A Systematic Evaluation of High-Throughput Sequencing Approaches to Identify Low-Frequency Single Nucleotide Variants in Viral Populations

 ,
, 
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of DNA and RNA Populations
2.2. Polymerase Chain Reaction (PCR) Optimisation
2.3. Illumina Sequencing
2.4. Bioinformatic Analysis
2.4.1 Sequence Alignment and Assessment of High-Throughput Sequencing (HTS) Pipeline Performance
3. Results
3.1. PCR Cycle Optimisation and Illumina Sequencing
3.1.1 Coverage
3.2. The Effect of Input Nucleic Acid and Aligner Choice on the Accuracy of Single-Nucleotide Variant (SNV) Calling
3.3. The Effect of Replicate Combinations and qScore Choice on the Accuracy of SNV Calling
3.3.1. RNA Input
3.3.2. DNA Input
3.4. The Effect of Replicate Combinations and Read Length Choice on the Accuracy of SNV Calling
3.4.1. RNA Input
3.4.2. DNA Input
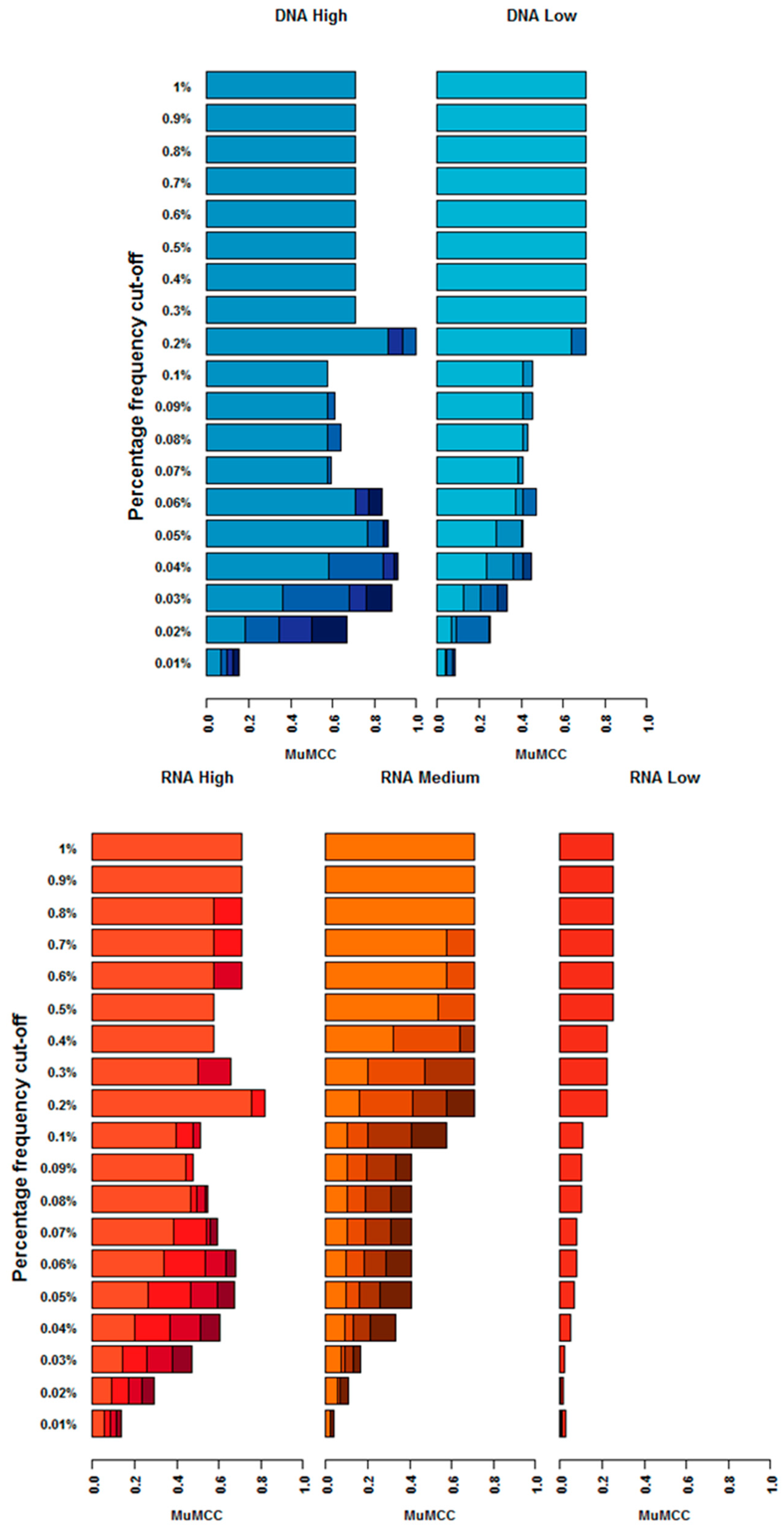
3.5. How Does Frequency Cut-Off Impact the Accuracy of Variant Calling?
3.5.1. RNA Input
3.5.2. DNA Input
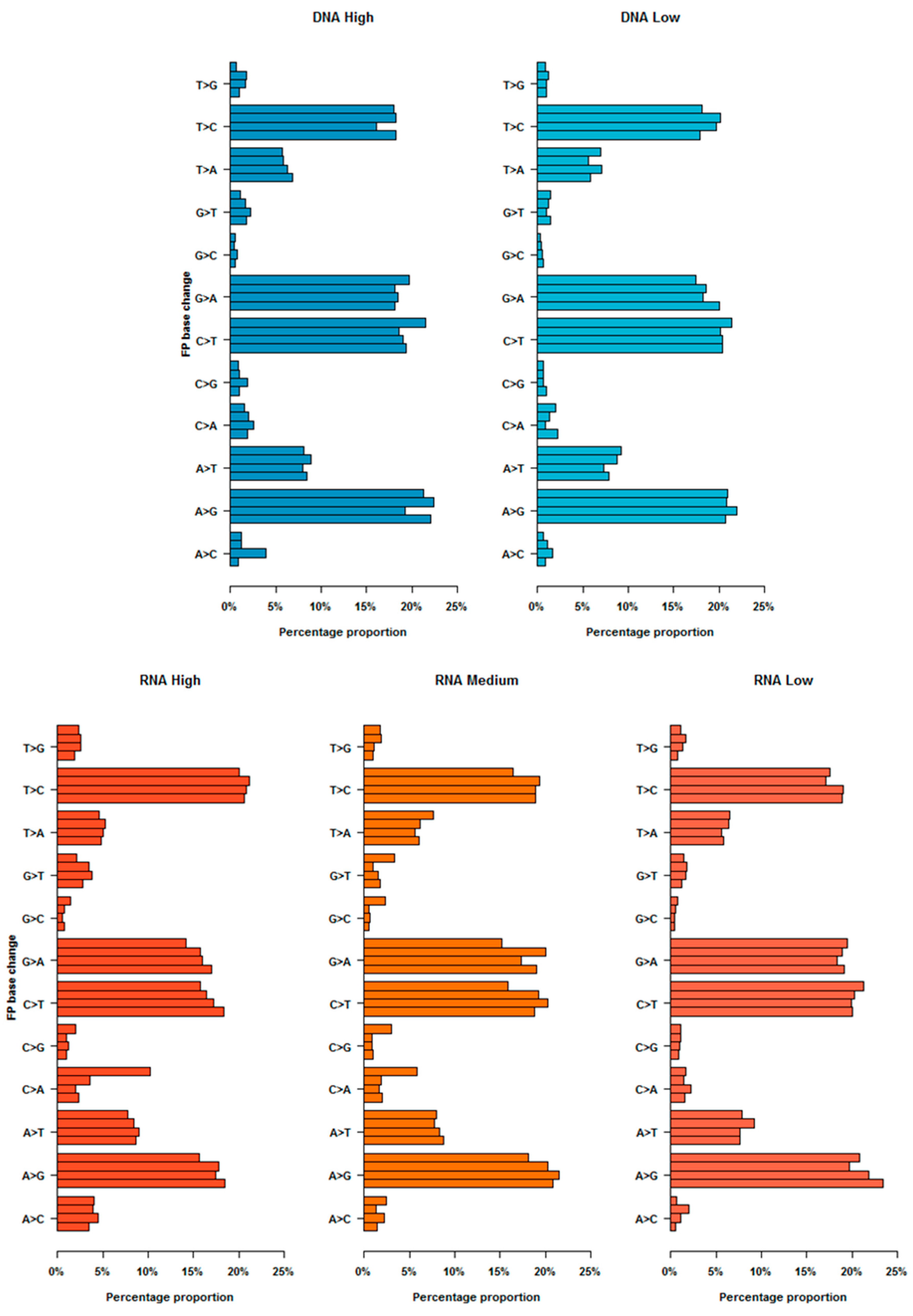
3.6. False Positive Patterns and Distrubutions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Koboldt, D.C.; Steinberg, K.M.; Larson, D.E.; Wilson, R.K.; Mardis, E. The Next-Generation Sequencing Revolution and Its Impact on Genomics. Cell 2014, 155, 27–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, B.; Kang, J.; Kibukawa, M.; Chen, L.; Qiu, P.; Lahser, F.; Marton, M.; Levitan, D. Development and Validation of a Template-Independent Next-Generation Sequencing Assay for Detecting Low-Level Resistance-Associated Variants of Hepatitis C Virus. J. Mol. Diagn. 