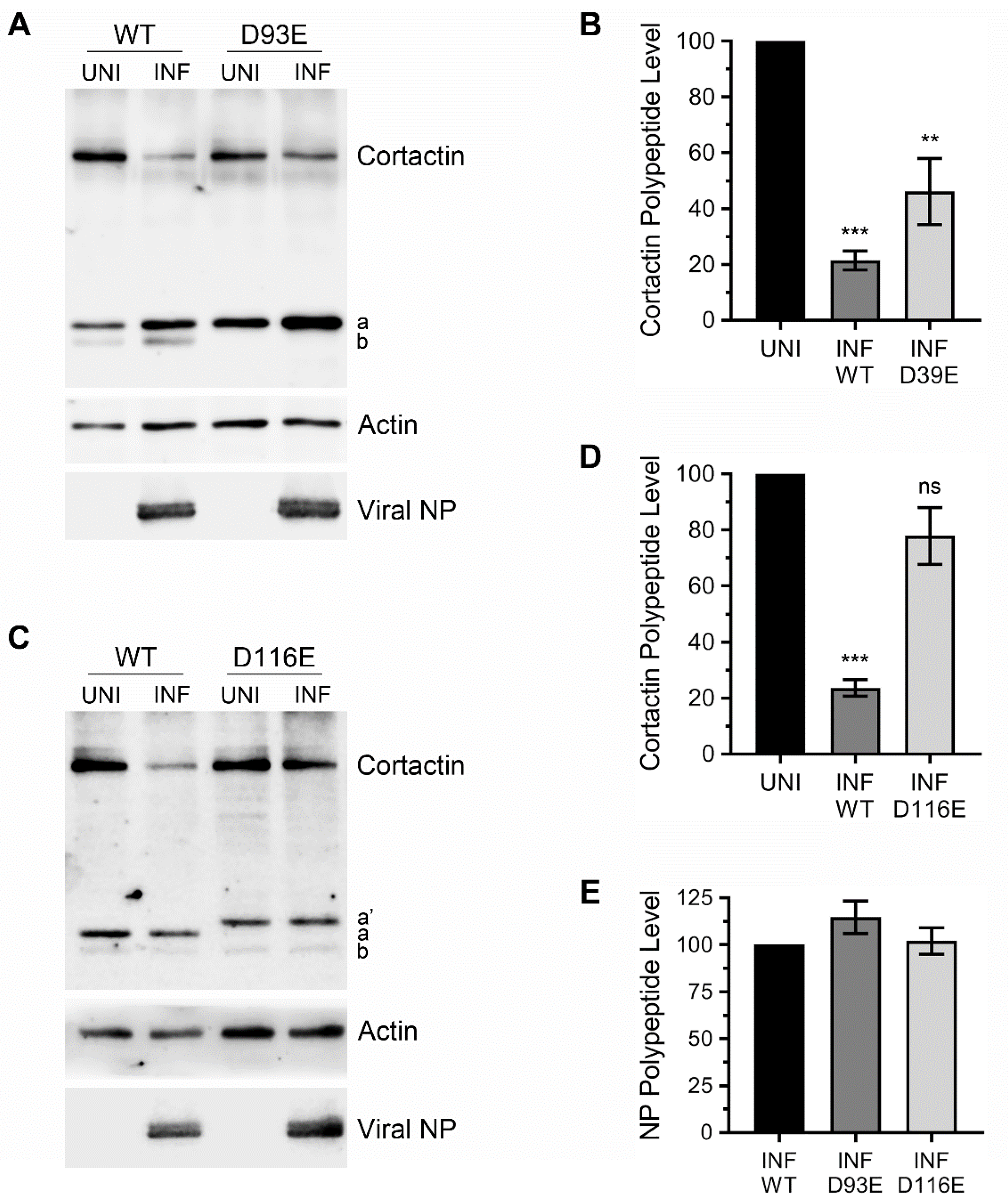
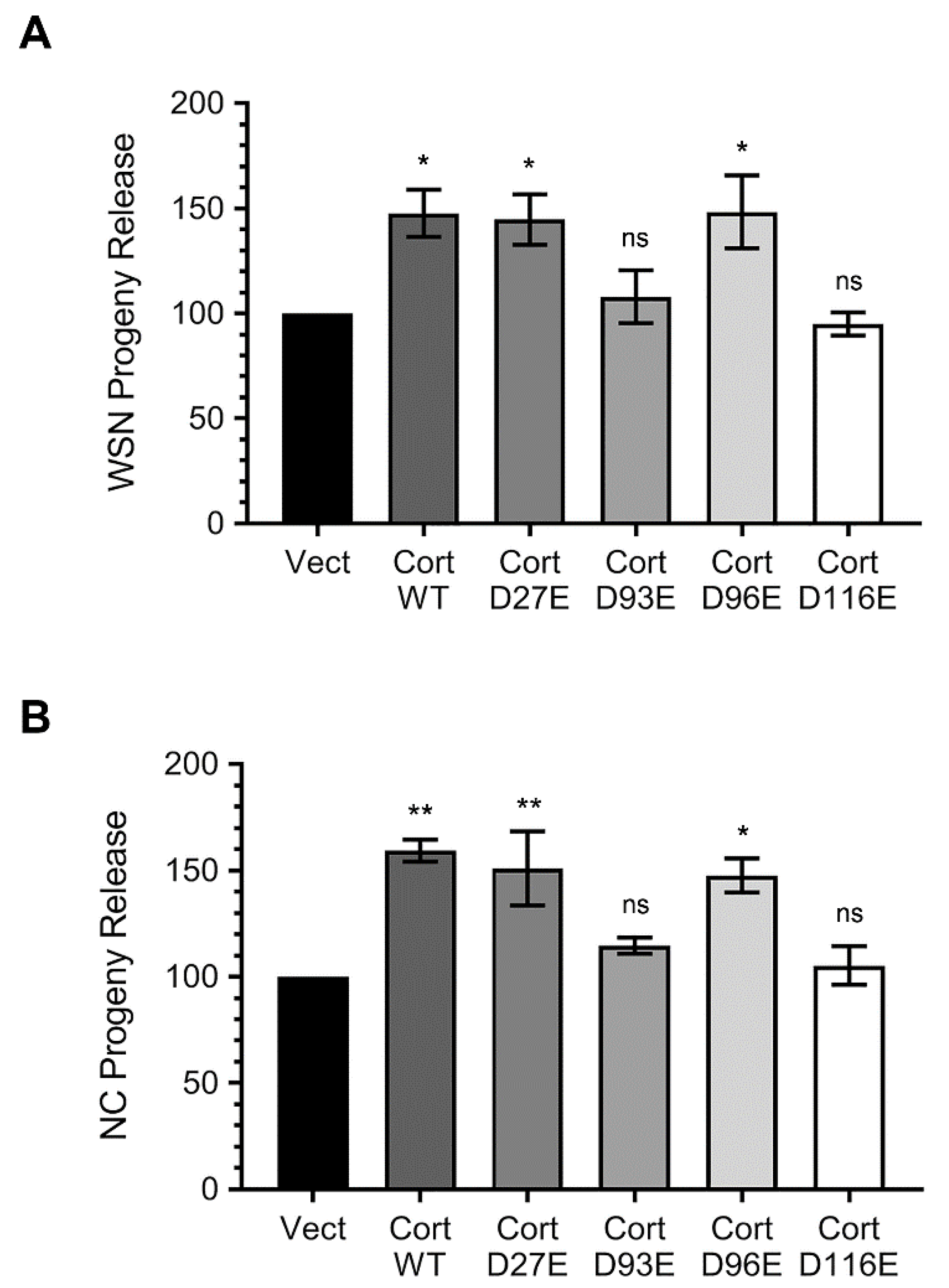
3.4. Ectopically Expressed Cortactin D93E and D116E Mutants Decrease the Amount of IAV Progeny Released from Infected Cells that is Enhanced by Cortactin WT
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bresee, J.; Fitzner, J.; Campbell, H.; Cohen, C.; Cozza, V.; Jara, J.; Krishnan, A.; Lee, V. Progress and Remaining Gaps in Estimating the Global Disease Burden of Influenza. Emerg. Infect. Dis. 2018, 24, 1173–1177. [Google Scholar] [CrossRef]
- Sellers, S.A.; Hagan, R.S.; Hayden, F.G.; Fischer, W.A. The hidden burden of influenza: A review of the extra-pulmonary complications of influenza infection. Influenza Other Respir. Viruses 2017, 11, 372–393. [Google Scholar] [CrossRef]
- Sutton, T.C. The Pandemic Threat of Emerging H5 and H7 Avian Influenza Viruses. Viruses 2018, 10, 461. [Google Scholar] [CrossRef] [Green Version]
- Xue, K.S.; Moncla, L.H.; Bedford, T.; Bloom, J.D. Within-Host Evolution of Human Influenza Virus. Trends Microbiol. 2018, 26, 781–793. [Google Scholar] [CrossRef]
- Sautto, G.A.; Kirchenbaum, G.A.; Ross, T.M. Towards a universal influenza vaccine: different approaches for one goal. Virol. J. 2018, 15, 17. [Google Scholar] [CrossRef] [Green Version]
- Hussain, M.; Galvin, H.D.; Haw, T.Y.; Nutsford, A.N.; Husain, M. Drug resistance in influenza A virus: the epidemiology and management. Infect. Drug Resist. 2017, 10, 121–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.Y.; Husain, M. Caspase-mediated degradation of host cortactin that promotes influenza A virus infection in epithelial cells. Virology 2016, 497, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Schnoor, M.; Stradal, T.E.; Rottner, K. Cortactin: Cell Functions of A Multifaceted Actin-Binding Protein. Trends Cell Biol. 2018, 28, 79–98. [Google Scholar] [CrossRef] [PubMed]
