Transcriptional Landscape of Staphylococcus aureus Kayvirus Bacteriophage vB_SauM-515A1

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Growth Conditions
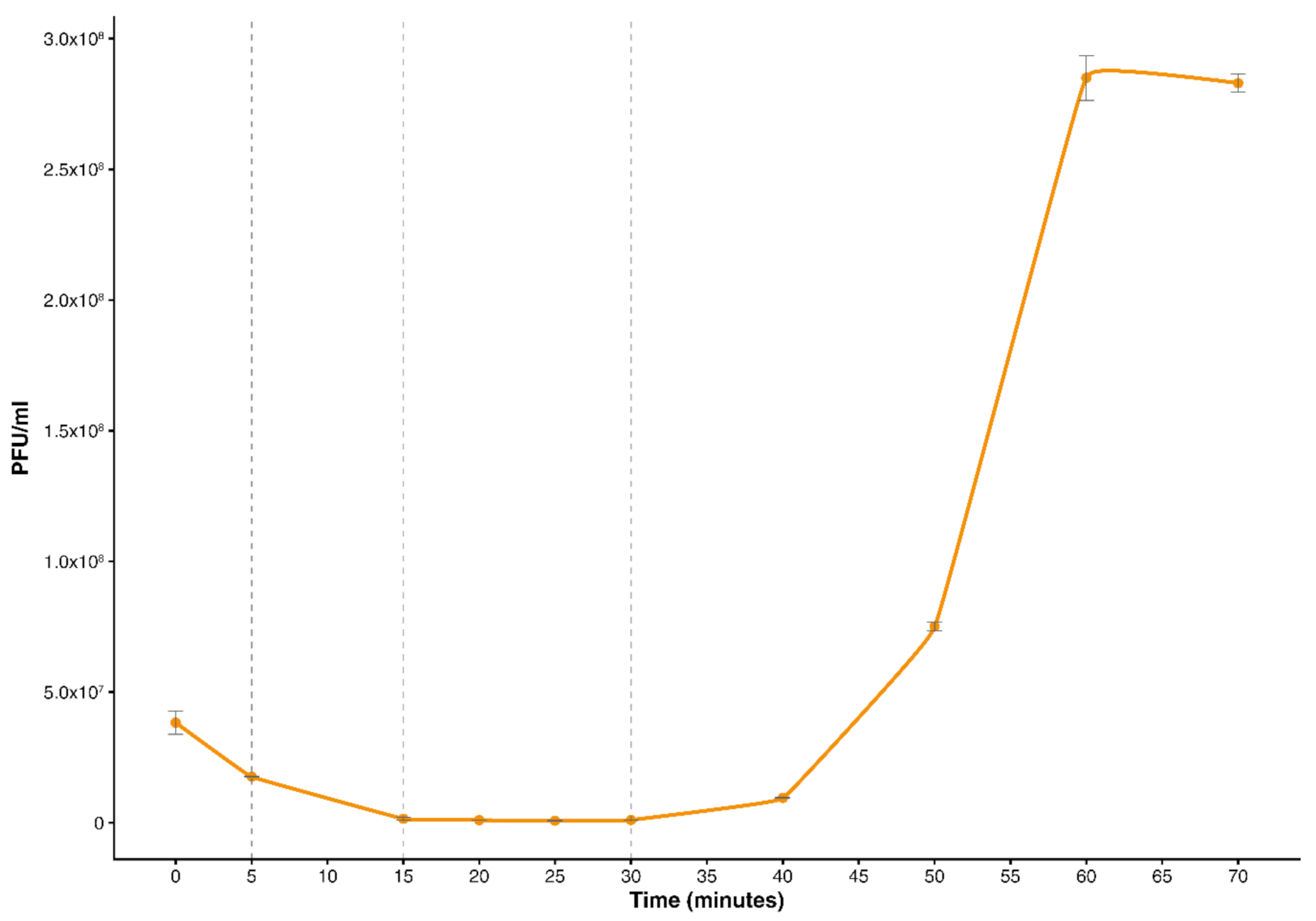
2.2. The One-Step Growth Curve of vB_SauM-515A1 Phage
2.3. Total RNA Extraction
2.4. RNA Sequencing and Analysis
3. Results
3.1. One-Step Growth Curve of vB_SauM-515A1
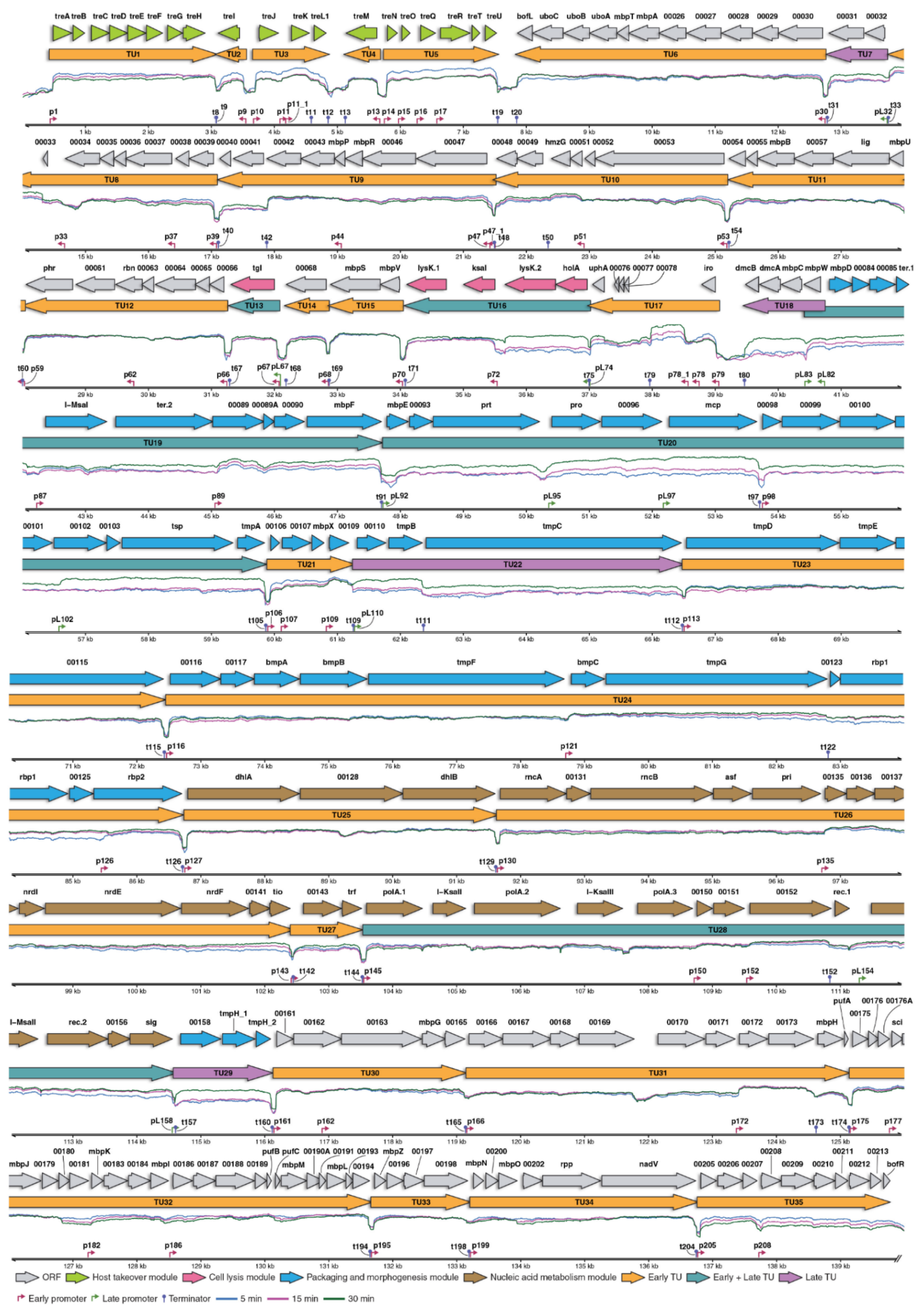
3.2. Global Transcriptional Changes during Bacteriophage Infection
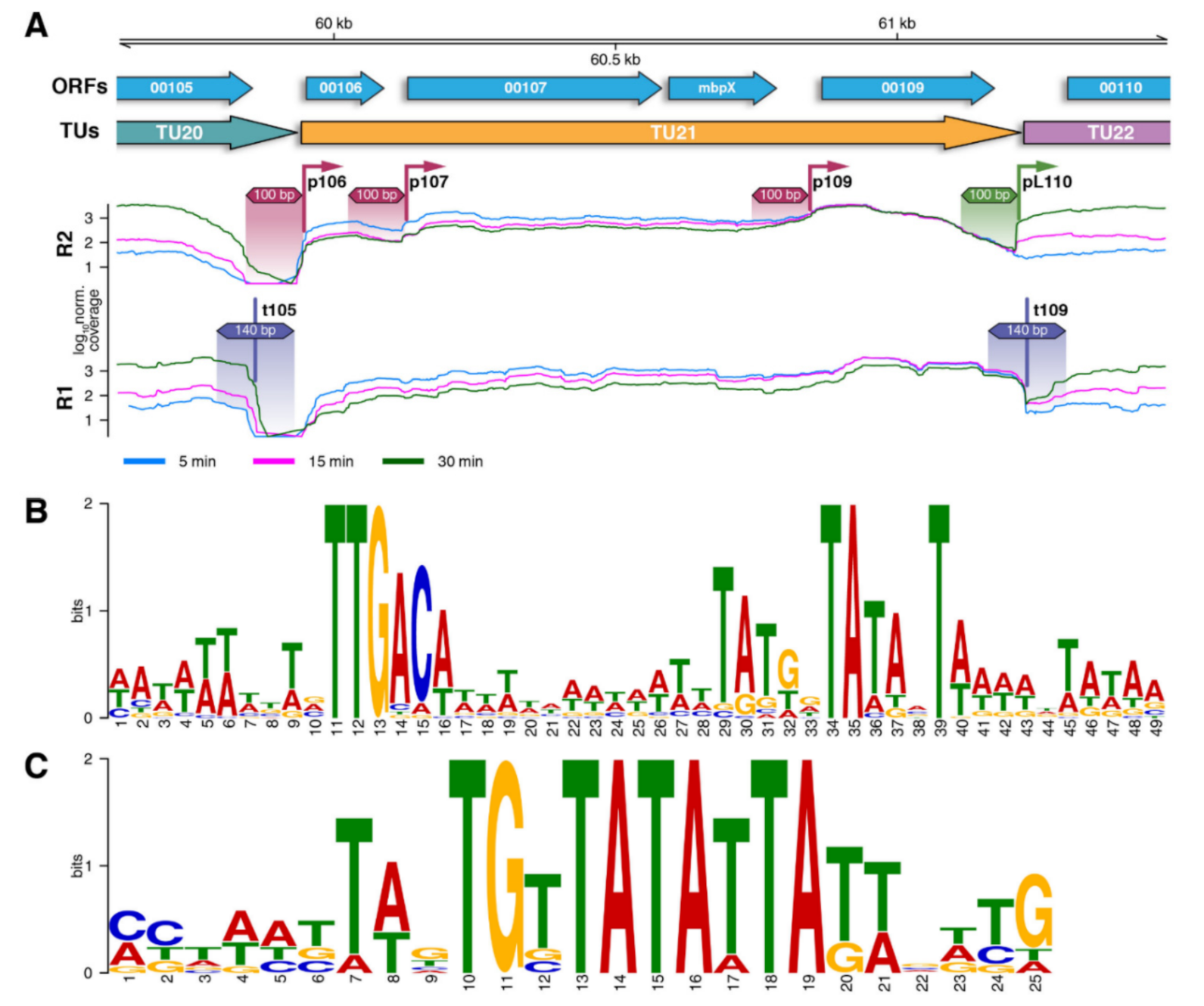
3.3. Analysis of Bacteriophage Promoters and Terminators
3.4. Transcriptional Landscape of vB_SauM-515A1 Phage
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Letarov, A.V. History of Early Bacteriophage Research and Emergence of Key Concepts in Virology. Biochemistry 2020, 85, 1093–1112. [Google Scholar] [PubMed]
- Perros, M. A sustainable model for antibiotics. Science 2015, 347, 1062–1064. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.H.; Letarov, A.V.; Clokie, M. Neat Science in a Messy World: The Global Impact of Human Behavior on Phage Therapy, Past and Present. PHAGE 2020, 1, 16–22. [Google Scholar] [CrossRef] [Green Version]
- Gordillo Altamirano, F.L.; Barr, J.J. Phage therapy in the postantibiotic era. Clin. Microbiol. Rev. 2019, 32, e00066-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinbauer, M.G. Ecology of prokaryotic viruses. FEMS Microbiol. Rev. 2004, 28, 127–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyman, P.; Abedon, S.T. Bacteriophage host range and bacterial resistance. Adv. Appl. Microbiol. 2010, 70, 217–248. [Google Scholar] [PubMed]
