Rhinovirus-Induced Modulation of Epithelial Phenotype: Role in Asthma


Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
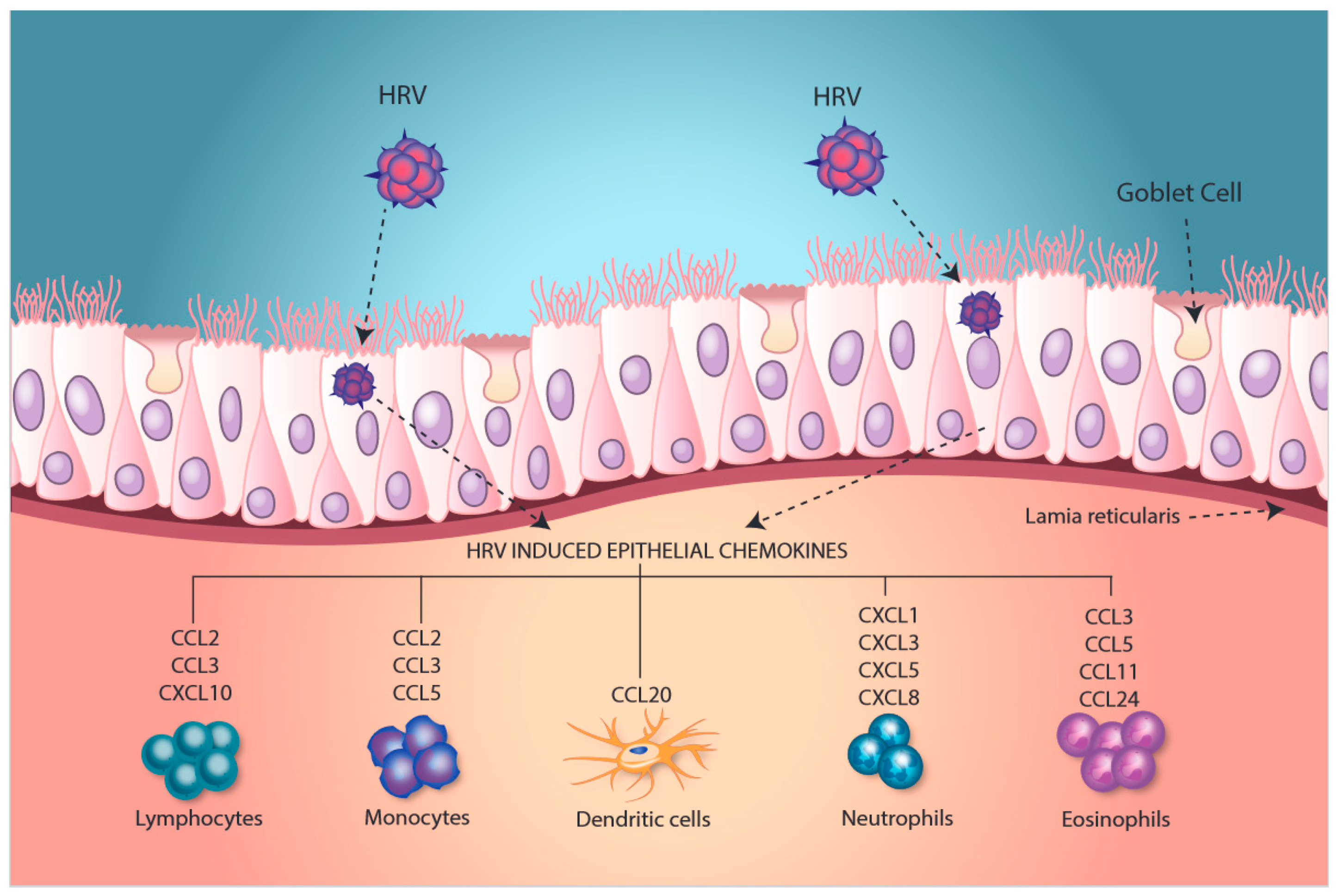
2. Modulation of an Inflammatory Phenotype
3. Induction of Epithelial Antiviral Responses
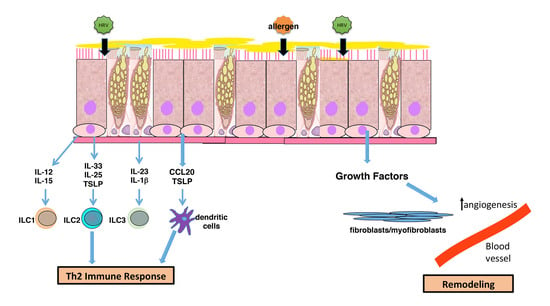
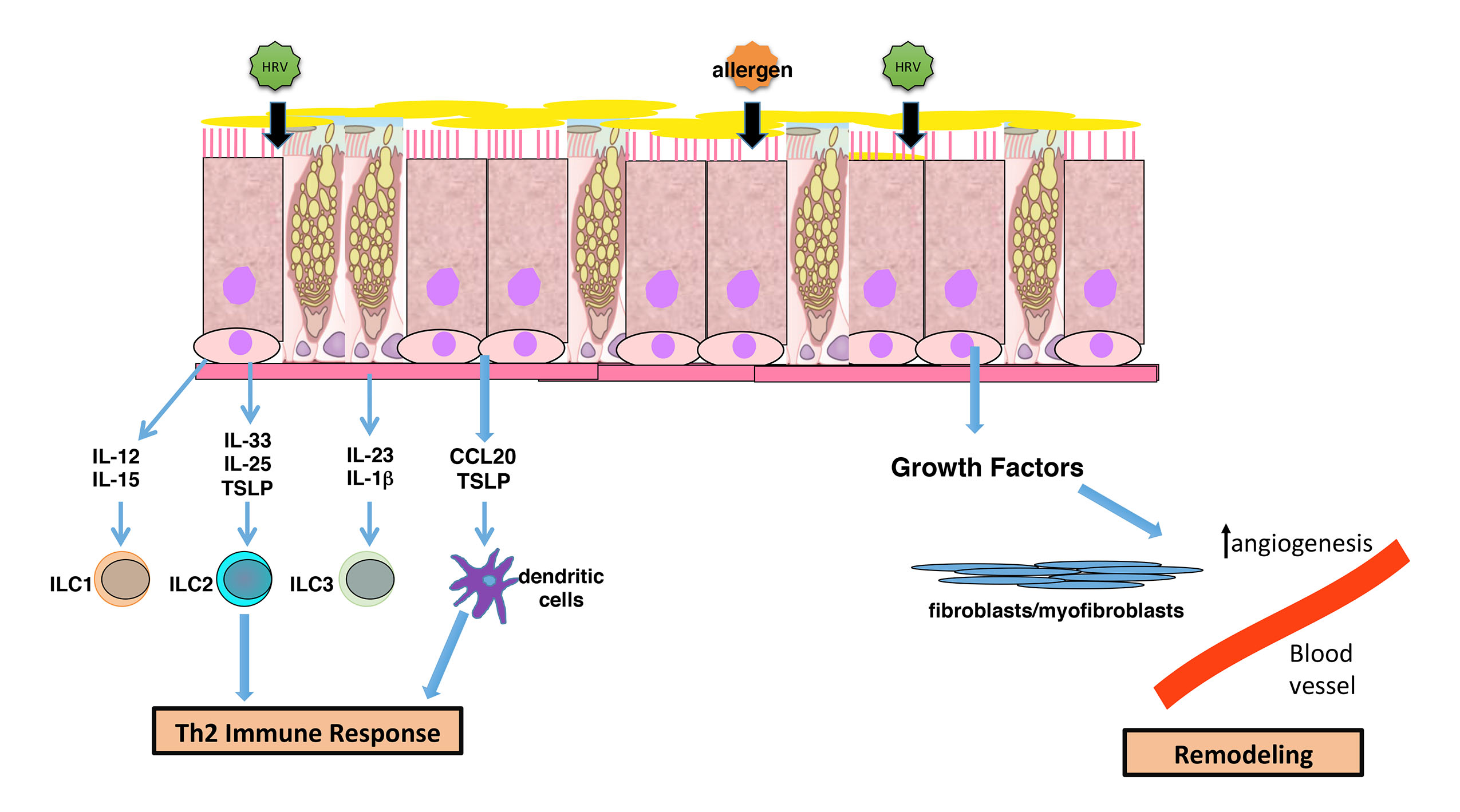
4. Airway Remodeling and Altered Epithelial Barrier
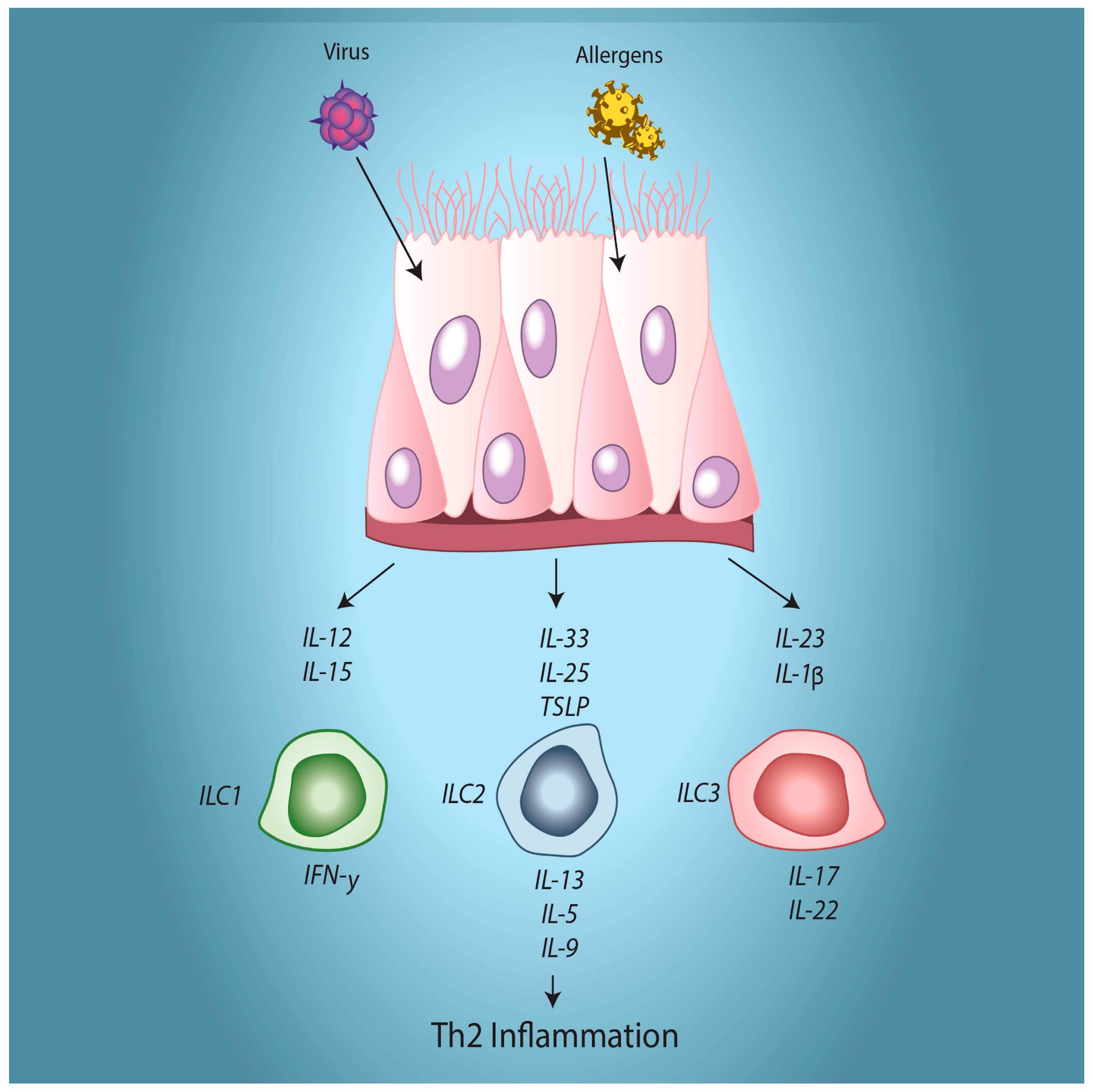
5. Modulation of Immune Interactions
6. Summary and Implications for Asthma Pathogenesis
Author Contributions
Funding
Conflicts of Interest
References
- Global Initiative for Asthma. Global Strategy for Asthma Management and Prevention; GINA Reports; Global Initiative for Asthma: Fontana, WI, USA, 2012. [Google Scholar]
- Masoli, M.; Fabian, D.; Holt, S.; Beasley, R. The global burden of asthma: Executive summary of the GINA dissemination committee report. Allergy 2004, 59, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, K.C.; Warner, S.M.; Leigh, R.; Proud, D. Rhinovirus in the pathogenesis and clinical course of asthma. Chest 2015, 148, 1508–1516. [Google Scholar] [CrossRef] [PubMed]
- Proud, D. Role of rhinovirus infections in asthma. Asian Pac. J. Allergy Immunol. 2011, 29, 201–208. [Google Scholar] [PubMed]
- Kotaniemi-Syrjänen, A.; Vainionpää, R.; Reijonen, T.M.; Waris, M.; Korhonen, K.; Korppi, M. Rhinovirus-induced wheezing in infancy-the first sign of childhood asthma? J. Allergy Clin. Immunol. 2003, 111, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.J.; Gangnon, R.E.; Evans, M.D.; Roberg, K.A.; Anderson, E.L.; Pappas, T.E.; Printz, M.C.; Lee, W.-M.; Shult, P.A.; Reisdorf, E.; et al. Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am. J. Respir. Crit. Care Med. 2008, 178, 667–672. [Google Scholar] [CrossRef]
- Arruda, E.; Boyle, T.R.; Winther, B.; Pevear, D.C.; Gwaltney, J.M., Jr.; Hayden, F.G. Localization of human rhinovirus replication in the upper respiratory tract by in situ hybridization. J. Infect. Dis. 1995, 171, 1329–1333. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, N.G.; Bates, P.J.; Bardin, P.G.; Papi, A.; Leir, S.H.; Fraenkel, D.J.; Meyer, J.; Lackie, P.M.; Sanderson, G.; Holgate, S.T.; et al. Rhinoviruses infect the lower airways. J. Infect. Dis. 2000, 181, 1875–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosser, A.G.; Brockman-Schneider, R.; Amineva, S.; Burchell, L.; Sedgewick, J.B.; Busse, W.W.; Gern, J.E. Similar frequency of rhinovirus-infectable cells in upper and lower airway epithelium. J. Infect. Dis. 2002, 185, 734–743. [Google Scholar] [CrossRef]
- Lachowicz-Scroggins, M.E.; Boushey, H.A.; Finkbeiner, W.E.; Widdicombe, J.H. Interleukin-13 induced mucous metaplasia increases susceptibility of human airway epithelium to rhinovirus infection. Am. J. Respir. Cell Mol. Biol. 2010, 43, 652–661. [Google Scholar] [CrossRef] [Green Version]
- Jakiela, B.; Gielicz, A.; Plutecka, H.; Hubalewska-Mazgaj, M.; Mastalerz, L.; Bochenek, G.; Soja, J.; Januszek, R.; Aab, A.; Musial, M.; et al. Th2-type cytokine-induced mucus metaplasia decreases susceptibility of human bronchial epithelium to rhinovirus infection. Am. J. Respir. Cell Mol. Biol. 2014, 51, 229–241. [Google Scholar]
- Griggs, T.F.; Bochkov, Y.A.; Basnet, S.; Pasic, T.R.; Brockman-Schneider, R.A.; Palmenberg, A.C.; Gern, J.E. Rhinovirus c targets ciliated airway epithelial cells. Respir. Res. 2017, 18, 84. [Google Scholar] [CrossRef] [PubMed]
- Warner, S.M.; Wiehler, S.; Michi, A.N.; Proud, D. Rhinovirus replication and innate immunity in highly differentiated human airway epithelial cells. Respir. Res. 2019, 20, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winther, B. Rhinovirus infections in the upper airway. Proc. Am. Thorac. Soc. 2011, 8, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Subauste, M.C.; Jacoby, D.B.; Richards, S.M.; Proud, D. Infection of a human respiratory epithelial cell line with rhinovirus. Induction of cytokine release and modulation of susceptibility to infection by cytokine exposure. J. Clin. Invest. 1995, 96, 549–557. [Google Scholar] [CrossRef] [Green Version]