- Van Rossum, A.G.; Schuuring-Scholtes, E.; van Buuren-van Seggelen, V.; Kluin, P.M.; Schuuring, E. Comparative genome analysis of cortactin and HS1: the significance of the F-actin binding repeat domain. BMC Genom. 2005, 6, 15. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Parsons, J.T. Cortactin, an 80/85-kilodalton pp60src substrate, is a filamentous actin-binding protein enriched in the cell cortex. J. Cell Biol. 1993, 120, 1417–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkbride, K.C.; Sung, B.H.; Sinha, S.; Weaver, A.M. Cortactin A multifunctional regulator of cellular invasiveness. Cell Adhes. Migr. 2011, 5, 187–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selbach, M.; Backert, S. Cortactin: an Achilles’ heel of the actin cytoskeleton targeted by pathogens. Trends Microbiol. 2005, 13, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Yuan, Z.G.; Zhang, Y.T.; Yong, S.; Salas-Burgos, A.; Koomen, J.; Olashaw, N.; Parsons, J.T.; Yang, X.J.; Dent, S.R.; et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol. Cell 2007, 27, 197–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artym, V.V.; Matsumoto, K.; Mueller, S.C.; Yamada, K.M. Dynamic membrane remodeling at invadopodia differentiates invadopodia from podosomes. Eur. J. Cell Biol. 2011, 90, 172–180. [Google Scholar] [CrossRef] [Green Version]
- Galvin, H.D.; Husain, M. Influenza A virus-induced host caspase and viral PA-X antagonise the antiviral host factor, histone deacetylase 4. J. Biol. Chem. 2019. [Google Scholar] [CrossRef]
- Chiu, J.; March, P.E.; Lee, R.; Tillett, D. Site-directed, Ligase-Independent Mutagenesis (SLIM): A single-tube methodology approaching 100% efficiency in 4 h. Nucleic. Acids Res. 2004, 32, e174. [Google Scholar] [CrossRef]
- Ferri, K.F.; Kroemer, G. Organelle-specific initiation of cell death pathways. Nat. Cell Biol. 2001, 3, E255–E263. [Google Scholar] [CrossRef]
- Turk, B.; Stoka, V. Protease signalling in cell death: caspases versus cysteine cathepsins. FEBS Lett. 2007, 581, 2761–2767. [Google Scholar] [CrossRef] [Green Version]
- Foghsgaard, L.; Wissing, D.; Mauch, D.; Lademann, U.; Bastholm, L.; Boes, M.; Elling, F.; Leist, M.; Jaattela, M. Cathepsin B acts as a dominant execution protease in tumor cell apoptosis induced by tumor necrosis factor. J. Cell Biol. 2001, 153, 999–1010. [Google Scholar] [CrossRef] [Green Version]
