The Role of EBV-Induced Hypermethylation in Gastric Cancer Tumorigenesis


Abstract
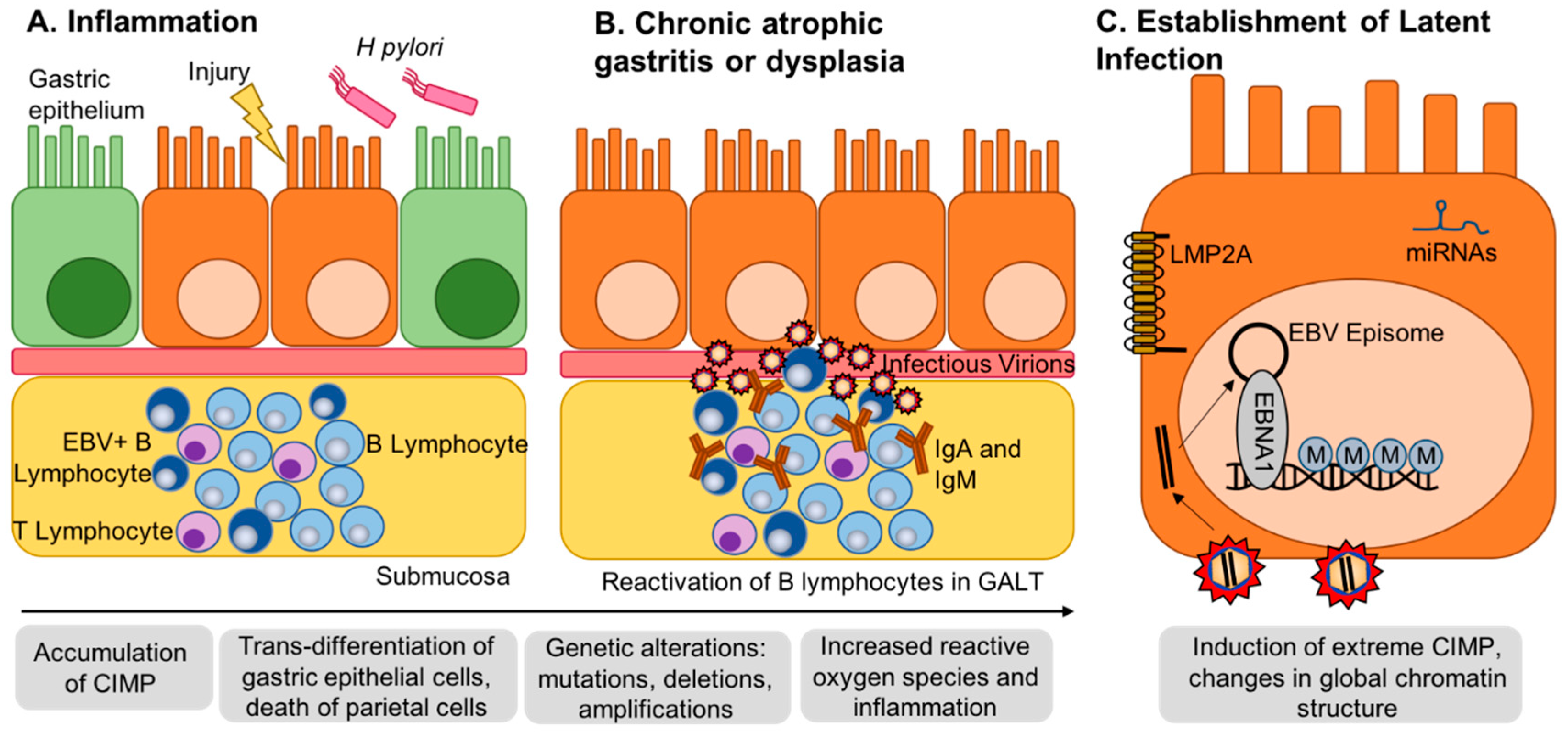
:1. Introduction
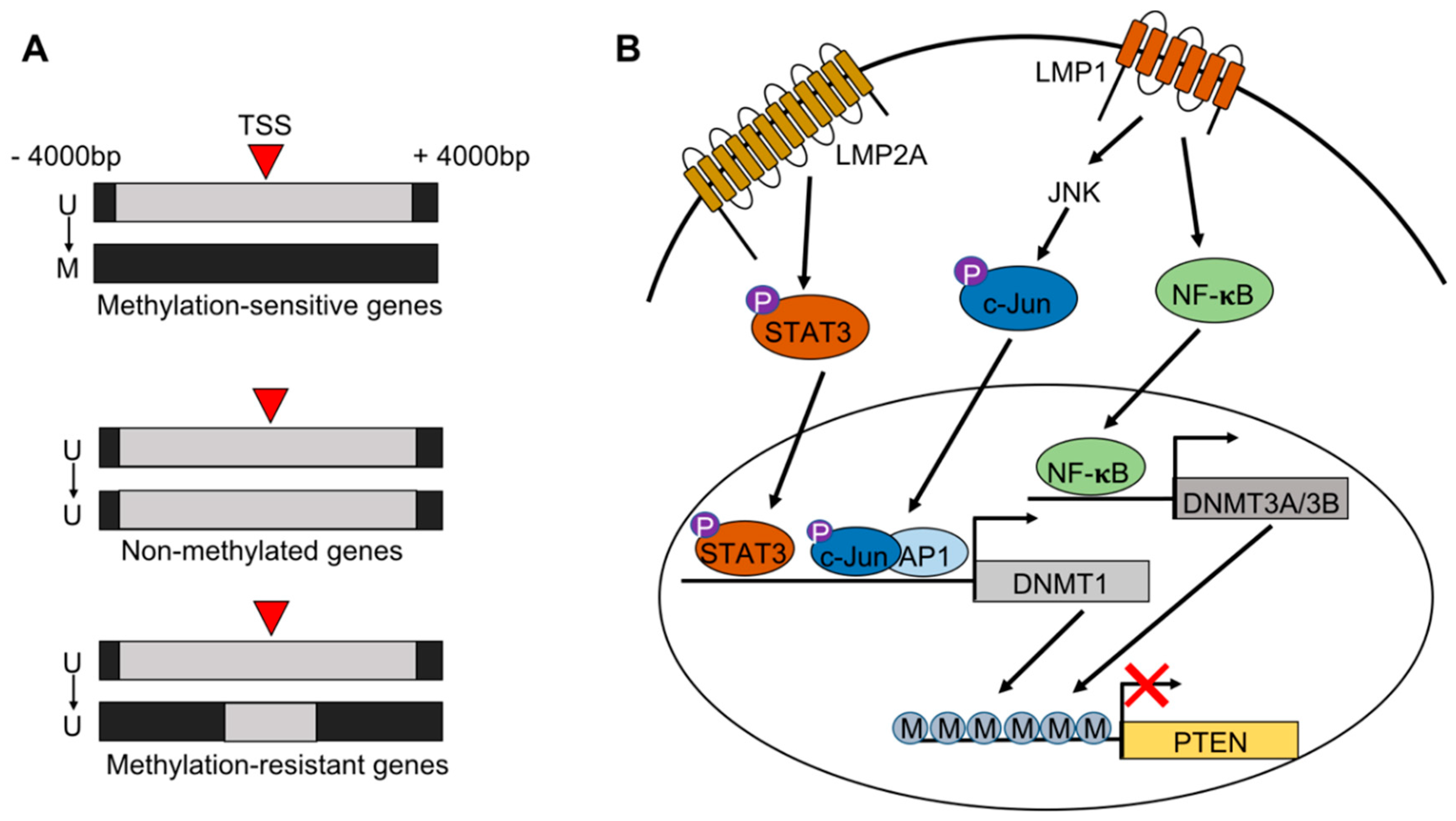
2. EBV Infection of Gastric Epithelial Cells and Induction of Hypermethylation
3. Alternative Epigenetic Mechanisms of Gene Silencing
4. Tumor Suppressors
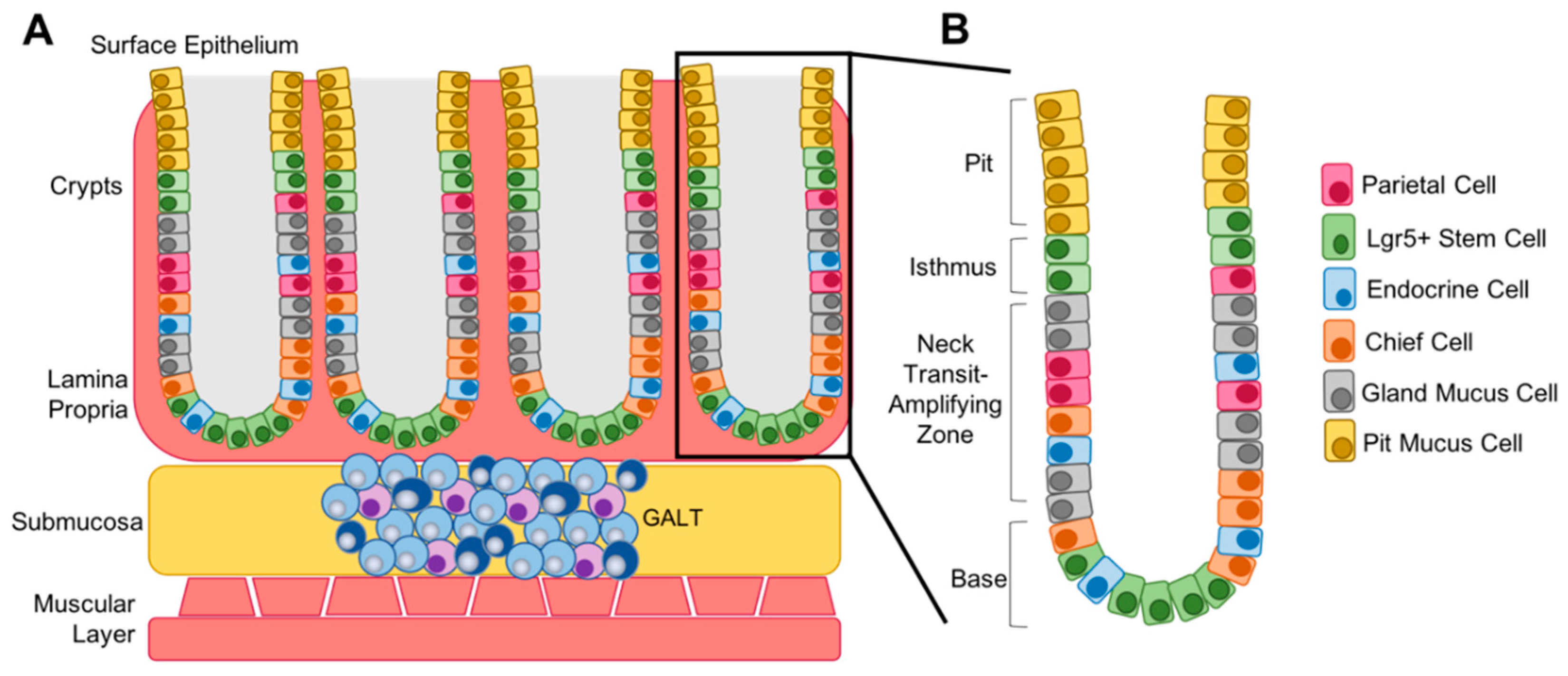
5. Differentiation Status
6. Methylation of the Viral Genome
7. Methylation in Other EBV-Infected Cell Lines and Tumors
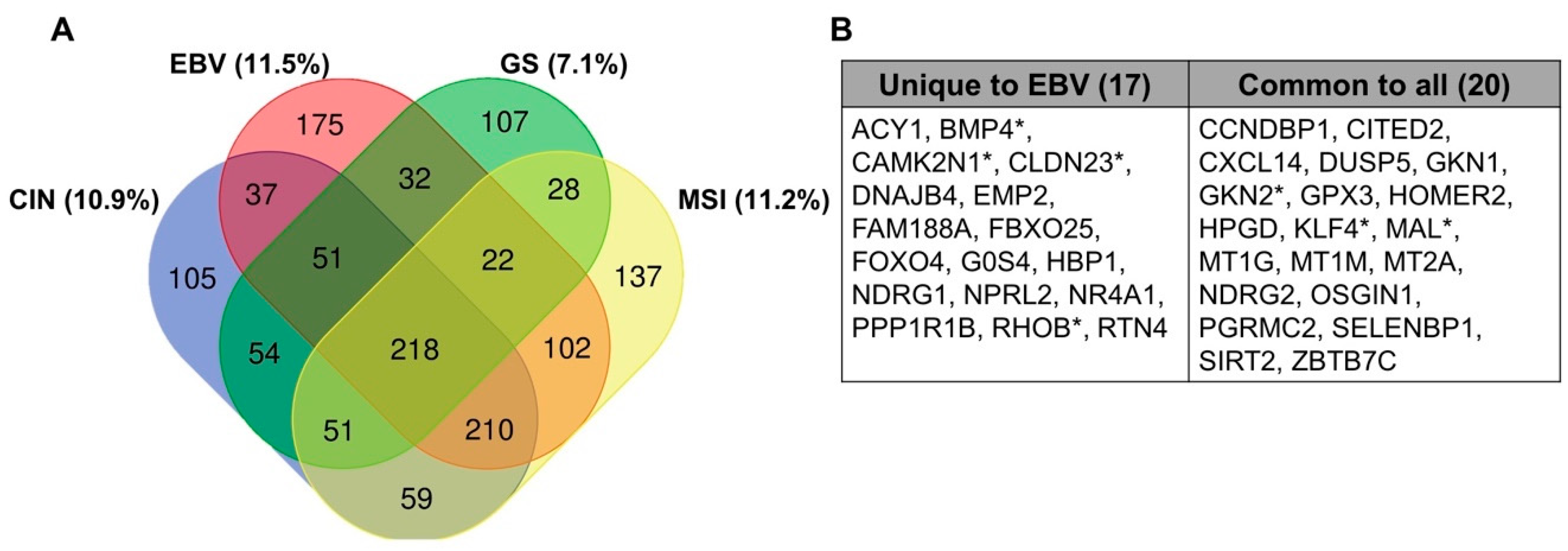
8. Comparison to Other Heavily Methylated Tumor Types
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.P.; Yen, T.S.; Shekitka, K.M.; Sobin, L.H. Lymphoepithelial carcinoma of the stomach with Epstein-Barr virus demonstrated by polymerase chain reaction. Mod. Pathol. 1990, 3, 377–380. [Google Scholar] [PubMed]
- Truong, C.D.; Feng, W.; Li, W.; Khoury, T.; Li, Q.; Alrawi, S.; Yu, Y.; Xie, K.; Yao, J.; Tan, D. Characteristics of Epstein-Barr virus-associated gastric cancer: A study of 235 cases at a comprehensive cancer center in U.S.A. J. Exp. Clin. Cancer Res. 2009, 28, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, G.H.; Lee, S.; Kim, W.H.; Lee, H.W.; Kim, J.C.; Rhyu, M.G.; Ro, J.Y. Epstein-barr virus-positive gastric carcinoma demonstrates frequent aberrant methylation of multiple genes and constitutes CpG island methylator phenotype-positive gastric carcinoma. Am. J. Pathol. 2002, 160, 787–794. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.M.; Sakuma, K.; Sudo, M.; Ushiku, T.; Uozaki, H.; Shibahara, J.; Nagai, H.; Funata, N.; Taniguchi, H.; Aburatani, H.; et al. Global and non-random CpG-island methylation in gastric carcinoma associated with Epstein-Barr virus. Cancer Sci. 2003, 94, 76–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, J.L.; Jones, R.J.; Kenney, S.C.; Rivenbark, A.G.; Tang, W.; Knight, E.R.; Coleman, W.B.; Gulley, M.L. Epstein-Barr virus-specific methylation of human genes in gastric cancer cells. Infect. Agents Cancer 2010, 5, 27. [Google Scholar] [CrossRef] [Green Version]
- Liang, Q.; Yao, X.; Tang, S.; Zhang, J.; Yau, T.O.; Li, X.; Tang, C.M.; Kang, W.; Lung, R.W.; Li, J.W.; et al. Integrative identification of Epstein-Barr virus-associated mutations and epigenetic alterations in gastric cancer. Gastroenterology 2014, 147, 1350–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, W.; Cheung, A.K.; Ko, J.M.; Cheng, Y.; Zheng, H.; Ngan, R.K.; Ng, W.T.; Lee, A.W.; Yau, C.C.; Lee, V.H.; et al. Comparative methylome analysis in solid tumors reveals aberrant methylation at chromosome 6p in nasopharyngeal carcinoma. Cancer Med. 2015, 4, 1079–1090. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Matsusaka, K.; Funata, S.; Fukuyo, M.; Seto, Y.; Aburatani, H.; Fukayama, M.; Kaneda, A. Epstein-Barr virus infection induces genome-wide de novo DNA methylation in non-neoplastic gastric epithelial cells. J. Pathol. 2017, 242, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Namba-Fukuyo, H.; Funata, S.; Matsusaka, K.; Fukuyo, M.; Rahmutulla, B.; Mano, Y.; Fukayama, M.; Aburatani, H.; Kaneda, A. TET2 functions as a resistance factor against DNA methylation acquisition during Epstein-Barr virus infection. Oncotarget 2016, 7, 81512–81526. [Google Scholar] [CrossRef] [Green Version]