2016, 18, 643–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrier, M.; Desire, N.; Storto, A.; Todesco, E.; Rodriguez, C.; Bertine, M.; Le Hingrat, Q.; Visseaux, B.; Calvez, V.; Descamps, D.; et al. Evaluation of different analysis pipelines for the detection of HIV-1 minority resistant variants. PLoS ONE 2018, 13, e0198334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dilcher, M.; Barratt, K.; Douglas, J.; Strathdee, A.; Anderson, T.; Werno, A. Monitoring Viral Genetic Variation as a Tool To Improve Molecular Diagnostics for Mumps Virus. J. Clin. Microbiol. 2018, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, W.; Ganusov, V.V.; Giorgi, E.E.; Hraber, P.T.; Keele, B.F.; Leitner, T.; Han, C.S.; Gleasner, C.D.; Green, L.; Lo, C.C.; et al. Transmission of single HIV-1 genomes and dynamics of early immune escape revealed by ultra-deep sequencing. PLoS ONE 2010, 5, e12303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon-Loriere, E.; Faye, O.; Faye, O.; Koivogui, L.; Magassouba, N.; Keita, S.; Thiberge, J.M.; Diancourt, L.; Bouchier, C.; Vandenbogaert, M.; et al. Distinct lineages of Ebola virus in Guinea during the 2014 West African epidemic. Nature 2015, 524, 102–104. [Google Scholar] [CrossRef] [Green Version]
- Wohl, S.; Metsky, H.C.; Schaffner, S.F.; Piantadosi, A.; Burns, M.; Lewnard, J.A.; Chak, B.; Krasilnikova, L.A.; Siddle, K.J.; Matranga, C.B.; et al. Combining genomics and epidemiology to track mumps virus transmission in the United States. PLoS Biol. 2020, 18, e3000611. [Google Scholar] [CrossRef] [Green Version]
- Wright, C.F.; Morelli, M.J.; Thebaud, G.; Knowles, N.J.; Herzyk, P.; Paton, D.J.; Haydon, D.T.; King, D.P. Beyond the consensus: Dissecting within-host viral population diversity of foot-and-mouth disease virus by using next-generation genome sequencing. J. Virol. 2011, 85, 2266–2275. [Google Scholar] [CrossRef] [Green Version]
- King, D.J.; Freimanis, G.L.; Orton, R.J.; Waters, R.A.; Haydon, D.T.; King, D.P. Investigating intra-host and intra-herd sequence diversity of foot-and-mouth disease virus. Infect. Genet. Evol. 2016, 44, 286–292. [Google Scholar] [CrossRef] [Green Version]