- Winther, B.; Brofeldt, S.; Christensen, B.; Mygind, N. Light and scanning electron microscopy of nasal biopsy material from patients with naturally acquired common colds. Acta. Otolaryngol. (Stockh) 1984, 97, 309–318. [Google Scholar] [CrossRef]
- Terajima, M.; Yamaya, M.; Sekizawa, K.; Okinaga, S.; Suzuki, T.; Yamada, N.; Nakayama, K.; Ohrui, T.; Oshima, T.; Numazaki, Y.; et al. Rhinovirus infection of primary cultures of human tracheal epithelium: Role of ICAM-1 and IL-1β. Am. J. Physiol. 1997, 273, L749–L759. [Google Scholar] [CrossRef]
- Proud, D.; Sanders, S.P.; Wiehler, S. Human rhinovirus infection induces airway epithelial cell production of human β-defensin-2 both in vitro and in vivo. J. Immunol. 2004, 172, 4637–4645. [Google Scholar] [CrossRef]
- Spurrell, J.C.L.; Wiehler, S.; Zaheer, R.S.; Sanders, S.P.; Proud, D. Human airway epithelial cells produce IP-10 (CXCL10) in vitro and in vivo upon rhinovirus infection. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L85–L95. [Google Scholar] [CrossRef] [Green Version]
- Leigh, R.; Oyelusi, W.; Wiehler, S.; Koetzler, R.; Zaheer, R.S.; Newton, R.; Proud, D. Human rhinovirus infection enhances airway epithelial cell production of growth factors involved in airway remodeling. J. Allergy Clin. Immunol. 2008, 121, 1238–1245. [Google Scholar] [CrossRef]
- Zaheer, R.S.; Wiehler, S.; Hudy, M.H.; Traves, S.L.; Pelikan, J.B.; Leigh, R.; Proud, D. Human rhinovirus-induced ISG15 selectively modulates epithelial antiviral immunity. Mucosal Immunol. 2014, 7, 1127–1138. [Google Scholar] [CrossRef]
- Proud, D.; Gwaltney, J.M., Jr.; Hendley, J.O.; Dinarello, C.A.; Gillis, S.; Schleimer, R.P. Increased levels of interleukin-1 are detected in nasal secretions of volunteers during experimental rhinovirus colds. J. Infect. Dis. 1994, 169, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Loxham, M.; Davies, D.E.; Blume, C. Epithelial function and dysfunction in asthma. Clin. Exp. Allergy 2014, 44, 1299–1313. [Google Scholar] [CrossRef] [PubMed]
- Carsin, A.; Mazenq, J.; Ilstad, A.; Dubus, J.-C.; Chanez, P.; Gras, D. Bronchial epithelium in children: A key player in asthma. Eur. Respir. Rev. 2016, 25, 158–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holgate, S.T. Epithelium dysfunction in asthma. J. Allergy Clin. Immunol. 2007, 120, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Puddicombe, S.M.; Field, S.; Haywood, J.; Broughton-Head, V.; Puxeddu, I.; Haitchi, H.M.; Vernon-Wilson, E.; Sammut, D.; Bedke, N.; et al. Defective epithelial barrier function in asthma. J Allergy Clin. Immunol. 2011, 128, 549–556. [Google Scholar] [CrossRef]
- Montefort, S.; Roberts, J.A.; Beasley, R.; Holgate, S.T.; Roche, W.R. The site of disruption of the bronchial epithelium in asthmatic and non-asthmatic subjects. Thorax 1992, 47, 499–503. [Google Scholar] [CrossRef] [Green Version]
- Singhania, A.; Rupani, H.; Jayasekera, N.; Lumb, S.; Hales, P.; Gozzard, N.; Davies, D.E.; Woelk, C.H.; Howarth, P.H. Altered epithelial gene expression in peripheral airways of severe asthma. PLoS ONE 2017, 12, e0168680. [Google Scholar] [CrossRef]
- Kicic, A.; Sutanto, E.N.; Stevens, P.T.; Knight, D.A.; Stick, S.M. Intrinsic biochemical and functional differences in bronchial epithelial cells of children with asthma. Am. J. Respir. Crit. Care Med. 2006, 174, 1110–1118. [Google Scholar] [CrossRef] [Green Version]
- Hackett, T.-L.; Singhera, G.K.; Shaheen, F.; Hayden, P.; Jackson, G.R.; Hegele, R.G.; Van Eeden, S.; Bai, T.R.; Dorscheid, D.R.; Knight, D.A. Intrinsic phenotypic differences of asthmatic epithelium and its inflammatory responses to respiratory syncytial virus and air pollution. Am. J. Respir. Cell Mol. Biol. 2011, 45, 1090–1100. [Google Scholar] [CrossRef]
- Lopez-Guisa, J.M.; Powers, C.; File, D.; Cochrane, E.; Jiminez, N.; Debley, J.S. Airway epithelial cells from asthmatic children differentially express proremodeling factors. J. Allergy Clin. Immunol. 2012, 129, 990–997. [Google Scholar] [CrossRef] [Green Version]
