Antiviral Effect of Epigallocatechin Gallate via Impairing Porcine Circovirus Type 2 Attachment to Host Cell Receptor

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Virus
2.2. Reagent and Antibodies
2.3. Cytotoxicity Assay
2.4. Determination of Virus Titer
2.5. Calculation of EC50
2.6. Infectivity Assay
2.7. Time of Addition and Dilution Experiment
2.8. Binding Assay and Flow Cytometry
2.9. Recombinant Protein Expression and Purification
2.10. Heparin Column Chromatography
2.11. Microscale Thermophoresis Assay
2.12. Construction of PCV2 Infectious Clone and Virus Rescue
2.13. Statistical Analysis
3. Results
3.1. EGCG Inhibits the Infectivity of PCV2
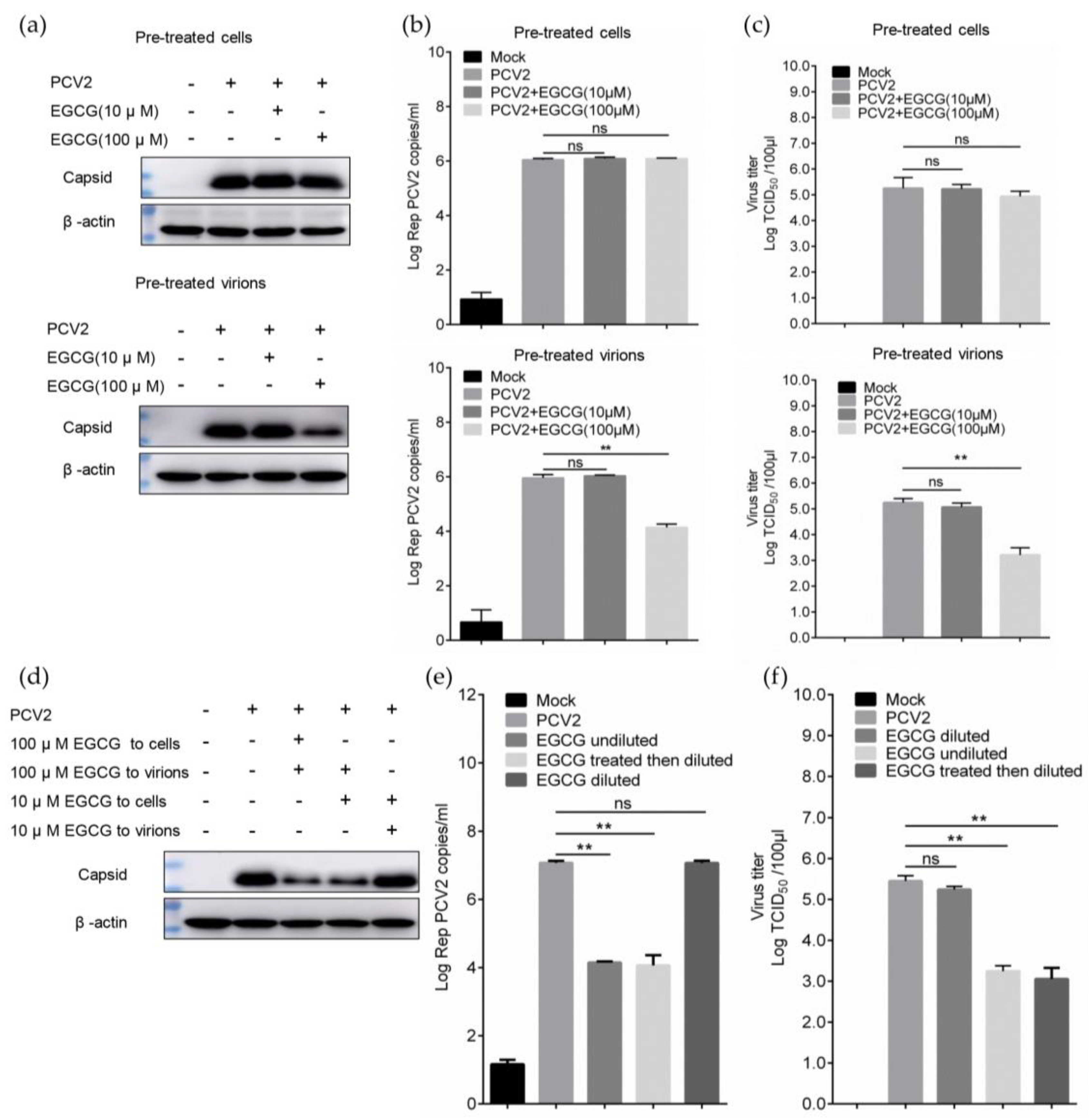
3.2. EGCG Exerts the Antiviral Effect via Directly Targeting Virions
3.3. EGCG Impairs the Attachment of PCV2 to Host Cells
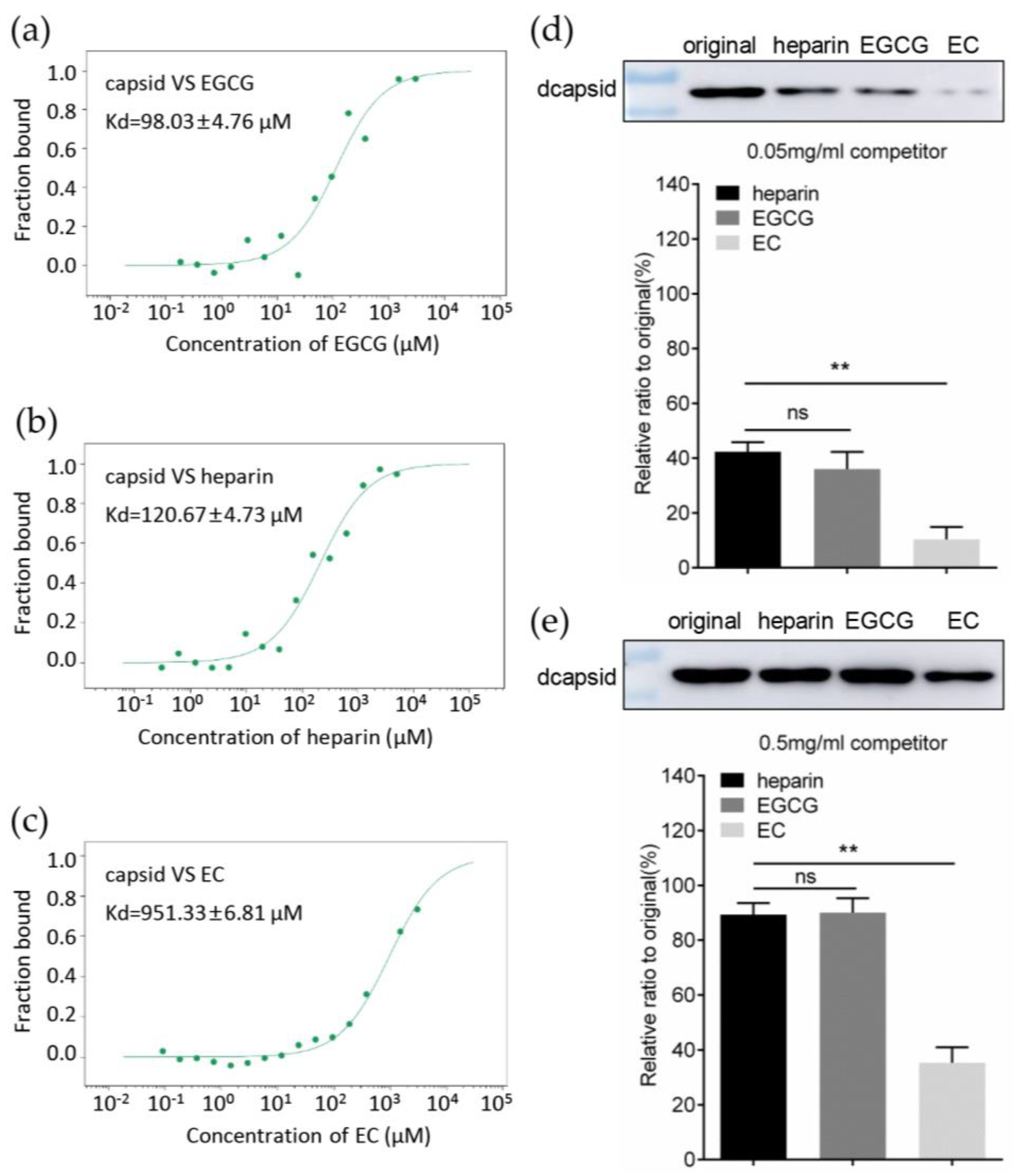
3.4. EGCG Competitively Inhibits the Interaction between Capsid and Heparan Sulfate