- McInerney, P.; Adams, P.; Hadi, M.Z. Error Rate Comparison during Polymerase Chain Reaction by DNA Polymerase. Mol. Biol. Int. 2014, 2014, 287430. [Google Scholar] [CrossRef] [Green Version]
- Orton, R.J.; Wright, C.F.; Morelli, M.J.; King, D.J.; Paton, D.J.; King, D.P.; Haydon, D.T. Distinguishing low frequency mutations from RT-PCR and sequence errors in viral deep sequencing data. BMC Genom. 2015, 16, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelbart, M.; Harari, S.; Ben-Ari, Y.A.; Kustin, T.; Wolf, D.; Mandelboim, M.; Mor, O.; Pennings, P.; Stern, A. AccuNGS: Detecting ultra-rare variants in viruses from clinical samples. bioRxiv 2019. [Google Scholar] [CrossRef]
- Acevedo, A.; Andino, R. Library preparation for highly accurate population sequencing of RNA viruses. Nat. Protoc. 2014, 9, 1760–1769. [Google Scholar] [CrossRef] [Green Version]
- Jabara, C.B.; Jones, C.D.; Roach, J.; Anderson, J.A.; Swanstrom, R. Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc. Natl. Acad. Sci. USA 2011, 108, 20166–20171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodin, J.; Hedskog, C.; Heddini, A.; Benard, E.; Neher, R.A.; Mild, M.; Albert, J. Challenges with using primer IDs to improve accuracy of next generation sequencing. PLoS ONE 2015, 10, e0119123. [Google Scholar] [CrossRef] [Green Version]
- Cacciabue, M.; Currá, A.; Carrillo, E.; König, G.; Gismondi, M.I. A beginner’s guide for FMDV quasispecies analysis: Sub-consensus variant detection and haplotype reconstruction using next-generation sequencing. Brief. Bioinform. 2020, 21, 1766–1775. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. Available online: https://arxiv.org/abs/1303.3997 (accessed on 15 October 2020).
- Marco-Sola, S.; Sammeth, M.; Guigo, R.; Ribeca, P. The GEM mapper: Fast, accurate and versatile alignment by filtration. Nat. Methods 2012, 9, 1185–1188. [Google Scholar] [CrossRef]