- Reeves, S.R.; Kolstad, T.; Lien, Y.-U.; Elliott, M.; Ziegler, S.F.; Wight, T.N.; Debley, J.S. Asthmatic airway epithelial cells differentially regulate fibroblast expression of extracellular matrix components. J. Allergy Clin. Immunol. 2014, 134, 663–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sajjan, U.; Wang, Q.; Zhao, Y.; Gruenert, D.C.; Hershenson, M.B. Rhinovirus disrupts the barrier function of polarized airway epithelial cells. Am. J. Respir. Crit. Care Med. 2008, 178, 1271–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holgate, S.T.; Davies, D.E.; Lackie, P.M.; Wilson, S.J.; Puddicombe, S.M.; Lordan, J.L. Epithelial-mesenchymal interactions in the pathogenesis of asthma. J. Allergy Clin. Immunol. 2000, 105, 193–204. [Google Scholar] [CrossRef]
- Holgate, S.T. The sentinal role of the airway epithelium in asthma pathogenesis. Immunol. Rev. 2011, 242, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lau, C.; Wiehler, S.; Pow, A.; Mazzulli, T.; Gutierrez, C.; Proud, D.; Chow, C.-W. Syk is downstream of intercellular adhesion molecule-1 and mediates human rhinovirus activation of p38 MAPK in airway epithelial cells. J. Immunol. 2006, 177, 6859–6870. [Google Scholar] [CrossRef] [Green Version]
- Lau, C.; Wang, X.; Song, L.; North, M.; Wiehler, S.; Proud, D.; Chow, C.-W. Syk associates with clathrin and mediates phosphatidylinositol 3-kinase activation during human rhinovirus internalization. J. Immunol. 2008, 180, 870–880. [Google Scholar] [CrossRef] [Green Version]
- Bentley, J.K.; Newcomb, D.C.; Goldsmith, A.M.; Jia, Y.; Sajjan, U.S.; Hershenson, M.B. Rhinovirus activates interleukin-8 expression via a Src/p110β phosphatylinositol 3-kinase pathway in human airway epithelial cells. J. Virol. 2007, 81, 1186–1194. [Google Scholar] [CrossRef] [Green Version]
- Sajjan, U.S.; Jia, Y.; Newcomb, D.C.; Bentley, J.K.; Lukacs, N.W.; LiPuma, J.J.; Hershenson, M.B. H. influenzae potentiates airway epithelial cell responses to rhinovirus by increasing ICAM-1 and TLR3 expression. FASEB J. 2006, 20, 2121–2123. [Google Scholar] [CrossRef] [Green Version]
- Jamieson, K.C.; Traves, S.L.; Kooi, C.; Wiehler, S.; Dumonceaux, C.J.; Maciejewski, B.A.; Arnason, J.W.; Michi, A.N.; Leigh, R.; Proud, D. Rhinovirus and bacteria synergistically induce IL-17C release from human airway epithelial cells to promote neutrophil recruitment. J. Immunol. 2019, 202, 160–170. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Guo, S.; Hibbert, J.M.; Jain, V.; Singh, N.; Wilson, N.O.; Stiles, J.K. CXCL10/IP10 in infectious diseases pathogenesis and potential therapeutic implications. Cytokine Growth Factor Rev. 2011, 22, 121–130. [Google Scholar]
- Loetscher, P.; Seitz, M.; Clark-Lewis, I.; Baggiolini, M.; Moser, B. Activation of NK cells by CC chemokines. Chemotaxis, Ca2+ mobilization, and enzyme release. J. Immunol. 1996, 156, 322–327. [Google Scholar] [PubMed]
- Schall, T.J.; Bacon, K.; Toy, K.J.; Goeddel, D.V. Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature 1990, 347, 669–671. [Google Scholar] [CrossRef] [PubMed]
- Rot, A.; Krieger, M.; Brunner, T.; Bischoff, S.C.; Schall, T.J.; Dahinden, C.A. RANTES and macrophage inflammatory protein 1α induce the migration and activation of normal human eosinophil granulocytes. J. Exp. Med. 1992, 176, 1489–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroth, M.K.; Grimm, E.; Frindt, P.; Galagan, D.M.; Konno, S.-I.; Love, R.; Gern, J.E. Rhinovirus replication causes RANTES production in primary bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 1999, 20, 1220–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slater, L.; Bartlett, N.W.; Haas, J.J.; Zhu, J.; Message, S.D.; Walton, R.P.; Sykes, A.; Dahdaleh, S.; Clarke, D.L.; Belvisi, M.G.; et al. Co-ordinated role of TLR-3, RIG-I and MDA5 in the innate response to rhinovirus in bronchial epithelium. PLoS Pathog. 2010, 6, e1001178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudy, M.H.; Traves, S.L.; Proud, D. Transcriptional and epigenetic modulation of human rhinovirus-induced CXCL10 production by cigarette smoke. Am. J. Respir. Cell Mol. Biol. 2014, 50, 571–582. [Google Scholar] [CrossRef]