- Oechslin, F. Resistance development to bacteriophages occurring during bacteriophage therapy. Viruses 2018, 10, 351. [Google Scholar] [CrossRef] [Green Version]
- Schooley, R.T.; Biswas, B.; Gill, J.J.; Hernandez-Morales, A.; Lancaster, J.; Lessor, L.; Barr, J.J.; Reed, S.L.; Rohwer, F.; Benler, S.; et al. Development and use of personalized bacteriophage-based therapeutic cocktails to treat a patient with a disseminated resistant Acinetobacter baumannii infection. Antimicrob. Agents Chemother. 2017, 61, e00954-17. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Batra, H.; Dong, J.; Chen, C.; Rao, V.B.; Tao, P. Genetic engineering of bacteriophages against infectious diseases. Front. Microbiol. 2019, 10, 954. [Google Scholar] [CrossRef]
- Ferriol-González, C.; Domingo-Calap, P. Phages for biofilm removal. Antibiotics 2020, 9, 268. [Google Scholar] [CrossRef]
- Kaur, S.; Harjai, K.; Chhibber, S. In Vivo Assessment of Phage and Linezolid Based Implant Coatings for Treatment of Methicillin Resistant S. aureus (MRSA) Mediated Orthopaedic Device Related Infections. PLoS ONE 2016, 11, e0157626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhibber, S.; Kaur, J.; Kaur, S. Liposome entrapment of bacteriophages improves wound healing in a diabetic mouse MRSA infection. Front. Microbiol. 2018, 9, 561. [Google Scholar] [CrossRef] [PubMed]