- Guicciardi, M.E.; Deussing, J.; Miyoshi, H.; Bronk, S.F.; Svingen, P.A.; Peters, C.; Kaufmann, S.H.; Gores, G.J. Cathepsin B contributes to TNF-alpha-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J. Clin. Investig. 2000, 106, 1127–1137. [Google Scholar] [CrossRef] [Green Version]
- Roberts, L.R.; Kurosawa, H.; Bronk, S.F.; Fesmier, P.J.; Agellon, L.B.; Leung, W.Y.; Mao, F.; Gores, G.J. Cathepsin B contributes to bile salt-induced apoptosis of rat hepatocytes. Gastroenterology 1997, 113, 1714–1726. [Google Scholar] [CrossRef] [PubMed]
- Jakos, T.; Pislar, A.; Jewett, A.; Kos, J. Cysteine Cathepsins in Tumor-Associated Immune Cells. Front. Immunol. 2019, 10, 2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamaraev, A.V.; Kopeina, G.S.; Prokhorova, E.A.; Zhivotovsky, B.; Lavrik, I.N. Post-translational Modification of Caspases: The Other Side of Apoptosis Regulation. Trends Cell Biol. 2017, 27, 322–339. [Google Scholar] [CrossRef] [PubMed]
- McStay, G.P.; Salvesen, G.S.; Green, D.R. Overlapping cleavage motif selectivity of caspases: implications for analysis of apoptotic pathways. Cell Death Differ. 2008, 15, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; van Raam, B.J.; Salvesen, G.S.; Cieplak, P. Caspase Cleavage Sites in the Human Proteome: CaspDB, a Database of Predicted Substrates. PLoS ONE 2014, 9, e110539. [Google Scholar] [CrossRef] [Green Version]
- Weed, S.A.; Karginov, A.V.; Schafer, D.A.; Weaver, A.M.; Kinley, A.W.; Cooper, J.A.; Parsons, J.T. Cortactin localization to sites of actin assembly in lamellipodia requires interactions with F-actin and the Arp2/3 complex. J. Cell Biol. 2000, 151, 29–40. [Google Scholar] [CrossRef]
- Korkmaz, B.; Caughey, G.H.; Chapple, I.; Gauthier, F.; Hirschfeld, J.; Jenne, D.E.; Kettritz, R.; Lalmanach, G.; Lamort, A.S.; Lauritzen, C.; et al. Therapeutic targeting of cathepsin C: from pathophysiology to treatment. Pharmacol. Ther. 2018, 190, 202–236. [Google Scholar] [CrossRef]
- Loison, F.; Zhu, H.; Karatepe, K.; Kasorn, A.; Liu, P.; Ye, K.; Zhou, J.; Cao, S.; Gong, H.; Jenne, D.E.; et al. Proteinase 3-dependent caspase-3 cleavage modulates neutrophil death and inflammation. J. Clin. Investig. 2014, 124, 4445–4458. [Google Scholar] [CrossRef] [Green Version]