- Silkoff, P.E.; Flavin, S.; Gordon, R.; Loza, M.J.; Sterk, P.J.; Lutter, R.; Diamant, Z.; Turner, R.B.; Lipworth, B.J.; Proud, D.; et al. Toll-like receptor 3 blockade in rhinovirus-induced experimental asthma exacerbations: A randomized controlled study. J. Allergy Clin. Immunol. 2017, 141, 1220–1230. [Google Scholar] [CrossRef] [Green Version]
- Triantafilou, K.; Vakakis, E.; Richer, E.A.J.; Evans, G.L.; Villiers, J.P.; Triantafilou, M. Human rhinovirus recognition in non-immune cells is mediated by Toll-like receptors and MDA-5, which trigger a synergetic pro-inflammatory immune response. Virulence 2011, 2, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, S.; Pham, D.; Jing, Y.; Farazuddin, M.; Hudy, M.H.; Unger, B.; Comstock, A.T.; Proud, D.; Lauring, A.S.; Sajjan, U.S. TLR2 activation limits rhinovirus-srimulated CXCL-10 by attenuating IRAK-1-dependent IL-33 receptor signaling in human bronchial epithelial cells. J. Immunol. 2016, 197, 2409–2420. [Google Scholar] [CrossRef] [Green Version]
- Triantafilou, K.; Kar, S.; van Kuppeveld, F.L.M.; Triantafilou, M. Rhinovirus-induced calcium flux triggers NLRP3 and NLRC5 activation in bronchiall cells. Am. J. Respir. Cell Mol. Biol. 2013, 49, 923–934. [Google Scholar] [CrossRef]
- Unger, B.L.; Ganesan, S.; Comstock, A.T.; Faris, A.M.; Hershenson, M.B.; Sajjan, U.S. Nod-like receptor X-1 is required for rhinovirus-induced barrier dysfunction in airway epithelial cells. J. Virol. 2014, 88, 3705–3718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaheer, R.S.; Proud, D. Human rhinovirus-induced epithelial production of CXCL10 is dependent upon IFN Regulatory Factor-1. Am. J. Respir. Cell Mol. Biol. 2010, 43, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Bosco, A.; Wiehler, S.; Proud, D. Interferon regulatory factor 7 regulates airway epithelial cell responses to human rhinovirus infection. BMC Genom. 2016, 17, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaheer, R.S.; Koetzler, R.; Holden, N.S.; Wiehler, S.; Proud, D. Selective transcriptional down-regulation of human rhinovirus-induced production of CXCL10 from airway epithelial cells via the MEK1 pathway. J. Immunol. 2009, 182, 4854–4864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, R.B.; Weingand, K.W.; Yeh, C.H.; Leedy, D.W. Association between interleukin-8 concentration in nasal secretions and severity of symptoms of experimental rhinovirus colds. Cli. Infect. Dis. 1998, 26, 840–846. [Google Scholar] [CrossRef] [Green Version]
- Teran, L.M.; Seminario, M.C.; Shute, J.K.; Papi, A.; Compton, S.J.; Low, J.L.; Gleich, G.J.; Johnston, S.L. RANTES, macrophage-inhibitory protein 1α, and the eosinophil product major basic protein are released into upper respiratory secretions during virus-induced asthma exacerbations in children. J. Infect. Dis. 1999, 179, 677–681. [Google Scholar] [CrossRef] [Green Version]
- Teran, L.M.; Johnston, S.L.; Schröder, J.-M.; Church, M.K.; Holgate, S.T. Role of nasal interleukin-8 in neutrophil recruitment and activation in children with virus-induced asthma. Am. J. Respir. Crit. Care Med. 1997, 155, 1362–1366. [Google Scholar] [CrossRef]
- Ordoñez, C.L.; Shaugnessy, T.E.; Matthay, M.A.; Fahy, J.V. Increased neutrophil numbers and IL-8 levels in airway secretions in acute severe asthma. Clinical and biologic significance. Am. J. Respir Crit. Care Med. 2000, 161, 1185–1190. [Google Scholar] [CrossRef]
- Proud, D.; Turner, R.B.; Winther, B.; Wiehler, S.; Tiesman, J.P.; Reichling, T.D.; Juhlin, K.D.; Fulmer, A.W.; Ho, B.Y.; Walanski, A.A.; et al. Gene expression profiles during in vivo human rhinovirus infection: Insights into the host response. Am. J. Respir. Crit. Care Med. 2008, 178, 962–968. [Google Scholar] [CrossRef] [Green Version]