- Okabe, A.; Funata, S.; Matsusaka, K.; Namba, H.; Fukuyo, M.; Rahmutulla, B.; Oshima, M.; Iwama, A.; Fukayama, M.; Kaneda, A. Regulation of tumour related genes by dynamic epigenetic alteration at enhancer regions in gastric epithelial cells infected by Epstein-Barr virus. Sci. Rep. 2017, 7, 7924. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.K.; Ramnarayanan, K.; Zhu, F.; Srivastava, S.; Xu, C.; Tan, A.L.K.; Lee, M.; Tay, S.; Das, K.; Xing, M.; et al. Genomic and Epigenomic Profiling of High-Risk Intestinal Metaplasia Reveals Molecular Determinants of Progression to Gastric Cancer. Cancer Cell 2018, 33, 137–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Sethi, N.S.; Hinoue, T.; Schneider, B.G.; Cherniack, A.D.; Sanchez-Vega, F.; Seoane, J.A.; Farshidfar, F.; Bowlby, R.; Islam, M.; et al. Comparative Molecular Analysis of Gastrointestinal Adenocarcinomas. Cancer Cell 2018, 33, 721–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichelberg, M.R.; Welch, R.; Guidry, J.T.; Ali, A.; Ohashi, M.; Makielski, K.R.; McChesney, K.; Van Sciver, N.; Lambert, P.F.; Keles, S.; et al. Epstein-Barr Virus Infection Promotes Epithelial Cell Growth by Attenuating Differentiation-Dependent Exit from the Cell Cycle. mBio 2019, 10, e01332-19. [Google Scholar] [CrossRef] [Green Version]
- Birdwell, C.E.; Queen, K.J.; Kilgore, P.C.; Rollyson, P.; Trutschl, M.; Cvek, U.; Scott, R.S. Genome-wide DNA methylation as an epigenetic consequence of Epstein-Barr virus infection of immortalized keratinocytes. J. Virol. 2014, 88, 11442–11458. [Google Scholar] [CrossRef] [Green Version]
- Edwards, R.H.; Dekroon, R.; Raab-Traub, N. Alterations in cellular expression in EBV infected epithelial cell lines and tumors. PLoS Pathog. 2019, 15, e1008071. [Google Scholar] [CrossRef]
- Zhao, J.; Liang, Q.; Cheung, K.F.; Kang, W.; Lung, R.W.; Tong, J.H.; To, K.F.; Sung, J.J.; Yu, J. Genome-wide identification of Epstein-Barr virus-driven promoter methylation profiles of human genes in gastric cancer cells. Cancer 2013, 119, 304–312. [Google Scholar] [CrossRef]
- Matsusaka, K.; Kaneda, A.; Nagae, G.; Ushiku, T.; Kikuchi, Y.; Hino, R.; Uozaki, H.; Seto, Y.; Takada, K.; Aburatani, H.; et al. Classification of Epstein-Barr virus-positive gastric cancers by definition of DNA methylation epigenotypes. Cancer Res. 2011, 71, 7187–7197. [Google Scholar] [CrossRef] [Green Version]
- Funata, S.; Matsusaka, K.; Yamanaka, R.; Yamamoto, S.; Okabe, A.; Fukuyo, M.; Aburatani, H.; Fukayama, M.; Kaneda, A. Histone modification alteration coordinated with acquisition of promoter DNA methylation during Epstein-Barr virus infection. Oncotarget 2017, 8, 55265–55279. [Google Scholar] [CrossRef] [Green Version]
