First Phylogenetic Analysis of Malian SARS-CoV-2 Sequences Provides Molecular Insights into the Genomic Diversity of the Sahel Region

 , ,
, ,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Sampling
2.2. RNA Extraction and SARS-CoV-2 Detection
2.3. Library Preparation and Sequencing
2.4. Phylogenetic Analysis
2.5. Ethical Statement
3. Results
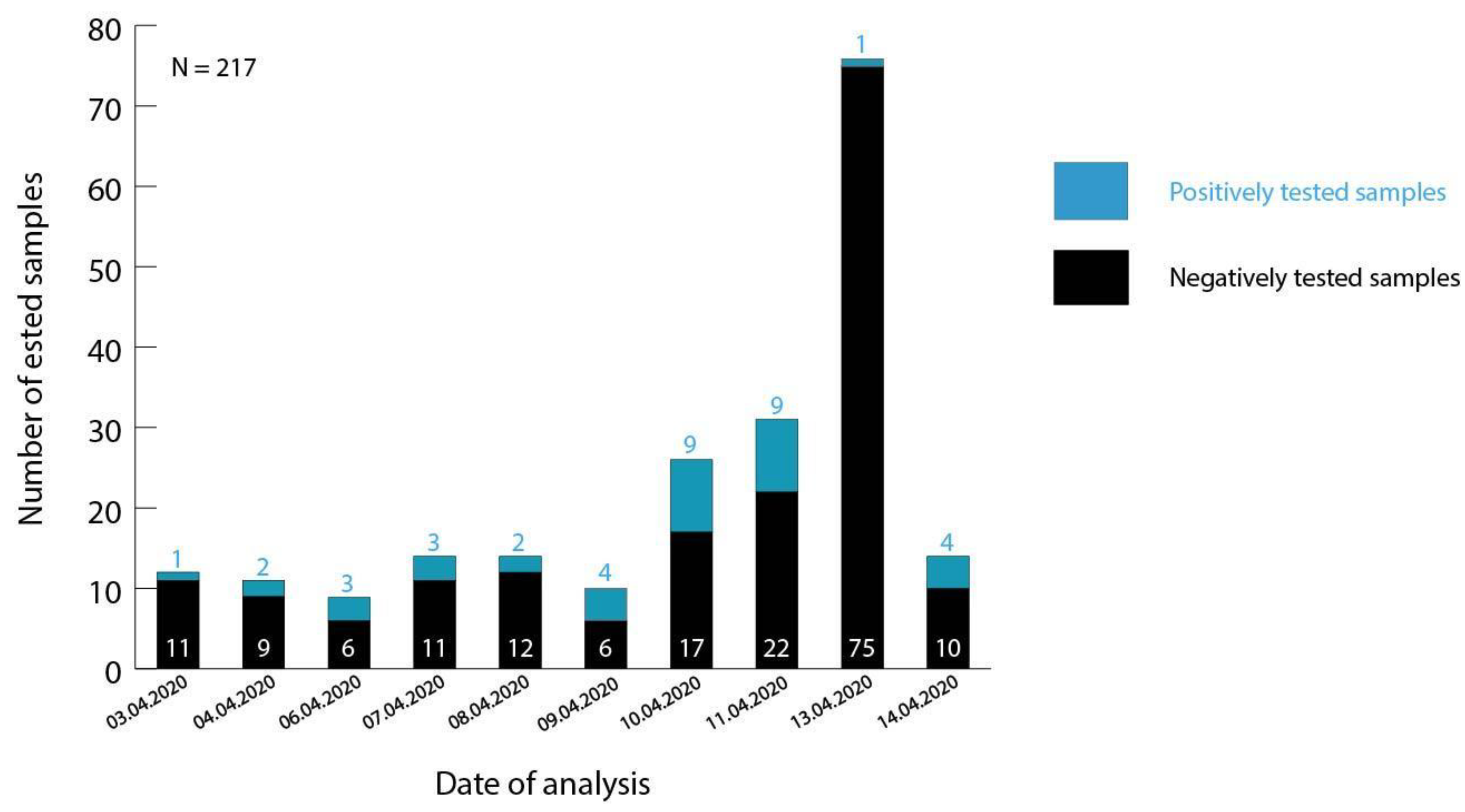
3.1. Testing of Malian Patient Samples
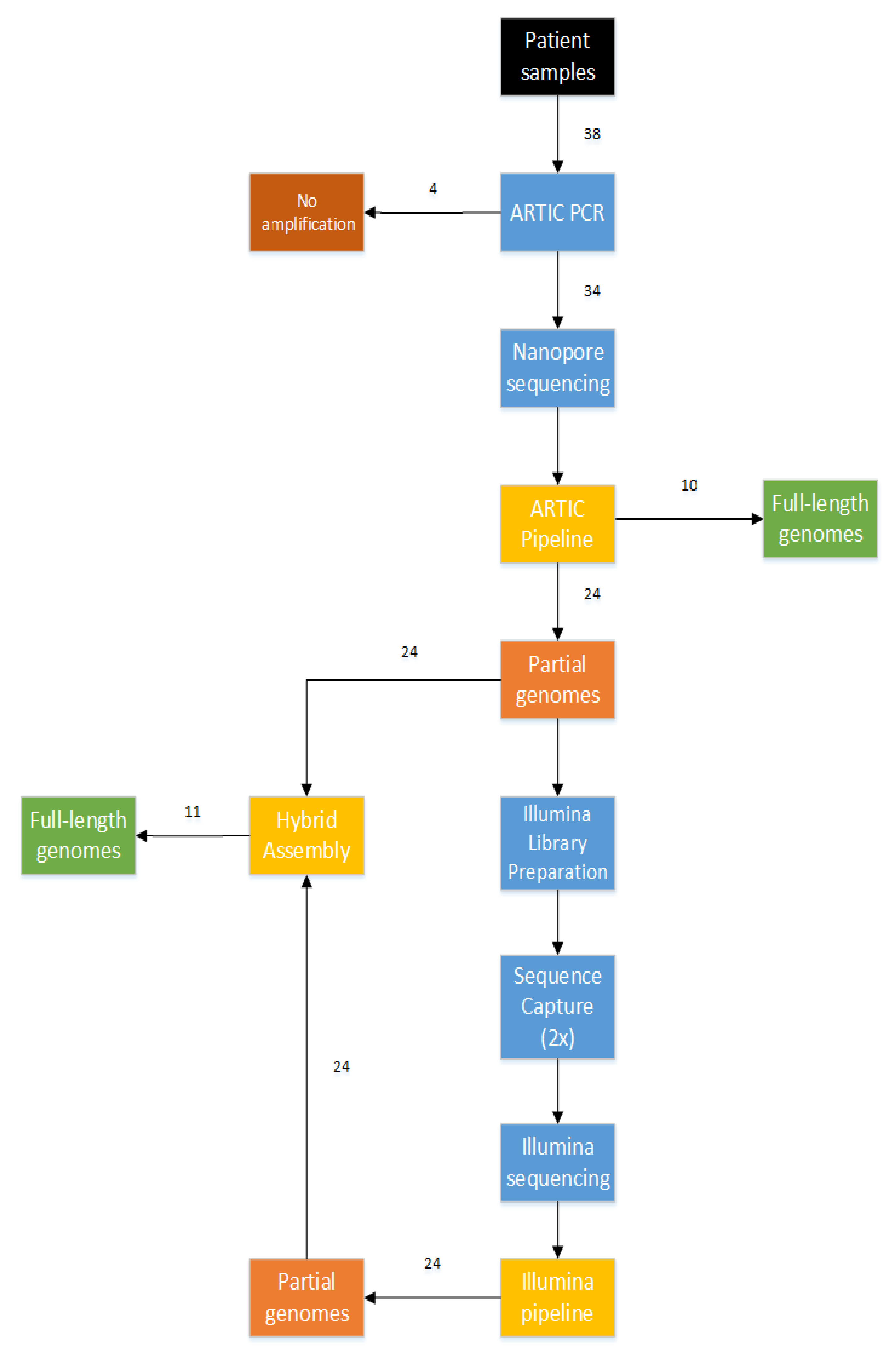
3.2. Combination of Short-Read and Long-Read Sequencing Technologies, Resulting in the First 21 Full-Length Genomes of Mali