- Ampomah, P.B.; Lim, L.H.K. Influenza A virus-induced apoptosis and virus propagation. Apoptosis 2019. [Google Scholar] [CrossRef]
- Weaver, A.M.; Karginov, A.V.; Kinley, A.W.; Weed, S.A.; Li, Y.; Parsons, J.T.; Cooper, J.A. Cortactin promotes and stabilizes Arp2/3-induced actin filament network formation. Curr. Biol. 2001, 11, 370–374. [Google Scholar] [CrossRef] [Green Version]
- Helgeson, L.A.; Nolen, B.J. Mechanism of synergistic activation of Arp2/3 complex by cortactin and N-WASP. Elife 2013, 2, e00884. [Google Scholar] [CrossRef] [PubMed]
- Meiler, E.; Nieto-Pelegrin, E.; Martinez-Quiles, N. Cortactin tyrosine phosphorylation promotes its deacetylation and inhibits cell spreading. PLoS ONE 2012, 7, e33662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edinger, T.O.; Pohl, M.O.; Stertz, S. Entry of influenza A virus: host factors and antiviral targets. J. Gen. Virol. 2014, 95, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Bedi, S.; Ono, A. Friend or Foe: The Role of the Cytoskeleton in Influenza A Virus Assembly. Viruses 2019, 11, 46. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Domain | P1 | P5–P5′ | Score | P5–P5′ (Post Mutation) | Remark |
|---|---|---|---|---|---|---|
| 1 | NTA | D15 | SIAQD-DAGAD | 0.795 | SIAQE-DAGAD | |
| 2 | D20 | DAGAD-DWETD | 0.510 | DAGAE-DWETD | ||
| 3 | D27 | ETDPD-FVNDV | 0.898 | ETDPE-FVNDV | ||
| 4 | Actin-Binding Repeats | D93 | GVEQD-RMDKS | 0.851 | GVEQE-RMDKS | Full-length recovery |
| 5 | D96 | QDRMD-KSAVG | 0.927 | QDRME-KSAVG | ||
| 6 | D116 | CSQVD-SVRGF | 0.948 | CSQVE-SVRGF | Full-length recovery | |
| 7 | D130 | GVQMD-RVDQS | 0.828 | GVQME-RVDQS | ||
| 8 | D133 | MDRVD-QSAVG | 0.851 | MDRVE-QSAVG | ||
| 9 | D153 | ASQKD-YSSGF | 0.881 | ASQKE-YSSGF | ||
| 10 | D167 | GVQAD-RVDKS | 0.775 | GVQAE-RVDKS | Failed to generate | |
| 11 | D170 | ADRVD-KSAVG | 0.904 | ADRVE-KSAVG | ||
| 12 | D177 | AVGFD-YQGKT | 0.916 | AVGFE-YQGKT | ||
| 13 | D190 | ESQRD-YSKGF | 0.890 | ESQRE-YSKGF | ||
| 14 | D207 | KDKVD-KSAVG | 0.925 | KDKVE-KSAVG | ||
| 15 | D227 | ESQKD-YVKGF | 0.800 | ESQKE-YVKGF | ||