- Proud, D.; Hudy, M.H.; Wiehler, S.; Zaheer, R.S.; Amin, M.A.; Pelikan, J.B.; Tacon, C.E.; Tonsaker, T.O.; Walker, B.L.; Kooi, C.; et al. Cigarette smoke modulates expression of human rhinovirus-induced airway epithelial host defense genes. PLoS ONE 2012, 7, e40762. [Google Scholar] [CrossRef]
- Sanders, S.P.; Siekierski, E.S.; Richards, S.M.; Porter, J.D.; Imani, F.; Proud, D. Rhinovirus infection induces expression of type 2 nitric oxide synthase in human respiratory epithelial cells in vitro and in vivo. J. Allergy Clin. Immunol. 2001, 107, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.P.; Siekierski, E.S.; Porter, J.D.; Richards, S.M.; Proud, D. Nitric oxide inhibits rhinovirus-induced cytokine production and viral replication in a human respiratory epithelial cell line. J. Virol. 1998, 72, 934–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koetzler, R.; Zaheer, R.S.; Wiehler, S.; Holden, N.S.; Giembycz, M.A.; Proud, D. Nitric oxide inhibits human rhinovirus-induced transcriptional activation of CXCL10 in airway epithelial cells. J. Allergy Clin. Immunol. 2009, 123, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.P.; Proud, D.; Siekierski, E.S.; Yachechko, R.; Liu, M.C. Role of nasal nitric oxide in the resolution of experimental rhinovirus infection. J. Allergy Clin. Immunol. 2004, 113, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Sykes, A.; Macintyre, J.; Edwards, M.R.; del Rosario, A.; Haas, J.; Gielen, V.; Kon, O.M.; McHale, M.; Johnston, S.L. Rhinovirus-induced interferon production is not deficient in well controlled asthma. Thorax 2013, 69, 240–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wark, P.A.B.; Johnston, S.L.; Bucchieri, F.; Powell, R.; Puddicombe, S.; Laza-Stanca, V.; Holgate, S.T.; Davies, D.E. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J. Exp. Med. 2005, 201, 937–947. [Google Scholar] [CrossRef] [Green Version]
- Contoli, M.; Message, S.D.; Laza-Stanca, V.; Edwards, M.R.; Wark, P.A.B.; Bartlett, N.W.; Kebadze, T.; Mallia, P.; Stanciu, L.A.; Parker, H.L.; et al. Role of deficient type-III interferon-λ production in asthma exacerbations. Nat. Med. 2006, 12, 1023–1026. [Google Scholar] [CrossRef]
- Edwards, M.R.; Regamey, N.; Vareille, M.; Kieninger, E.; Gupta, A.; Shoemark, A.; Saglani, S.; Sykes, A.; Macintyre, J.; Davies, J.; et al. Impaired innate interferon induction in severe therapy resistant atopic asthmatic children. Mucosal Immunol. 2013, 6, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Souza, N.; Favoreto, S.; Wong, H.; Ward, T.; Yagi, S.; Schnurr, D.; Finkbeiner, W.E.; Dolganov, G.M.; Widdicombe, J.H.; Boushey, H.A.; et al. In vitro susceptibility to rhinovirus infection is greater for bronchial than for nasal airway epithelial cells in human subjects. J. Allergy Clin. Immunol. 2009, 123, 1384–1390. [Google Scholar] [CrossRef] [Green Version]
- Bochkov, Y.A.; Hanson, K.M.; Keles, S.; Brockman-Schneider, R.A.; Jarjour, N.N.; Gern, J.E. Rhinovirus-induced modulation of gene expression in bronchial epithelial cells from subjects with asthma. Mucosal. Immunol. 2010, 3, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, J.L.; Shaker, M.; McMeen, V.; Gern, J.; Carper, H.; Deborah, M.; Lee, W.-M.; Bochkov, Y.A.; Vrtis, R.F.; Platts-Mills, T.; et al. Comparison of viral load in individuals with and without asthma during infections with rhinovirus. Am. J. Respir. Crit. Care Med. 2014, 189, 532–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djukanovic, R.; Harrison, T.; Johnston, S.L.; Gabbay, F.; Wark, P.; Thomson, N.C.; Niven, R.; Singh, D.; Reddel, H.K.; Davies, D.E.; et al. The effect of inhaled IFN-β on worsening of asthma symptoms caused by viral infections. A randomized trial. Am. J. Respir. Crit. Care Med. 2014, 190, 145–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asensio, V.C.; Maier, J.; Milner, R.; Boztug, K.; Kincaid, C.; Moulard, M.; Phillipson, C.; Lindsley, K.; Krucker, T.; Fox, H.S.; et al. Interferon-independent, human immunodeficiency virus type 1 gp120-mediated induction of CXCL10/IP-10 gene expression by astrocytes in vivo and in vitro. J. Virol. 2001, 75, 7067–7077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, S.; Mordstein, M.; Kochs, G.; García-Sastre, A.; tenOever, B.R. Transcription factor redundancy ensures induction of the antiviral state. J. Biol. Chem. 2010, 285, 42013–42022. [Google Scholar] [CrossRef] [Green Version]