- Queen, K.J.; Shi, M.; Zhang, F.; Cvek, U.; Scott, R.S. Epstein-Barr virus-induced epigenetic alterations following transient infection. Int. J. Cancer 2013, 132, 2076–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hino, R.; Uozaki, H.; Murakami, N.; Ushiku, T.; Shinozaki, A.; Ishikawa, S.; Morikawa, T.; Nakaya, T.; Sakatani, T.; Takada, K.; et al. Activation of DNA methyltransferase 1 by EBV latent membrane protein 2A leads to promoter hypermethylation of PTEN gene in gastric carcinoma. Cancer Res. 2009, 69, 2766–2774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, H.; Chen, Y.; Gong, P.; Cai, L.; Lyu, X.; Jiang, Q.; Wang, J.; Lu, J.; Yao, K.; Liu, K.; et al. Higher methylation intensity induced by EBV LMP1 via NF-kappaB/DNMT3b signaling contributes to silencing of PTEN gene. Oncotarget 2016, 7, 40025–40037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, C.L.; Li, H.P.; Lu, Y.J.; Hsueh, C.; Liang, Y.; Chen, C.L.; Tsao, S.W.; Tse, K.P.; Yu, J.S.; Chang, Y.S. Activation of DNA methyltransferase 1 by EBV LMP1 Involves c-Jun NH(2)-terminal kinase signaling. Cancer Res. 2006, 66, 11668–11676. [Google Scholar] [CrossRef] [Green Version]
- Saha, A.; Jha, H.C.; Upadhyay, S.K.; Robertson, E.S. Epigenetic silencing of tumor suppressor genes during in vitro Epstein-Barr virus infection. Proc. Natl. Acad. Sci. USA 2015, 112, E5199–E5207. [Google Scholar] [CrossRef] [Green Version]
- Leong, M.M.L.; Cheung, A.K.L.; Dai, W.; Tsao, S.W.; Tsang, C.M.; Dawson, C.W.; Mun Yee Ko, J.; Lung, M.L. EBV infection is associated with histone bivalent switch modifications in squamous epithelial cells. Proc. Natl. Acad. Sci. USA 2019, 116, 14144–14153. [Google Scholar] [CrossRef] [Green Version]
- Hernando, H.; Islam, A.B.; Rodriguez-Ubreva, J.; Forne, I.; Ciudad, L.; Imhof, A.; Shannon-Lowe, C.; Ballestar, E. Epstein-Barr virus-mediated transformation of B cells induces global chromatin changes independent to the acquisition of proliferation. Nucleic Acids Res. 2014, 42, 249–263. [Google Scholar] [CrossRef] [Green Version]
- Morrison, J.A.; Klingelhutz, A.J.; Raab-Traub, N. Epstein-Barr virus latent membrane protein 2A activates beta-catenin signaling in epithelial cells. J. Virol. 2003, 77, 12276–12284. [Google Scholar]
- Chen, J. Roles of the PI3K/Akt pathway in Epstein-Barr virus-induced cancers and therapeutic implications. World J. Virol. 2012, 1, 154–161. [Google Scholar] [CrossRef]
- Soutto, M.; Chen, Z.; Bhat, A.A.; Wang, L.; Zhu, S.; Gomaa, A.; Bates, A.; Bhat, N.S.; Peng, D.; Belkhiri, A.; et al. Activation of STAT3 signaling is mediated by TFF1 silencing in gastric neoplasia. Nat. Commun. 2019, 10, 3039. [Google Scholar] [CrossRef] [PubMed]
- Portis, T.; Longnecker, R. Epstein-Barr virus (EBV) LMP2A mediates B-lymphocyte survival through constitutive activation of the Ras/PI3K/Akt pathway. Oncogene 2004, 23, 8619–8628. [Google Scholar] [CrossRef] [Green Version]