- Fish, R.; Kutter, E.; Bryan, D.; Wheat, G.; Kuhl, S. Resolving Digital Staphylococcal Osteomyelitis Using Bacteriophage—A Case Report. Antibiotics 2018, 7, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morozova, V.V.; Vlassov, V.V.; Tikunova, N.V. Applications of bacteriophages in the treatment of localized infections in humans. Front. Microbiol. 2018, 9, 1696. [Google Scholar] [CrossRef] [Green Version]
- Phage Products|Bacteriophage.News. Available online: https://www.bacteriophage.news/phage-products/ (accessed on 9 November 2020).
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/ (accessed on 9 November 2020).
- Leskinen, K.; Tuomala, H.; Wicklund, A.; Horsma-Heikkinen, J.; Kuusela, P.; Skurnik, M.; Kiljunen, S. Characterization of vB_SauM-fRuSau02, a twort-like bacteriophage isolated from a therapeutic phage cocktail. Viruses 2017, 9, 258. [Google Scholar] [CrossRef]
- Kwan, T.; Liu, J.; DuBow, M.; Gros, P.; Pelletier, J. The complete genomes and proteomes of 27 Staphylococcus aureus bacteriophages. Proc. Natl. Acad. Sci. USA 2005, 102, 5174–5179. [Google Scholar] [CrossRef] [Green Version]
- Xia, G.; Corrigan, R.M.; Winstel, V.; Goerke, C.; Gründling, A.; Peschel, A. Wall teichoic acid-dependent adsorption of staphylococcal siphovirus and myovirus. J. Bacteriol. 2011, 193, 4006–4009. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Gerlach, D.; Du, X.; Larsen, J.; Stegger, M.; Kuhner, P.; Peschel, A.; Xia, G.; Winstel, V. An accessory wall teichoic acid glycosyltransferase protects Staphylococcus aureus from the lytic activity of Podoviridae. Sci. Rep. 2015, 5, 1–10. [Google Scholar] [CrossRef]