- Ashley, C.L.; Abendroth, A.; McSharry, B.P.; Slobedman, B. Interferon-independent upregulation of interferon-stimulated genes during human cytolegalovirus infection is dependent upon IRF3 expression. Viruses 2019, 11, 246. [Google Scholar] [CrossRef] [Green Version]
- Jeffery, P.K. Remodeling and inflammation of bronchi in asthma and chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2004, 1, 176–183. [Google Scholar] [CrossRef]
- Saglani, S.; Payne, D.N.; Zhu, J.; Nicholson, A.G.; Bush, A.; Jeffery, P.K. Early detection of airway wall remodeling and eosinophilic inflammation in preschool wheezers. Am. J. Respir. Crit. Care Med. 2007, 176, 858–864. [Google Scholar] [CrossRef]
- O’Reilly, R.; Ullmann, N.; Irving, S.; Bossley, C.J.; Sonnappa, S.; Zhu, J.; Oates, T.; Banya, W.; Jeffery, P.K.; Bush, A.; et al. Increased airway smooth muscle in preschool wheezers who have asthma at school age. J. Allergy Clin. Immunol. 2013, 131, 1024–1032. [Google Scholar] [CrossRef]
- Jartti, T.; Lee, W.-M.; Pappas, T.; Evans, M.; Lemanske, R.F., Jr.; Gern, J.E. Serial viral infections in infants with recurrent respiratory illnesses. Eur. Respir. J. 2008, 32, 314–320. [Google Scholar] [CrossRef] [Green Version]
- Jackson, D.J.; Evans, M.D.; Gangnon, R.E.; Tisler, C.J.; Pappas, T.E.; Lee, W.-M.; Gern, J.E.; Lemanske, R.F., Jr. Evidence for a causal relationship between allergic sensitization and rhinovirus wheezing in early life. Am. J. Respir. Crit. Care Med. 2012, 185, 281–285. [Google Scholar] [CrossRef] [Green Version]
- Kuo, C.; Lim, S.; King, N.J.C.; Bartlett, N.W.; Walton, R.P.; Zhu, J.; Glanville, N.; Aniscenko, J.; Johnston, S.L.; Burgess, J.K.; et al. Rhinovirus infection induces expression of airway remodelling factors in vitro and in vivo. Respirology 2011, 16, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Parikh, V.; Scala, J.; Patel, R.; Corbi, C.; Lo, D.; Bochkov, Y.A.; Kennedy, J.L.; Kurten, R.C.; Liggett, S.B.; Gern, J.E.; et al. Rhinovirus C15 induces airway hyperresponsiveness via calcium mobilization in airway smooth muscle. Am. J. Respir. Cell Mol. Biol. 2020, 62, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Tacon, C.E.; Wiehler, S.; Holden, N.S.; Newton, R.; Proud, D.; Leigh, R. Human rhinovirus infection of airway epithelial cells upregulates MMP-9 production via NF-κB. Am. J. Respir. Cell Mol. Biol. 2010, 43, 201–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Psarras, S.; Volonaki, E.; Skevaki, C.L.; Xatzipsalti, M.; Bossios, A.; Pratsinis, H.; Tsigkos, S.; Gourgiotis, D.; Constantopoulos, A.G.; Papapetropoulos, A.; et al. Vascular endothelial growth factor-mediated induction of angiogenesis by human rhinovirus. J. Allergy Clin. Immunol. 2006, 117, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Shelfoon, C.; Shariff, S.; Traves, S.L.; Kooi, C.; Leigh, R.; Proud, D. Chemokine release from human rhinovirus-infected airway epithelial cells promotes fibroblast migration. J. Allergy Clin. Immunol. 2016, 138, 110–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shariff, S.; Shelfoon, C.; Holden, N.S.; Traves, S.L.; Wiehler, S.; Kooi, C.; Proud, D.; Leigh, R. Human rhinovirus infection of epithelial cells modulates airway smooth muscle migration. Am. J. Respir. Cell Mol. Biol. 2017, 56, 796–803. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Lee, P.K.; Lee, W.-M.; Zhao, Y.; Yu, D.; Chen, Y. Rhinovirus-induced major airway mucin production involves a novel TLR3-EGFR-dependent pathway. Am. J. Respir. Cell Mol. Biol. 2009, 40, 610–619. [Google Scholar] [CrossRef]