| 16 | D241 | GVQTD-RQDKC | 0.788 | GVQTE-RQDKC | ||
| 17 | D244 | TDRQD-KCALG | 0.642 | TDRQE-KCALG | ||
| 18 | D264 | ESQKD-YKTGF | 0.794 | ESQKE-YKTGF | ||
| 19 | D281 | SERQD-SAAVG | 0.963 | SERQE-SAAVG | ||
| 20 | D288 | AVGFD-YKEKL | 0.914 | AVGFE-YKEKL | ||
| 21 | D301 | ESQQD-YSKGF | 0.955 | ESQQE-YSKGF | ||
| 22 | D315 | GVQKD-RMDKN | 0.536 | GVQKE-RMDKN | ||
| 23 | D318 | KDRMD-KNAST | 0.884 | KDRME-KNAST | ||
| 24 | Helical | D326 | STFED-VTQVS | 0.636 | STFEE-VTQVS | |
| 25 | D365 | KEQED-RRKAE | 0.595 | KEQEE-RRKAE | ||
| 26 | Proline-Rich | D423 | PVYED-AASFK | 0.792 | PVYEE-AASFK | |
| 27 | D452 | MEAAD-YREAS | 0.751 | MEAAE-YREAS | ||
| 28 | D483 | YPAED-STYDE | 0.711 | YPAEE-STYDE | ||
| 29 | D487 | DSTYD-EYEND | 0.767 | DSTYE-EYEND | ||
| 30 | D492 | EYEND-LGITA | 0.581 | EYENE-LGITA | Full-length mobility shift ↓ | |
| 31 | SH3 | D502 | VALYD-YQAAG | 0.901 | VALYE-YQAAG | |
| 32 | D508 | QAAGD-DEISF | 0.578 | QAAGE-DEISF | ||
| 33 | D516 | SFDPD-DIITN | 0.572 | SFDPE-DIITN | Full-length mobility shift ↑ | |
| 34 | D526 | IEMID-DGWWR | 0.829 | IEMIE-DGWWR |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, D.-Y.; Husain, M. Caspase-Mediated Cleavage of Human Cortactin during Influenza A Virus Infection Occurs in Its Actin-Binding Domains and Is Associated with Released Virus Titres. Viruses 2020, 12, 87. https://doi.org/10.3390/v12010087
Chen D-Y, Husain M. Caspase-Mediated Cleavage of Human Cortactin during Influenza A Virus Infection Occurs in Its Actin-Binding Domains and Is Associated with Released Virus Titres. Viruses. 2020; 12(1):87. https://doi.org/10.3390/v12010087
Chicago/Turabian StyleChen, Da-Yuan, and Matloob Husain. 2020. "Caspase-Mediated Cleavage of Human Cortactin during Influenza A Virus Infection Occurs in Its Actin-Binding Domains and Is Associated with Released Virus Titres" Viruses 12, no. 1: 87. https://doi.org/10.3390/v12010087
APA StyleChen, D. -Y., & Husain, M. (2020). Caspase-Mediated Cleavage of Human Cortactin during Influenza A Virus Infection Occurs in Its Actin-Binding Domains and Is Associated with Released Virus Titres. Viruses, 12(1), 87. https://doi.org/10.3390/v12010087