- O’Flaherty, S.; Coffey, A.; Edwards, R.; Meaney, W.; Fitzgerald, G.F.; Ross, R.P. Genome of Staphylococcal Phage K: A New Lineage of Myoviridae Infecting Gram-Positive Bacteria with a Low G+C Content. J. Bacteriol. 2004, 186, 2862–2871. [Google Scholar] [CrossRef] [Green Version]
- Botka, T.; Pantůček, R.; Mašlaňová, I.; Benešík, M.; Petráš, P.; Růžičková, V.; Havlíčková, P.; Varga, M.; Žemličková, H.; Koláčková, I.; et al. Lytic and genomic properties of spontaneous host-range Kayvirus mutants prove their suitability for upgrading phage therapeutics against staphylococci. Sci. Rep. 2019, 9, 5475. [Google Scholar] [CrossRef]
- Mojardín, L.; Salas, M. Global Transcriptional Analysis of Virus-Host Interactions between Phage ϕ29 and Bacillus subtilis. J. Virol. 2016, 90, 9293–9304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacher, J.C.; Flint, A.; Butcher, J.; Blasdel, B.; Reynolds, H.M.; Lavigne, R.; Stintzi, A.; Szymanski, C.M. Transcriptomic analysis of the campylobacter Jejuni response to T4-like phage NCTC 12673 infection. Viruses 2018, 10, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceyssens, P.-J.; Minakhin, L.; Van den Bossche, A.; Yakunina, M.; Klimuk, E.; Blasdel, B.; De Smet, J.; Noben, J.-P.; Blasi, U.; Severinov, K.; et al. Development of Giant Bacteriophage KZ Is Independent of the Host Transcription Apparatus. J. Virol. 