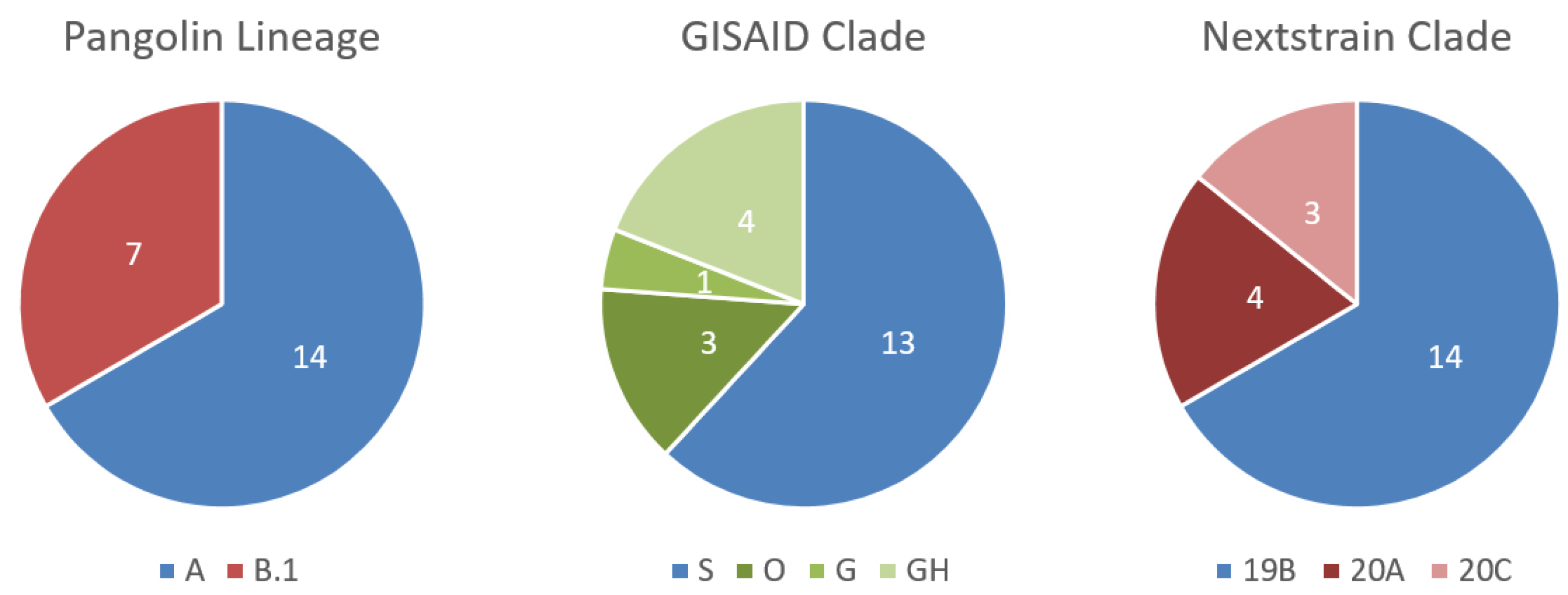
3.3. Testing of Malian Patient Samples, Where Malian SARS-CoV-2 Genomes Can Be Assigned to Two Different Lineages
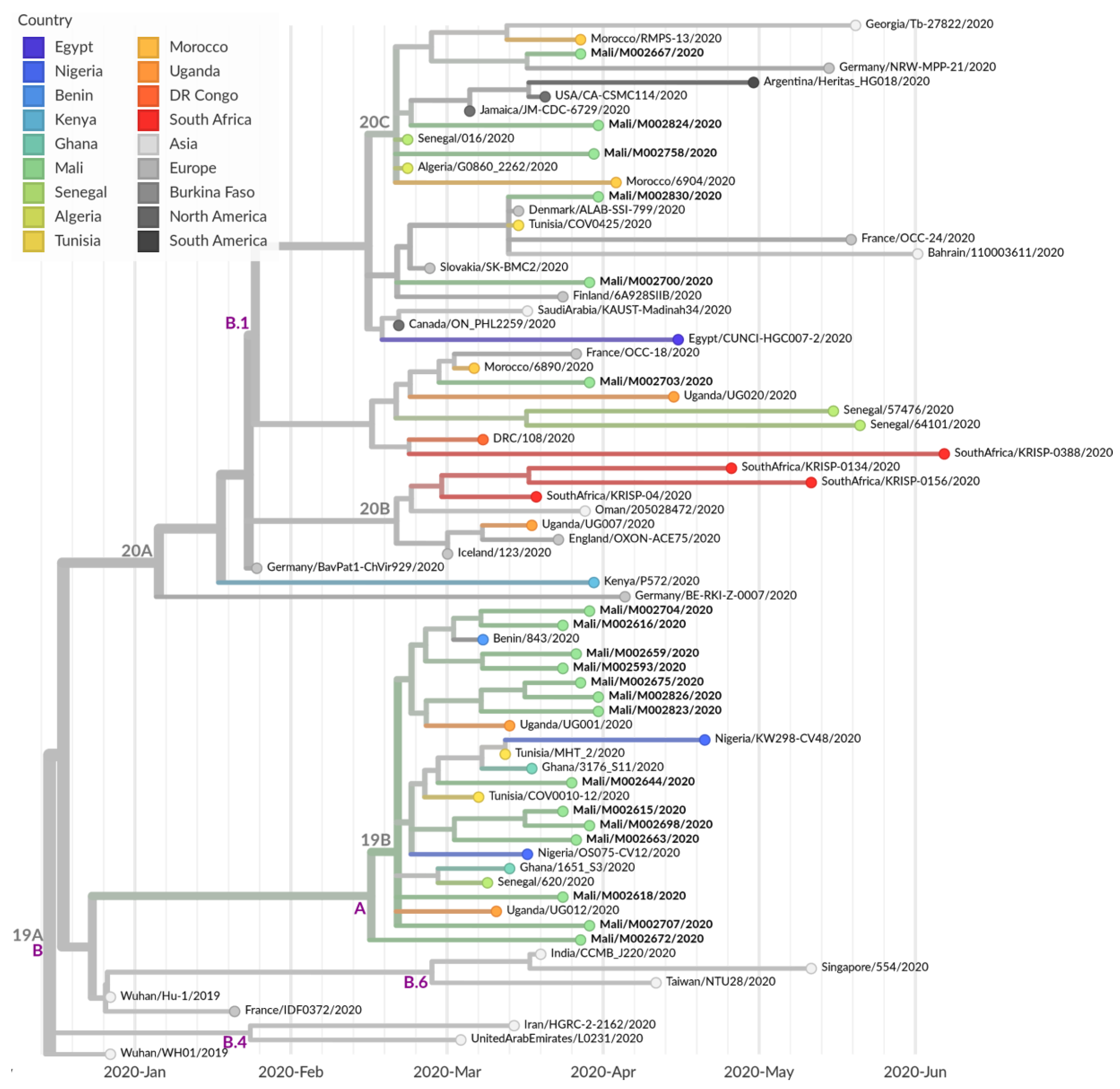
3.4. Further Characterization of SARS-CoV-2 Genomes Derived from Malian Patients
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early transmission dynamics in Wuhan, China, of Novel Coronavirus–infected pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- WHO Regional Office for Europe. World Health Organization Europe Coronavirus (COVID-19). Available online: https://www.euro.who.int/en/health-topics/health-emergencies/coronavirus-covid-19/novel-coronavirus-2019-ncov (accessed on 10 July 2020).
- WHO Regional Office for Africa. World Health Organization Africa Coronavirus (COVID-19). Available online: https://www.afro.who.int/health-topics/coronavirus-covid-19 (accessed on 10 July 2020).
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Mali Government Sur les Premieres cas de Coronavirus au Mali—Communique du Gouvernement de la Republique du Mali; Mali Government: Bamako, Mali, 2020. Available online: http://www.sante.gov.ml/index.php/actualites/communiques/item/3459-urgent-communique-du-gouvernement-de-la-republique-du-mali-sur-les-premiers-cas-de-coronavirus-au-mali (accessed on 2 October 2020).
- The Food Crisis Prevention Network Food and Nutrition Crisis 2020. Available online: http://www.food-security.net/en/topic/food-and-nutrition-crisis-2020/ (accessed on 10 July 2020).
- OECD Coronavirus-West-Africa—Sahel and West Africa Club Secretariat. Available online: http://www.oecd.org/swac/coronavirus-west-africa/ (accessed on 10 July 2020).