- Hino, R.; Uozaki, H.; Inoue, Y.; Shintani, Y.; Ushiku, T.; Sakatani, T.; Takada, K.; Fukayama, M. Survival advantage of EBV-associated gastric carcinoma: Survivin up-regulation by viral latent membrane protein 2A. Cancer Res. 2008, 68, 1427–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iizasa, H.; Nanbo, A.; Nishikawa, J.; Jinushi, M.; Yoshiyama, H. Epstein-Barr Virus (EBV)-associated gastric carcinoma. Viruses 2012, 4, 3420–3439. [Google Scholar] [CrossRef] [Green Version]
- Caglar, H.O.; Avci, C.B. Alterations of cell cycle genes in cancer: Unmasking the role of cancer stem cells. Mol. Biol. Rep. 2020, 47, 3065–3076. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Kang, M.S.; Kim, K.M. Epstein-Barr Virus-Associated Gastric Carcinoma and Specific Features of the Accompanying Immune Response. J. Gastric Cancer 2016, 16, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Becker, J.; Leser, U.; Marschall, M.; Langford, A.; Jilg, W.; Gelderblom, H.; Reichart, P.; Wolf, H. Expression of proteins encoded by Epstein-Barr virus trans-activator genes depends on the differentiation of epithelial cells in oral hairy leukoplakia. Proc. Natl. Acad. Sci. USA 1991, 88, 8332–8336. [Google Scholar] [CrossRef] [Green Version]
- Young, L.S.; Lau, R.; Rowe, M.; Niedobitek, G.; Packham, G.; Shanahan, F.; Rowe, D.T.; Greenspan, D.; Greenspan, J.S.; Rickinson, A.B.; et al. Differentiation-associated expression of the Epstein-Barr virus BZLF1 transactivator protein in oral hairy leukoplakia. J. Virol. 1991, 65, 2868–2874. [Google Scholar] [CrossRef] [Green Version]
- Young, L.S.; Rickinson, A.B. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef]
- Price, A.M.; Dai, J.; Bazot, Q.; Patel, L.; Nikitin, P.A.; Djavadian, R.; Winter, P.S.; Salinas, C.A.; Barry, A.P.; Wood, K.C.; et al. Epstein-Barr virus ensures B cell survival by uniquely modulating apoptosis at early and late times after infection. eLife 2017, 6, e22509. [Google Scholar] [CrossRef]
- Koboziev, I.; Karlsson, F.; Grisham, M.B. Gut-associated lymphoid tissue, T cell trafficking, and chronic intestinal inflammation. Ann. N. Y. Acad. Sci. 2010, 1207 (Suppl. 1), E86–E93. [Google Scholar] [CrossRef]
- McCracken, K.W.; Cata, E.M.; Crawford, C.M.; Sinagoga, K.L.; Schumacher, M.; Rockich, B.E.; Tsai, Y.H.; Mayhew, C.N.; Spence, J.R.; Zavros, Y.; et al. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature 2014, 516, 400–404. [Google Scholar] [CrossRef] [Green Version]
- Willet, S.G.; Mills, J.C. Stomach Organ and Cell Lineage Differentiation: From Embryogenesis to Adult Homeostasis. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 546–559. [Google Scholar] [CrossRef] [Green Version]
- Burclaff, J.; Mills, J.C. Plasticity of differentiated cells in wound repair and tumorigenesis, part I: Stomach and pancreas. Dis. Models Mech. 2018, 11, dmm033373. [Google Scholar] [CrossRef] [Green Version]