2014, 88, 10501–10510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornienko, M.; Kuptsov, N.; Gorodnichev, R.; Bespiatykh, D.; Guliaev, A.; Letarova, M.; Kulikov, E.; Veselovsky, V.; Malakhova, M.; Ilina, E.; et al. Contribution of Podoviridae and Myoviridae bacteriophages to the effectiveness of anti-staphylococcal therapeutic cocktails. Sci. Rep. 2020, 10, 18612. [Google Scholar] [CrossRef]
- Bespyatykh, J.; Shitikov, E.; Bespiatykh, D.; Guliaev, A.; Klimina, K.; Veselovsky, V.; Arapidi, G.; Dogonadze, M.; Zhuravlev, V.; Ilina, E.; et al. Metabolic Changes of Mycobacterium tuberculosis during the Anti-Tuberculosis Therapy. Pathogens 2020, 9, 131. [Google Scholar] [CrossRef] [Green Version]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Smid, M.; van den Braak, R.R.J.C.; van de Werken, H.J.G.; van Riet, J.; van Galen, A.; de Weerd, V.; van der Vlugt-Daane, M.; Bril, S.I.; Lalmahomed, Z.S.; Kloosterman, W.P.; et al. Gene length corrected trimmed mean of M-values (GeTMM) processing of RNA-seq data performs similarly in intersample analyses while improving intrasample comparisons. BMC Bioinform. 2018, 19, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahne, F.; Ivanek, R. Visualizing genomic data using Gviz and bioconductor. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2016; Volume 1418, pp. 335–351. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 16 October 2020).