- Communique N°238 du Ministere de la Sante et des Affaires Sociales Sur le Suivi des Actions de Prevention et de Riposte Face a la Maladie a Coronavirus. Available online: http://www.sante.gov.ml/index.php/actualites/communiques/item/5857-communique-n-238-du-ministere-de-la-sante-et-du-developpement-social-sur-le-suivi-des-actions-de-prevention-et-de-riposte-face-a-la-maladie-a-coronavirus (accessed on 29 October 2020).
- Africa CDC—COVID-19 Daily Updates 7 March 2020. Available online: https://africacdc.org/news-item/africa-cdc-leads-continental-response-to-covid-19-outbreak-in-africa-statement-by-the-director-of-africa-cdc/ (accessed on 2 October 2020).
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 2020, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreier, J.; Störmer, M.; Kleesiek, K. Use of bacteriophage MS2 as an internal control in viral reverse transcription-PCR assays. J. Clin. Microbiol. 2005, 43, 4551–4557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quick, J. nCoV-2019 Sequencing Protocol. 2020. Available online: https://www.protocols.io/view/ncov-2019-sequencing-protocol-bbmuik6w (accessed on 2 October 2020).
- Quick, J.; Lohman, N. hCoV-2019/nCoV-2019 Version 3 Amplicon Set. Available online: https://artic.network/resources/ncov/ncov-amplicon-v3.pdf (accessed on 28 April 2020).
- Wick, R.R. Porechop. 2017. Available online: https://github.com/rrwick/Porechop (accessed on 2 October 2020).
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Maio, N.; Gozashti, L.; Turakhia, Y.; Walker, C.; Lanfear, R.; Corbett-Detig, R.; Goldman, N. Issues with SARS-CoV-2 Sequencing Data. Available online: https://virological.org/t/issues-with-sars-cov-2-sequencing-data/473 (accessed on 24 July 2020).
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Freunde von GISAID, e.V. Clade and Lineage Nomenclature Aids in Genomic Epidemiology Studies of Active hCoV-19 Viruses. Available online: https://www.gisaid.org/references/statements-clarifications/clade-and-lineage-nomenclature-aids-in-genomic-epidemiology-of-active-hcov-19-viruses/ (accessed on 22 July 2020).
- Year-Letter Genetic Clade Naming for SARS-CoV-2 on Nextstain.org. Available online: https://nextstrain.org//blog/2020-06-02-SARSCoV2-clade-naming (accessed on 25 June 2020).
- Chrisman, B.; Paskov, K.; Stockham, N.; Jung, J.-Y.; Varma, M.; Washington, P.; Wall, D.P. Common Microdeletions in SARS-CoV-2 Sequences. Available online: https://virological.org/t/common-microdeletions-in-sars-cov-2-sequences/485 (accessed on 13 July 2020).
- Van Dorp, L.; Acman, M.; Richard, D.; Shaw, L.P.; Ford, C.E.; Ormond, L.; Owen, C.J.; Pang, J.; Tan, C.C.S.; Boshier, F.A.T.; et al. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect. Genet. Evol. 2020, 83, 104351. [Google Scholar] [CrossRef] [PubMed]
- Giandhari, J.; Pillay, S.; Wilkinson, E.; Tegally, H.; Sinayskiy, I.; Schuld, M.; Lourenço, J.; Chimukangara, B.; Lessells, R.J.; Moosa, Y.; et al. Early transmission of SARS-CoV-2 in South Africa: An epidemiological and phylogenetic report. medRxiv 2020. [Google Scholar] [CrossRef]
- Githinji, G. Introduction and Local Transmission of SARS-CoV-2 Cases in Kenya. Available online: https://virological.org/t/introduction-and-local-transmission-of-sars-cov-2-cases-in-kenya/497 (accessed on 10 July 2020).
- Onuliyi, P. SARS-CoV-2 Genomes from Nigeria Reveal Community Transmission, Multiple Virus Lineages and Spike Protein Mutation Associated with Higher Transmission and Pathogenicity. Available online: https://virological.org/t/sars-cov-2-genomes-from-nigeria-reveal-community-transmission-multiple-virus-lineages-and-spike-protein-mutation-associated-with-higher-transmission-and-pathogenicity/494 (accessed on 10 July 2020).