- Tsang, C.M.; Yip, Y.L.; Lo, K.W.; Deng, W.; To, K.F.; Hau, P.M.; Lau, V.M.; Takada, K.; Lui, V.W.; Lung, M.L.; et al. Cyclin D1 overexpression supports stable EBV infection in nasopharyngeal epithelial cells. Proc. Natl. Acad. Sci. USA 2012, 109, E3473–E3482. [Google Scholar] [CrossRef] [Green Version]
- Tsang, C.M.; Zhang, G.; Seto, E.; Takada, K.; Deng, W.; Yip, Y.L.; Man, C.; Hau, P.M.; Chen, H.; Cao, Y.; et al. Epstein-Barr virus infection in immortalized nasopharyngeal epithelial cells: Regulation of infection and phenotypic characterization. Int. J. Cancer 2010, 127, 1570–1583. [Google Scholar] [CrossRef]
- Yoon, J.H.; Min, K.; Lee, S.K. Epstein-Barr Virus miR-BART17-5p Promotes Migration and Anchorage-Independent Growth by Targeting Kruppel-Like Factor 2 in Gastric Cancer. Microorganisms 2020, 8, 258. [Google Scholar] [CrossRef] [Green Version]
- Marquitz, A.R.; Mathur, A.; Shair, K.H.; Raab-Traub, N. Infection of Epstein-Barr virus in a gastric carcinoma cell line induces anchorage independence and global changes in gene expression. Proc. Natl. Acad. Sci. USA 2012, 109, 9593–9598. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, J.; Imai, S.; Oda, T.; Kojima, T.; Okita, K.; Takada, K. Epstein-Barr virus promotes epithelial cell growth in the absence of EBNA2 and LMP1 expression. J. Virol. 1999, 73, 1286–1292. [Google Scholar] [CrossRef] [Green Version]
- Nawandar, D.M.; Wang, A.; Makielski, K.; Lee, D.; Ma, S.; Barlow, E.; Reusch, J.; Jiang, R.; Wille, C.K.; Greenspan, D.; et al. Differentiation-Dependent KLF4 Expression Promotes Lytic Epstein-Barr Virus Infection in Epithelial Cells. PLoS Pathog. 2015, 11, e1005195. [Google Scholar] [CrossRef]
- Wille, C.K.; Nawandar, D.M.; Henning, A.N.; Ma, S.; Oetting, K.M.; Lee, D.; Lambert, P.; Johannsen, E.C.; Kenney, S.C. 5-hydroxymethylation of the EBV genome regulates the latent to lytic switch. Proc. Natl. Acad. Sci. USA 2015, 112, E7257–E7265. [Google Scholar] [CrossRef] [Green Version]
- Fukayama, M.; Abe, H.; Kunita, A.; Shinozaki-Ushiku, A.; Matsusaka, K.; Ushiku, T.; Kaneda, A. Thirty years of Epstein-Barr virus-associated gastric carcinoma. Virchows Arch. 2019. [Google Scholar] [CrossRef] [PubMed]
- Tempera, I.; Lieberman, P.M. Epigenetic regulation of EBV persistence and oncogenesis. Semin. Cancer Biol. 2014, 26, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Scott, R.S. Epstein-Barr virus: A master epigenetic manipulator. Curr. Opin. Virol. 2017, 26, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Tao, Q.; Robertson, K.D.; Manns, A.; Hildesheim, A.; Ambinder, R.F. The Epstein-Barr virus major latent promoter Qp is constitutively active, hypomethylated, and methylation sensitive. J. Virol. 1998, 72, 7075–7083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, M.; Zuo, J. Immune responses to Epstein-Barr virus: Molecular interactions in the virus evasion of CD8+ T cell immunity. Microbes Infect. 2010, 12, 173–181. [Google Scholar] [CrossRef]