- Alioto, T.S.; Buchhalter, I.; Derdak, S.; Hutter, B.; Eldridge, M.D.; Hovig, E.; Heisler, L.E.; Beck, T.A.; Simpson, J.T.; Tonon, L.; et al. A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nat. Commun. 2015, 6, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbist, B.M.P.; Thys, K.; Reumers, J.; Wetzels, Y.; van der Borght, K.; Talloen, W.; Aerssens, J.; Clement, L.; Thas, O. VirVarSeq: A low-frequency virus variant detection pipeline for Illumina sequencing using adaptive base-calling accuracy filtering. Bioinformatics 2015, 31, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferretti, L.; Tennakoon, C.; Silesian, A.; Ribeca, G.F.A. SiNPle: Fast and Sensitive Variant Calling for Deep Sequencing Data. Genes 2019, 10, 561. [Google Scholar] [CrossRef] [Green Version]
- Ellard, F.M.; Drew, J.; Blakemore, W.E.; Stuart, D.I.; King, A.M.Q. Evidence for the role of His-142 of protein 1C in the acid-induced disassembly of foot-and-mouth disease virus capsids. J. Gen. Virol. 1999, 80 Pt 8, 1911–1918. [Google Scholar] [CrossRef]
- King, D.; The Pirbright Institute, Woking, Surrey, UK. Unpublished work. 2020.
- Cottam, E.M.; Haydon, D.T.; Paton, D.J.; Gloster, J.; Wilesmith, J.W.; Ferris, N.P.; Hutchings, G.H.; King, D.P. Molecular epidemiology of the foot-and-mouth disease virus outbreak in the United Kingdom in 2001. J. Virol. 2006, 80, 11274–11282. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Hurtle, W.; Rowland, J.M.; Casteran, K.A.; Bucko, S.M.; Grau, F.R.; Valdazo-Gonzalez, B.; Knowles, N.J.; King, D.P.; Beckham, T.R.; et al. Development of a universal RT-PCR for amplifying and sequencing the leader and capsid-coding region of foot-and-mouth disease virus. J. Virol. Methods 2013, 189, 70–76. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data (Version 0.11.8) [Software]. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 15 October 2020).