- Garanina, I.A.; Fisunov, G.Y.; Govorun, V.M. BAC-BROWSER: The Tool for Visualization and Analysis of Prokaryotic Genomes. Front. Microbiol. 2018, 9, 2827. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, M.; Rocha, M.; Oliveira, H.; DIas, O.; Valencia, A. Predicting promoters in phage genomes using PhagePromoter. Bioinformatics 2019, 35, 5301–5302. [Google Scholar] [CrossRef] [PubMed]
- Mazin, P.V.; Fisunov, G.Y.; Gorbachev, A.Y.; Kapitskaya, K.Y.; Altukhov, I.A.; Semashko, T.A.; Alexeev, D.G.; Govorun, V.M. Transcriptome analysis reveals novel regulatory mechanisms in a genome-reduced bacterium. Nucleic Acids Res. 2014, 42, 13254–13268. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Williams, N.; Misleh, C.; Li, W.W. MEME: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006, 34, W369–W373. [Google Scholar] [CrossRef]
- Choe, D.; Szubin, R.; Dahesh, S.; Cho, S.; Nizet, V.; Palsson, B.; Cho, B.K. Genome-scale analysis of Methicillin-resistant Staphylococcus aureus USA300 reveals a tradeoff between pathogenesis and drug resistance. Sci. Rep. 2018, 8, 2215. [Google Scholar] [CrossRef] [Green Version]
- Jeng, S.-T.; Gardner, J.F.; Gumport, R.I. Transcription Termination by Bacteriophage T7 RNA Polymerase at Rho-independent Terminators. J. Biol. Chem. 1990, 265, 3823–3830. [Google Scholar]
- Łobocka, M.; Hejnowicz, M.S.; Dabrowski, K.; Gozdek, A.; Kosakowski, J.; Witkowska, M.; Ulatowska, M.I.; Weber-Dabrowska, B.; Kwiatek, M.; Parasion, S.; et al. Genomics of Staphylococcal Twort-like Phages—Potential Therapeutics of the Post-Antibiotic Era. In Advances in Virus Research; Academic Press Inc.: Cambridge, MA, USA, 2012; Volume 83, pp. 143–216. [Google Scholar]
- Deghorain, M.; Van Melderen, L. The staphylococci phages family: An overview. Viruses 2012, 4, 3316–3335. [Google Scholar] [CrossRef] [Green Version]
- Mäder, U.; Nicolas, P.; Depke, M.; Pané-Farré, J.; Debarbouille, M.; van der Kooi-Pol, M.M.; Guérin, C.; Dérozier, S.; Hiron, A.; Jarmer, H.; et al. Staphylococcus aureus Transcriptome Architecture: From Laboratory to Infection-Mimicking Conditions. PLoS Genet. 2016, 12, e1005962. [Google Scholar] [CrossRef]
- Hinton, D.M. Transcriptional control in the prereplicative phase of T4 development. Virol. J. 2010, 7, 289. [Google Scholar] [CrossRef] [Green Version]
- Osmundson, J.; Montero-Diez, C.; Westblade, L.F.; Hochschild, A.; Darst, S.A. Promoter-specific transcription inhibition in staphylococcus aureus by a phage protein. Cell 2012, 151, 1005–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osmundson, J.; Darst, S.A. Biochemical insights into the function of phage G1 gp67 in Staphylococcus aureus. Bacteriophage 2013, 3, e24767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, K.A.; Breaker, R.R. Large Noncoding RNAs in Bacteria. In Regulating with RNA in Bacteria and Archaea; American Society of Microbiology: Washington, DC, USA, 2018; Volume 6, pp. 515–526. [Google Scholar]
- Bloch, S.; Lewandowska, N.; Węgrzyn, G.; Nejman-Faleńczyk, B. Bacteriophages as sources of small non-coding RNA molecules. Plasmid 2020, 102527. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kornienko, M.; Fisunov, G.; Bespiatykh, D.; Kuptsov, N.; Gorodnichev, R.; Klimina, K.; Kulikov, E.; Ilina, E.; Letarov, A.; Shitikov, E. Transcriptional Landscape of Staphylococcus aureus Kayvirus Bacteriophage vB_SauM-515A1. Viruses 2020, 12, 1320. https://doi.org/10.3390/v12111320
Kornienko M, Fisunov G, Bespiatykh D, Kuptsov N, Gorodnichev R, Klimina K, Kulikov E, Ilina E, Letarov A, Shitikov E. Transcriptional Landscape of Staphylococcus aureus Kayvirus Bacteriophage vB_SauM-515A1. Viruses. 2020; 12(11):1320. https://doi.org/10.3390/v12111320
Chicago/Turabian StyleKornienko, Maria, Gleb Fisunov, Dmitry Bespiatykh, Nikita Kuptsov, Roman Gorodnichev, Ksenia Klimina, Eugene Kulikov, Elena Ilina, Andrey Letarov, and Egor Shitikov. 2020. "Transcriptional Landscape of Staphylococcus aureus Kayvirus Bacteriophage vB_SauM-515A1" Viruses 12, no. 11: 1320. https://doi.org/10.3390/v12111320
APA StyleKornienko, M., Fisunov, G., Bespiatykh, D., Kuptsov, N., Gorodnichev, R., Klimina, K., Kulikov, E., Ilina, E., Letarov, A., & Shitikov, E. (2020). Transcriptional Landscape of Staphylococcus aureus Kayvirus Bacteriophage vB_SauM-515A1. Viruses, 12(11), 1320. https://doi.org/10.3390/v12111320