- Masucci, M.G.; Contreras-Salazar, B.; Ragnar, E.; Falk, K.; Minarovits, J.; Ernberg, I.; Klein, G. 5-Azacytidine up regulates the expression of Epstein-Barr virus nuclear antigen 2 (EBNA-2) through EBNA-6 and latent membrane protein in the Burkitt’s lymphoma line rael. J. Virol. 1989, 63, 3135–3141. [Google Scholar] [CrossRef] [Green Version]
- Niller, H.H.; Banati, F.; Salamon, D.; Minarovits, J. Epigenetic Alterations in Epstein-Barr Virus-Associated Diseases. Adv. Exp. Med. Biol. 2016, 879, 39–69. [Google Scholar] [CrossRef]
- Hernandez-Vargas, H.; Gruffat, H.; Cros, M.P.; Diederichs, A.; Sirand, C.; Vargas-Ayala, R.C.; Jay, A.; Durand, G.; Le Calvez-Kelm, F.; Herceg, Z.; et al. Viral driven epigenetic events alter the expression of cancer-related genes in Epstein-Barr-virus naturally infected Burkitt lymphoma cell lines. Sci. Rep. 2017, 7, 5852. [Google Scholar] [CrossRef]
- Kretzmer, H.; Bernhart, S.H.; Wang, W.; Haake, A.; Weniger, M.A.; Bergmann, A.K.; Betts, M.J.; Carrillo-de-Santa-Pau, E.; Doose, G.; Gutwein, J.; et al. DNA methylome analysis in Burkitt and follicular lymphomas identifies differentially methylated regions linked to somatic mutation and transcriptional control. Nat. Genet. 2015, 47, 1316–1325. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, I.; Iwaya, C.; Ohnaka, K.; Shibata, H.; Yamamoto, K. Genome-wide DNA methylation analysis reveals hypomethylation in the low-CpG promoter regions in lymphoblastoid cell lines. Hum. Genom. 2017, 11, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estecio, M.R.; Issa, J.P. Dissecting DNA hypermethylation in cancer. FEBS Lett. 2011, 585, 2078–2086. [Google Scholar] [CrossRef] [Green Version]
- Hinoue, T.; Weisenberger, D.J.; Lange, C.P.; Shen, H.; Byun, H.M.; Van Den Berg, D.; Malik, S.; Pan, F.; Noushmehr, H.; van Dijk, C.M.; et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2012, 22, 271–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author | Dataset | Tissue | Array | Platform | Samples |
|---|---|---|---|---|---|
| Matsusaka 2011 [19] | GSE31789 | Gastric | Methylation and Expression | GPL370 GPL8490 | Gastric cell lines and primary tumors |
| Zhao 2013 [18] | Gastric | Methylation | Gastric cancer cell line ± EBV infection | ||
| Birdwell 2014 [16] | GSE59843 | NOKs | HT Seq | GPL16791 | Normalized oral keratinocytes |
| Bass 2014 [9] | TCGA Portal | Gastric | WGS, Methylation, RNA-seq | GPL8490 GPL13534 | Gastric tumors and normal gastric tissues |
| Liang 2014 [7] | SRA67982 | Gastric | WGS | GPL9115 | Gastric cancer cell line ± EBV infection |
| Dai 2015 [8] | GSE62336 | NPC | Methylation | GPL13534 | Matched NPC and normal tissue from same donor |
| Namba-Fukuyo 2016 [11] | GSE84897 | Gastric | RNA-seq, MeDIP-seq | GPL18460 GPL18573 | Gastric cancer cell line ± EBV infection |
| Okabe 2017 [12] | GSE97837 GSE97838 | Gastric | ChIP-Seq, FAIRE-seq | GPL10999 GPL18460 | Gastric cancer cell line ± EBV infection |