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [Green Version]
- Joshi, N.; Fass, J. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files (Version 1.33) [Software]. 2011. Available online: https://github.com/najoshi/sickle (accessed on 15 October 2020).
- Ponsting, N.; Ning, Z. SMALT Alignment Tool (Version 0.7.6) [Software]. 2012. Available online: https://www/sanger.ac.uk/tool/smalt-0/ (accessed on 15 October 2020).
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinform. 2014, 47, 11–12. [Google Scholar] [CrossRef]
- Liu, S.L.; Rodrigo, A.G.; Shankarappa, R.; Learn, G.H.; Hsu, L.; Davidov, O.; Zhao, L.P.; Mullins, J.I. HIV quasispecies and resampling. Science (New York) 1996, 273, 415–416. [Google Scholar] [CrossRef] [PubMed]
- Miranda, J.A.; Steward, G.F. Variables influencing the efficiency and interpretation of reverse transcription quantitative PCR (RT-qPCR): An empirical study using Bacteriophage MS2. J. Virol. Methods 2017, 241, 1–10. [Google Scholar] [CrossRef]
- Robasky, K.; Lewis, N.E.; Church, G.M. The role of replicates for error mitigation in next-generation sequencing. Nat. Rev. Genet. 2014, 15, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Dessilly, G.; Goeminne, L.; Vandenbroucke, A.T.; Dufrasne, F.E.; Martin, A.; Kabamba-Mukabi, B. First evaluation of the next-generation sequencing platform for the detection of HIV-1 drug resistance mutations in Belgium. PLoS ONE 2018, 13, e0209561. [Google Scholar] [CrossRef] [PubMed]
- Operario, D.J.; Koeppel, A.F.; Turner, S.D.; Bao, Y.; Pholwat, S.; Banu, S.; Foongladda, S.; Mpagama, S.; Gratz, J.; Ogarkov, O.; et al. Prevalence and extent of heteroresistance by next generation sequencing of multidrug-resistant tuberculosis. PLoS ONE 2017, 12, e0176522. [Google Scholar]
- Pfeiffer, F.; Gröber, C.; Blank, M.; Händler, K.; Beyer, M.; Schultze, J.L.; Mayer, G. Systematic evaluation of error rates and causes in short samples in next-generation sequencing. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Original | Site-Directed Mutagenesis | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| pT7S3 Wild Type | pT7S3 A | pT7S3 B | pT7S3 C | pT7S3 D | Nucleotide Frequency | |||||
| Plasmid Relative Abundance | 0.01% | 1.00% | 10.00% | 88.89% | 0.10% | A | T | C | G | |
| Amplicon position* | 1754 | C | T | T | T | C | 99.89% | 0.11% | ||
| 1932 | G | G | G | G | A | 0.10% | 99.90% | |||
| 2149 | G | A | A | G | G | 11.00% | 89.00% | |||
| 2297 | T | G | G | G | G | 0.01% | 99.99% | |||
| 2323 | A | G | G | G | G | 0.01% | 99.99% | |||
| 2505 | A | A | G | G | A | 1.11% | 98.89% | |||
| 2507 | T | T | G | G | T | 1.11% | 98.89% | |||
| 2755 | G | A | A | A | A | 99.99% | 0.01% | |||
| 2761 | A | T | T | T | A | 0.11% | 99.89% | |||
| 2767 | G | A | A | A | A | 99.99% | 0.01% | |||
| 2791 | C | T | T | T | T | 99.99% | 0.01% | |||
| 2843 | A | C | C | C | C | 0.01% | 99.99% | |||
| 2955 | G | A | A | A | A | 99.99% | 0.01% | |||
| 3106 | G | A | A | A | A | 99.99% | 0.01% | |||