- Hewson, C.A.; Haas, J.J.; Bartlett, N.W.; Message, S.D.; Laza-Stanca, V.; Kebadze, T.; Caramori, G.; Zhu, J.; Edbrooke, M.R.; Stanciu, L.A.; et al. Rhinovirus induces MUC5AC in a human infection model and in vitro via NF-κB and EGFR pathways. Eur. Respir. J. 2010, 36, 1425–1435. [Google Scholar] [CrossRef] [Green Version]
- Bai, J.; Smock, S.L.; Jackson, G.R.J.; MacIsaac, K.D.; Huang, Y.; Mankus, C.; Oldach, J.; Roberts, B.; Ma, Y.-L.; Klappenbach, J.A.; et al. Phenotypic responses of differentiated asthmatic human airway epithelial cultures to rhinovirus. PLoS ONE 2015, 10, e0118286. [Google Scholar] [CrossRef]
- Schneeberger, E.E.; Lynch, R.D. The tight junction: A multifunctional complex. Am. J. Physiol. Cell. Physiol. 2004, 286, C1213–C1228. [Google Scholar] [CrossRef]
- Looi, K.; Buckley, A.G.; Rigby, P.J.; Garratt, L.W.; Iosifidis, T.; Zosky, G.R.; Larcombe, A.N.; Lannigan, F.J.; Ling, K.-M.; Martinovich, K.M.; et al. Effects of human rhinovirus on epithelial barrier integrity and function in children with asthma. Clin. Exp. Allergy 2018, 48, 513–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagome, K.; Bochkov, Y.A.; Ashraf, S.; Brockman-Schneider, R.A.; Evans, M.D.; Pasic, T.R.; Gern, J.E. Effects of rhinovirus species on viral replication and cytokine production. J. Allergy Clin. Immunol. 2014, 134, 332–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comstock, A.T.; Ganesan, S.; Chattoraj, A.; Faris, A.N.; Margolis, B.L.; Hershenson, M.B.; Sajjan, U.S. Rhinovirus-induced barrier dysfunction in polarized airway epithelial cells is mediated by NADPH oxidase 1. J. Virol. 2011, 85, 6795–6808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, A.; Favoreto, S.; Avila, P.C.; Schleimer, R.P. TLR-3 and Th2 cytokine-dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J. Immunol. 2007, 179, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Beale, J.; Jayaraman, A.; Jackson, D.J.; Macintyre, J.D.R.; Edwards, M.R.; Walton, R.P.; Zhu, J.; Ching, Y.M.; Shamji, B.; Edwards, M.; et al. Rhinovirus-induced IL-25 in asthma exacerbation drives type 2 immunity and allergic pulmonary inflammation. Sci. Transl. Med. 2014, 6, 256ra134. [Google Scholar] [CrossRef] [Green Version]
- Jackson, D.J.; Makrinioti, H.; Rana, B.M.J.; Shamji, B.W.H.; Trujillo-Torralbo, M.-B.; Footit, J.; del-Rosario, J.; Telcian, A.G.; Nikonova, A.; Zhu, J.; et al. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am. J. Respir. Crit. Care Med. 2014, 190, 1373–1382. [Google Scholar] [CrossRef] [Green Version]
- Maciejewski, B.A.; Jamieson, K.C.; Arnason, J.W.; Kooi, C.; Wiehler, S.; Traves, S.L.; Leigh, R.; Proud, D. Rhinovirus-bacteria coexposure synergistically induces CCL20 production from human bronchial epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L731–L740. [Google Scholar] [CrossRef]
- Zhu, Z.; Tang, W.; Ray, A.; Wu, Y.; Einarsson, O.; Landry, M.L.; Gwaltney, J.M., Jr.; Elias, J.A. Rhinovirus stimulation of interleukin-6 in vivo and in vitro. Evidence for nuclear factor κB-dependent transcriptional activation. J. Clin. Invest. 1996, 97, 421–430. [Google Scholar] [CrossRef]
- Jayaraman, A.; Jackson, D.J.; Message, S.D.; Pearson, R.M.; Aniscenko, J.; Caramori, G.; Mallia, P.; Papi, A.; Shamji, B.; Edwards, M.; et al. IL-15 complexes induce NK- and T-cell responses independent of type I IFN signaling during rhinovirus infection. Mucosal Immunol. 2014, 7, 1151–1164. [Google Scholar] [CrossRef] [Green Version]
- Upham, J.W.; Stick, S.M. Interactions between airway epithelial cells and dendritic cells: Implications for the regulation of airway inflammation. Curr. Drug Targets 2006, 7, 541–545. [Google Scholar] [CrossRef]