| Huang 2017 [13] | GSE103186 | IM | Methylation | GPL13534 | Intestinal metaplasias and normal gastric tissue |
| Matsusaka 2017 [10] | GSE89269 | Gastric | Methylation | GPL13534 | Gastric cell lines, tumors, and normal gastric tissue |
| Liu 2018 [14] | GDC Legacy Archive | Gastric | Methylation | GPL8490 GPL13534 | Gastrointestinal adenocarcinomas |
| Edwards 2019 [17] | PRJNA503182 PRJNA501807 | Gastric and NPC | RNA-seq | GPL20301 | Gastric and NPC cell lines ± EBV and xenograft |
| Eichelberg 2019 [15] | PRJNA555053 | NOKs | RNA-seq | GPL18573 | Normalized oral keratinocytes ± EBV infection, and methylcellulose |
| Gene | Location | Full Name | Function | References |
|---|---|---|---|---|
| TGFBR3 | 1p33–p32 | Transforming growth factor beta receptor type 3 | TGF-β signaling | [9,12] |
| SFN | 1p36.11 | Stratifin | Cell cycle checkpoint, p53 activator | [4,12] |
| CAMK2N1 | 1p36.12 | Calcium/calmodulin dependent protein kinase II inhibitor 1 | Cell cycle, apoptosis | [9,14] |
| TP73 | 1p36.3 | Tumor protein p73 | Apoptosis, DNA damage response | [10] |
| GKN2 | 2p13.3 | Gastric motility protein 2 | Proliferation, apoptosis, invasion | [9,12] |
| RHOB | 2p24 | Ras homolog gene family member B | Negative regulator of intracellular signaling, cell-to-cell adhesion | [9,12] |
| MAL | 2q11.1 | Myelin and lymphocyte protein | Integral membrane protein, anti-metastatic in epithelial cancers | [9,12] |
| RASSF1A | 3p21.3 | Ras association domain family member 1 | Apoptosis, cell cycle checkpoint | [4] |
| PLCD1 | 3p22.3 | Phospholipase C delta 1 | Motility, migration and invasion, cytoskeleton reorganization | [9,14] |
| APC | 5q21–q22 | Adenomatous polyposis coli | Negative regulator of β-catenin, cell adhesion | [4] |
| SPINK5 | 5q32 | Serine protease inhibitor Kazal-type 5 | Inhibitor of Wnt signaling, anti-proliferation, migration and invasion | [12,17] |
| AKAP12 | 6q24–q25 | A-kinase anchor protein 12 | PKA scaffold protein, anti-metastatic | [10,12] |
| DFNA5 | 7p15 | Deafness associated tumor suppressor | Apoptosis, regulated by p53 | [10] |
| SFRP1 | 8p11.21 | Secreted frizzled related protein 1 | Growth inhibitory, anti-proliferative | [9,10] |
| SOX7 | 8p21.3 | SRY-box transcription factor 7 | Antagonism of Wnt signaling, anti-proliferation | [10] |
| CLDN23 | 8p23.1 | Claudin 23 | Cell polarity | [9,12] |
| CDKN2A | 9p21 | Cyclin dependent kinase inhibitor 2A | Cell cycle | [4,5,6,9,14,18] |
| TUSC1 | 9p21.2 | Tumor suppressor candidate 1 | Cell growth | [9,14] |
| WNK2 | 9q22.3 | WNK lysine deficient protein kinase 2 | Proliferation, negative regulator of ERK/MAPK pathway | [12,14] |
| PTEN | 10q23.3 | Phosphatase and tensin homolog | Negative regulator of PI3K/Akt signaling | [4,22] |
| CDKN1C | 11p15.5 | Cyclin dependent kinase inhibitor 1C | Cell cycle, anti-proliferation | [7,9,12] |
| AHNAK | 11q12.2 | AHNAK nucleoprotein | Invasion and migration, EMT | [9,12] |