| 3376 | C | A | C | A | C | 89.89% | 10.11% | |||
| 3645 | G | G | G | G | A | 0.10% | 99.90% | |||
| 3661 | G | A | A | A | G | 99.89% | 0.11% | |||
| 3691 | T | G | G | G | T | 0.11% | 99.89% | |||
| 3695 | G | T | T | T | G | 99.89% | 0.11% | |||
| 3697 | T | C | C | C | T | 0.11% | 99.89% | |||
| Input | Replicate Combinations | Aligner | qScore | Read Length (bp) | Suggested Frequency Cut-Off | MuMCC | |
|---|---|---|---|---|---|---|---|
| RNA | High | Singlet | GEM3 | 38 | 70 | 0.20% | 0.756 |
| Duplicate | 0.20% | 0.816 | |||||
| Triplicate | 0.20% | 0.816 | |||||
| Quadruplicate | 0.20% | 0.816 | |||||
| Medium | Singlet | GEM3 | 38 | 70 | 0.80% | 0.707 | |
| Duplicate | 0.50% | 0.707 | |||||
| Triplicate | 0.30% | 0.707 | |||||
| Quadruplicate | 0.20% | 0.707 | |||||
| DNA | High | Singlet | GEM3 | 35 | 70 | 0.20% | 1.000 |
| Duplicate | 0.20% | 0.933 | |||||
| Triplicate | 0.04% | 0.891 | |||||
| Quadruplicate | 0.04% | 0.913 | |||||
| Low | Singlet | GEM3 | 35 | 70 | 0.20% | 0.707 | |
| Duplicate | 0.20% | 0.707 | |||||
| Triplicate | 0.20% | 0.707 | |||||
| Quadruplicate | 0.20% | 0.707 |
| Input Replicate | |||||||
|---|---|---|---|---|---|---|---|
| Input | Suggested Frequency Cut-Off | Amplicon Position | Base Change | 1 | 2 | 3 | 4 |
| DNA Low | 0.2% | 605 | G > T | 0.22% | |||
| RNA High | 0.2% | 1135 | T > C | 0.39% | 0.35% | 0.41% | 0.30% |
| 0.2% | 1915 | T > C | 0.21% | 0.28% | |||
| 0.2% | 2300 | T > C | 0.21% | ||||
| 0.2% | 3056 | T > C | 1.06% | 0.81% | 0.62% | 0.87% | |
| RNA Medium | 0.8% | 1135 | T > C | 1.14% | |||
| RNA Low | 1609 | T > C | 53.76% | ||||
| 2199 | G > T | 18.49% | |||||
| 2744 | G > C | 99.94% | |||||
| 2933 | T > C | 9.06% | |||||
| 3648 | T > C | 45.06% | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
King, D.J.; Freimanis, G.; Lasecka-Dykes, L.; Asfor, A.; Ribeca, P.; Waters, R.; King, D.P.; Laing, E. A Systematic Evaluation of High-Throughput Sequencing Approaches to Identify Low-Frequency Single Nucleotide Variants in Viral Populations. Viruses 2020, 12, 1187. https://doi.org/10.3390/v12101187
King DJ, Freimanis G, Lasecka-Dykes L, Asfor A, Ribeca P, Waters R, King DP, Laing E. A Systematic Evaluation of High-Throughput Sequencing Approaches to Identify Low-Frequency Single Nucleotide Variants in Viral Populations. Viruses. 2020; 12(10):1187. https://doi.org/10.3390/v12101187
Chicago/Turabian StyleKing, David J., Graham Freimanis, Lidia Lasecka-Dykes, Amin Asfor, Paolo Ribeca, Ryan Waters, Donald P. King, and Emma Laing. 2020. "A Systematic Evaluation of High-Throughput Sequencing Approaches to Identify Low-Frequency Single Nucleotide Variants in Viral Populations" Viruses 12, no. 10: 1187. https://doi.org/10.3390/v12101187
APA StyleKing, D. J., Freimanis, G., Lasecka-Dykes, L., Asfor, A., Ribeca, P., Waters, R., King, D. P., & Laing, E. (2020). A Systematic Evaluation of High-Throughput Sequencing Approaches to Identify Low-Frequency Single Nucleotide Variants in Viral Populations. Viruses, 12(10), 1187. https://doi.org/10.3390/v12101187