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Mutation (nt) | Mutation (aa) | Counts | Samples |
|---|---|---|---|---|
| 5′UTR | C241T | - | 6 | M002667,M002672,M002703,M002758,M002824,M002830 |
| ORF1a | A361G | syn | 2 | M002644 |
| C1059T | T265I | 3 | M002667,M002758,M002824 | |
| C1968T | T563I | 2 | M002672,M002703 | |
| C2416T | syn | 2 | M002700,M002830 | |
| C3037T | syn | 7 | M002667,M002673,M002700,M002703,M002758,M002824,M002830 | |
| C8782T | syn | 14 | M002593,M002615,M002616,M002618,M002644,M002659,M002663,M002672,M002675,M002698,M002704,M002707,M002823,M002826 | |
| G11083T(hp) | L3606F | 2 | M002593,M002659 | |
| G11417T | V3718F | 3 | M002675,M002823,M002826 | |
| C11747T | syn | 2 | M002672,M002703 | |
| ORF1b | C14408T | P314L | 7 | M002667,M002673,M002700,M002703,M002758,M002824,M002830 |
| C15324T | syn | 2 | M002673,M002703 | |
| C16658T(het) | T1064I | 2 | M002675,M002707 | |
| S | G22468T | syn | 4 | M002615,M002644,M002663,M002698 |
| A23403G | D614G | 6 | M002667,M002700,M002703,M002758,M002824,M002830 | |
| ORF3a | G25563T | Q57H | 5 | M002667,M002700,M002758,M002824,M002830 |
| C25904T | S171L | 3 | M002615,M002663,M002698 | |
| ORF8 | T28144C | L84S | 13 | M002593,M002615,M002616,M002618,M002644,M002659,M002663,M002675,M002698,M002704,M002707,M002823,M002826 |
| N | G28878A | S202N | 14 | M002593,M002615,M002616,M002618,M002644,M002659,M002663,M002672,M002675,M002698,M002704,M002707,M002823,M002826 |
| 3′UTR | G29742A | - | 12 | M002593,M002615,M002616,M002618,M002644,M002659,M002663,M002672,M002675,M002704,M002707,M002826 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kouriba, B.; Dürr, A.; Rehn, A.; Sangaré, A.K.; Traoré, B.Y.; Bestehorn-Willmann, M.S.; Ouedraogo, J.; Heitzer, A.; Sogodogo, E.; Maiga, A.; et al. First Phylogenetic Analysis of Malian SARS-CoV-2 Sequences Provides Molecular Insights into the Genomic Diversity of the Sahel Region. Viruses 2020, 12, 1251. https://doi.org/10.3390/v12111251
Kouriba B, Dürr A, Rehn A, Sangaré AK, Traoré BY, Bestehorn-Willmann MS, Ouedraogo J, Heitzer A, Sogodogo E, Maiga A, et al. First Phylogenetic Analysis of Malian SARS-CoV-2 Sequences Provides Molecular Insights into the Genomic Diversity of the Sahel Region. Viruses. 2020; 12(11):1251. https://doi.org/10.3390/v12111251
Chicago/Turabian StyleKouriba, Bourema, Angela Dürr, Alexandra Rehn, Abdoul Karim Sangaré, Brehima Y. Traoré, Malena S. Bestehorn-Willmann, Judicael Ouedraogo, Asli Heitzer, Elisabeth Sogodogo, Abderrhamane Maiga, and et al. 2020. "First Phylogenetic Analysis of Malian SARS-CoV-2 Sequences Provides Molecular Insights into the Genomic Diversity of the Sahel Region" Viruses 12, no. 11: 1251. https://doi.org/10.3390/v12111251
APA StyleKouriba, B., Dürr, A., Rehn, A., Sangaré, A. K., Traoré, B. Y., Bestehorn-Willmann, M. S., Ouedraogo, J., Heitzer, A., Sogodogo, E., Maiga, A., Walter, M. C., Zimmermann, F., Wölfel, R., & Antwerpen, M. H. (2020). First Phylogenetic Analysis of Malian SARS-CoV-2 Sequences Provides Molecular Insights into the Genomic Diversity of the Sahel Region. Viruses, 12(11), 1251. https://doi.org/10.3390/v12111251