| BMP4 | 14q22–q23 | Bone morphogenetic protein 4 | TGF-β signaling | [5,9,10,12] |
| YPEL3 | 16p11.2 | Yippee-like 3 | P53 inducible, anti-proliferation, cellular senescence | [9,12,14] |
| FBLN1 | 22q13.31 | Fibulin 1 | Cell morphology, growth, adhesion, motility | [9,10,12] |
| Gene | Location | Full Name | Function | References |
|---|---|---|---|---|
| VANGL1 | 1p13.1 | VANGL cell polarity protein 1 | Planar cell polarity, columnar epithelial structure | [14] |
| VANGL2 | 1q23.2 | VANGL cell polarity protein 2 | Planar cell polarity, columnar epithelial structure | [9,10,14] |
| DVL | 1p36.33 | Disheveled | Cell adhesion, cell polarity | [12] |
| CD109 | 6q13 | CD109 molecule | Negative regulator of EMT, invasion and migration | [10,12] |
| MAP7 | 6q23.3 | Microtubule-associated protein 7 | Microtubule structure, cell polarization | [12,14] |
| CAV1 | 7q31.1 | Caveolin 1 | Proliferation, migration, differentiation | [9,10] |
| KLF4 | 9q31 | Kruppel-like factor 4 | Proliferation, differentiation, apoptosis | [9] |
| OVOL1 | 11q13.1 | Ovo like transcriptional repressor 1 | Terminal differentiation, anti-proliferative | [9,14] |
| TBX3 | 12q23–24.1 | T-box transcription factor 3 | Transcription factor activates differentiation genes, cell cycle inhibition | [9,14] |
| CDX2 | 13q12.2 | Caudal type homeobox 2 | Pro-differentiation of intestinal epithelial cells | [9,10,14] |
| SCEL | 13q22.3 | Sciellin | Terminal differentiation | [12,17] |
| CDH1 | 16q22.1 | E-cadherin | EMT, cell adhesion | [4,5,6,12] |
| RARA | 17q21.2 | Retinoic acid receptor alpha | Pro-differentiation and anti-proliferation | [7,12,18] |
| FUZ | 19q13.33 | Fuzzy planar cell protein | Planar cell polarity | [9,14] |
| FOXA2 | 20p11 | Forkhead Box A2 | Drives differentiation of mature gastric cell types | [7,12] |
| BMP7 | 20q13.31 | Bone morphogenetic protein 7 | Pro-differentiation and anti-inflammatory | [14,17,18] |
| CDH4 | 20q13.3 | Cadherin 4 | Cell adhesion, cell polarity | [10,18] |
| GATA5 | 20q13.33 | GATA binding protein 5 | Pro-differentiation | [7,17] |
| TFF1 | 21q22.3 | Trefoil factor 1 | Anti-inflammatory, pro-apoptotic, maintain gastric tissue health and integrity | [6,17] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stanland, L.J.; Luftig, M.A. The Role of EBV-Induced Hypermethylation in Gastric Cancer Tumorigenesis. Viruses 2020, 12, 1222. https://doi.org/10.3390/v12111222
Stanland LJ, Luftig MA. The Role of EBV-Induced Hypermethylation in Gastric Cancer Tumorigenesis. Viruses. 2020; 12(11):1222. https://doi.org/10.3390/v12111222
Chicago/Turabian StyleStanland, Lyla J., and Micah A. Luftig. 2020. "The Role of EBV-Induced Hypermethylation in Gastric Cancer Tumorigenesis" Viruses 12, no. 11: 1222. https://doi.org/10.3390/v12111222
APA StyleStanland, L. J., & Luftig, M. A. (2020). The Role of EBV-Induced Hypermethylation in Gastric Cancer Tumorigenesis. Viruses, 12(11), 1222. https://doi.org/10.3390/v12111222