- Huston, D.P.; Liu, Y.J. Thymic stromal lymphopoietin: A potential therapeutic target for allergy and asthma. Curr. Allergy Asthma Rep. 2006, 6, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Stanciu, L.A.; Papadopoulos, N.G.; Teran, L.M.; Holgate, S.T.; Johnston, S.L. Rhinovirus infection induces major histocompatability complex class I and costimulatory molecule upregulation on respiratory epithelial cells. J. Infect. Dis. 2000, 181, 1780–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Myers, A.C.; Chen, L.; Pardoll, D.M.; Truong-Tran, Q.-A.; Lane, A.P.; McDyer, J.F.; Fortuno, L.; Schleimer, R.P. Constitutive and inducible expression of B7 family of ligands by human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2005, 33, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Heinecke, L.; Proud, D.; Sanders, S.; Schleimer, R.P.; Kim, J. Induction of B7-H1 and B7-DC expression on airway epithelial cells by the Toll-like receptor 3 agonist double-stranded RNA and human rhinovirus infection: In vivo and in vitro studies. J. Allergy Clin. Immunol. 2008, 121, 1155–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salomon, B.; Bluestone, J.A. LFA-1 interaction with ICAM-1 and ICAM-2 regulates Th2 cytokine production. J. Immunol. 1998, 161, 5138–5142. [Google Scholar]
- Proud, D.; Leigh, R. Epithelial cells and airway diseases. Immunol. Rev. 2011, 242, 186–204. [Google Scholar] [CrossRef]
- Martinez, F.D. Respiratory syncytial virus bronchiolitis and the pathogenesis of childhood asthma. Pediatr. Infect. Dis. J. 2003, 22, S76–S82. [Google Scholar] [CrossRef]
- Bønnelykke, K.; Vissing, N.H.; Sevelsted, A.; Johnston, S.L.; Bisgaard, H. Association between respiratory infections in early life and later asthma is independent of virus type. J. Allergy Clin. Immunol. 2015, 136, 81–86. [Google Scholar] [CrossRef]
- Feldman, A.S.; He, Y.; Moore, M.L.; Hershenson, M.B.; Hartert, T.V. Toward primary prevention of asthma. Reviewing the evidence for early-life respiratory viral infections as modifiable risk factors to prevent childhood asthma. Am. J. Respir. Crit. Care Med. 2015, 191, 34–44. [Google Scholar] [CrossRef]
- Reeves, S.R.; Kolstad, T.; Lien, T.-Y.; Herrington-Shaner, S.; Debley, J.S. Fibroblast-myofibroblast transition is differentially regulated by bronchial epithelial cells from asthmatic children. Respir. Res. 2015, 16, 21. [Google Scholar] [CrossRef] [Green Version]
- Bizzintino, J.; Lee, W.-M.; Laing, I.A.; Vang, F.; Pappas, T.; Zhang, G.; Martin, A.C.; Khoo, S.-K.; Cox, D.W.; Geelhoed, G.C.; et al. Association between human rhinovirus C and severity of acute asthma in children. Eur. Respir. J. 2011, 37, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.W.; Bizzintino, J.; Ferrari, G.; Khoo, S.K.; Zhang, G.; Whelan, S.; Lee, W.M.; Bochkov, Y.A.; Geelhoed, G.C.; Goldblatt, J.; et al. Human rhinovirus species C infection in young children with acute wheeze is associated with increased acute respiratory hospital admissions. Am. J. Respir. Crit. Care Med. 2013, 188, 1358–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasagawa, K.; Jartti, T.; Bochkov, Y.A.; Gern, J.E.; Mansbach, J.M.; Piedra, P.A.; Toivonen, L.; Camargo, C.A. Rhinovirus species in children with severe bronchiolitis: Multicenter cohort studies in the United States and Finland. Pediatr. Infect. Dis. J. 2019, 38, e59–e62. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-M.; Lemanske, R.F., Jr.; Evans, M.D.; Vang, F.; Pappas, T.; Gangnon, R.; Jackson, D.J.; Gern, J.E. Human rhinovirus species and season of infection determine illness severity. Am. J. Respir. Crit. Care Med. 2012, 186, 886–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michi, A.N.; Love, M.E.; Proud, D. Rhinovirus-Induced Modulation of Epithelial Phenotype: Role in Asthma. Viruses 2020, 12, 1328. https://doi.org/10.3390/v12111328
Michi AN, Love ME, Proud D. Rhinovirus-Induced Modulation of Epithelial Phenotype: Role in Asthma. Viruses. 2020; 12(11):1328. https://doi.org/10.3390/v12111328
Chicago/Turabian StyleMichi, Aubrey N., Michelle E. Love, and David Proud. 2020. "Rhinovirus-Induced Modulation of Epithelial Phenotype: Role in Asthma" Viruses 12, no. 11: 1328. https://doi.org/10.3390/v12111328
APA StyleMichi, A. N., Love, M. E., & Proud, D. (2020). Rhinovirus-Induced Modulation of Epithelial Phenotype: Role in Asthma. Viruses, 12(11), 1328. https://doi.org/10.3390/v12111328