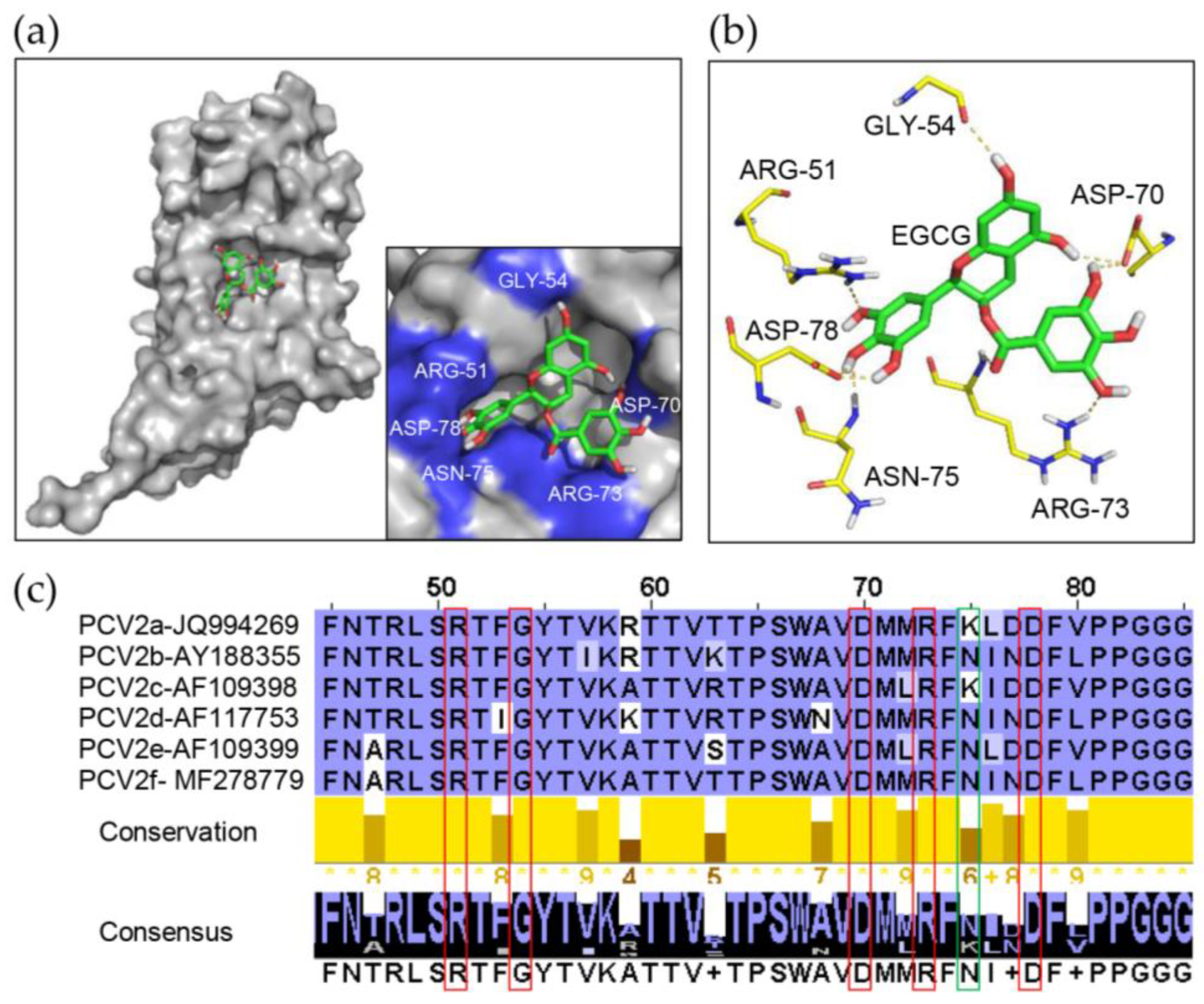
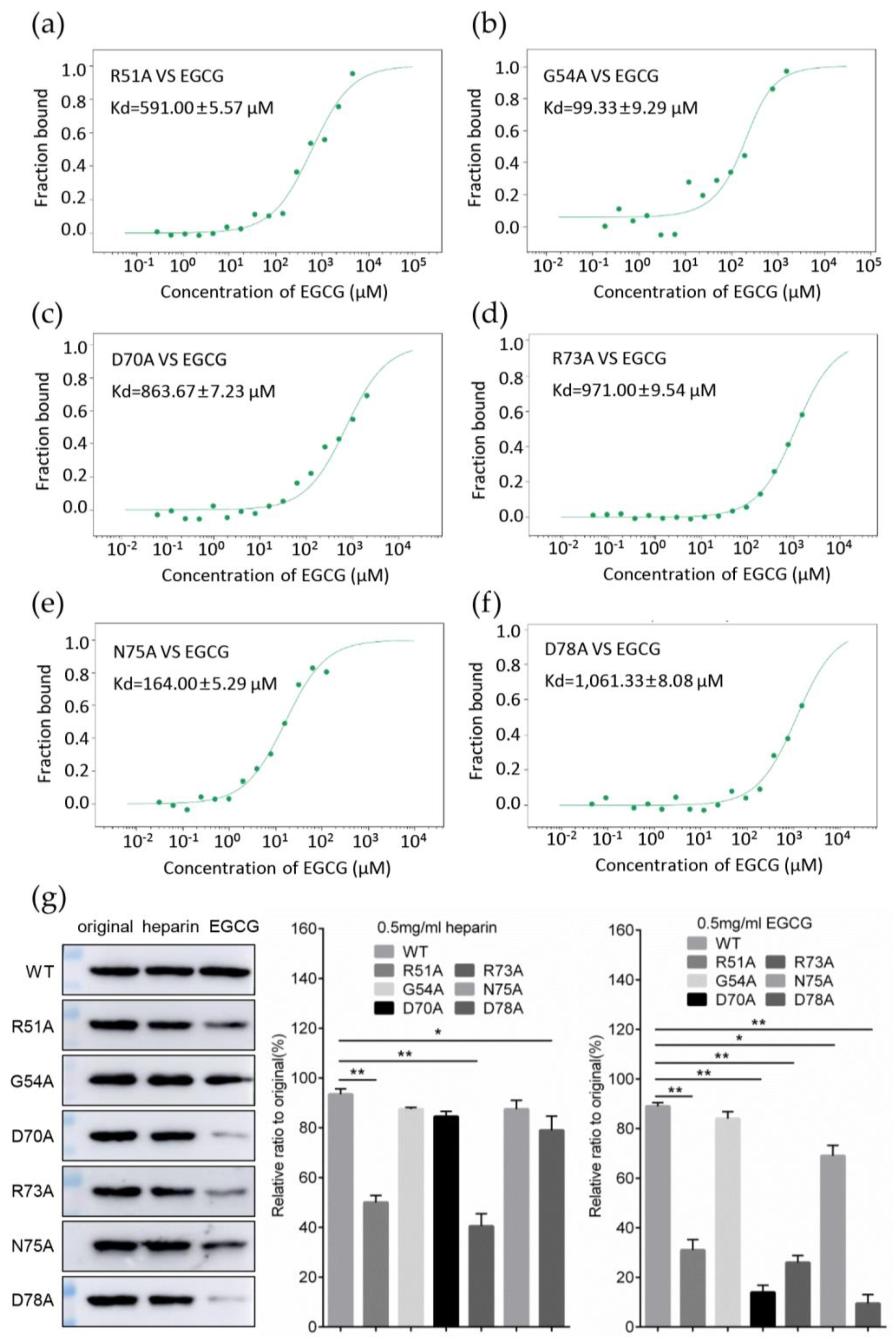
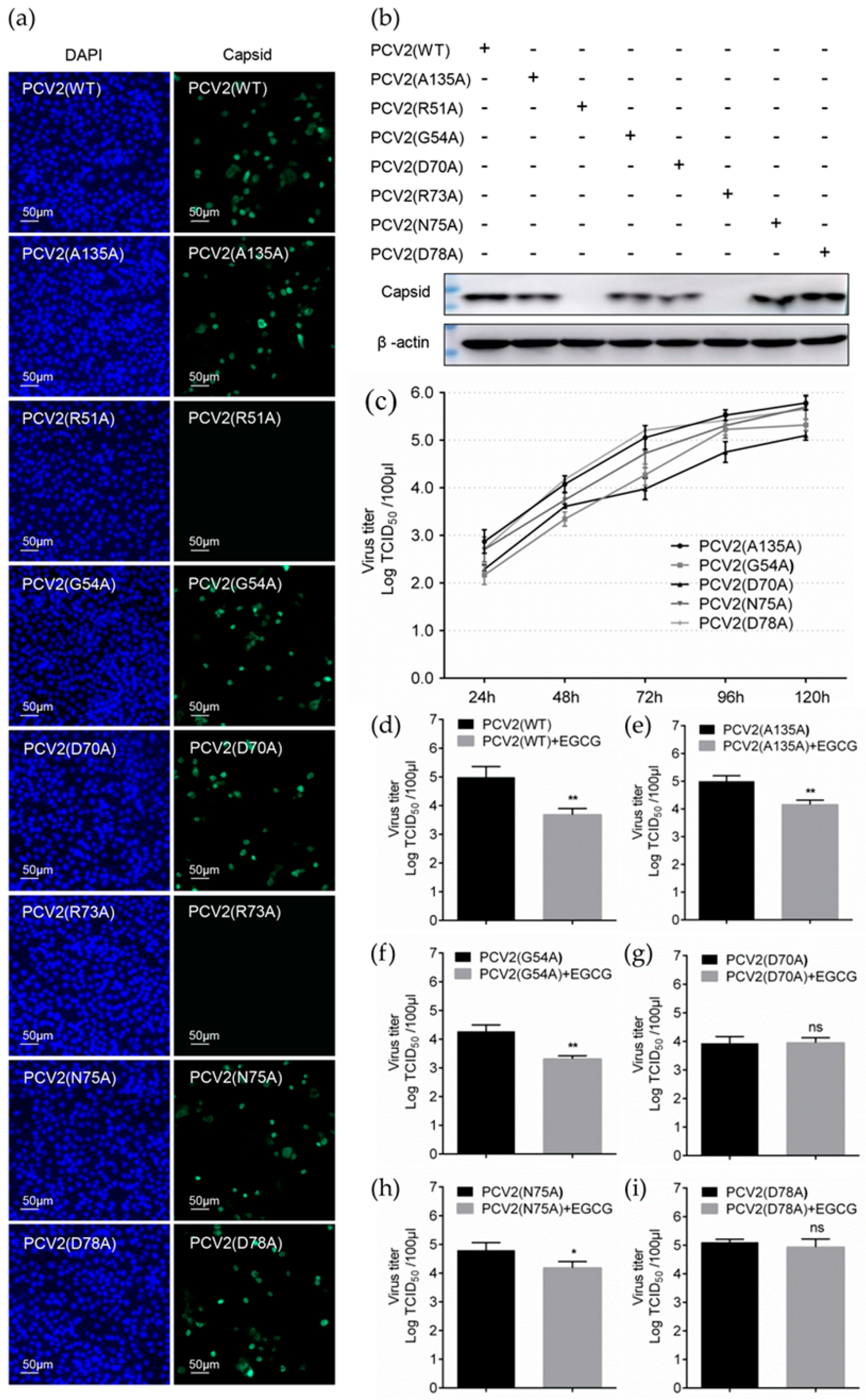
3.5. Identification of Key Amino Acids in PCV2 Capsid Contributing to the Interaction with EGCG
3.6. Replacement of Key Amino Acids in Capsid Weakens the Antiviral Effect of EGCG
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pringle, C.R. Virus taxonomy at the XIth International Congress of Virology, Sydney, Australia, 1999. Arch. Virol. 1999, 144, 2065–2070. [Google Scholar] [CrossRef]
- Li, J.; Xing, G.; Zhang, C.; Yang, H.; Li, G.; Wang, N.; Wang, R.; Sun, H.; Shi, Z.; Lei, J.; et al. Cross-species transmission resulted in the emergence and establishment of circovirus in pig. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2019, 75, 103973. [Google Scholar] [CrossRef]
- Finsterbusch, T.; Mankertz, A. Porcine circoviruses—Small but powerful. Virus Res. 2009, 143, 177–183. [Google Scholar] [CrossRef]
- Ouyang, T.; Zhang, X.; Liu, X.; Ren, L. Co-Infection of Swine with Porcine Circovirus Type 2 and Other Swine Viruses. Viruses 2019, 11, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afolabi, K.O.; Iweriebor, B.C.; Okoh, A.I.; Obi, L.C. Global Status of Porcine circovirus Type 2 and Its Associated Diseases in Sub-Saharan Africa. Adv. Virol. 2017, 2017, 6807964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamel, A.L.; Lin, L.L.; Nayar, G.P. Nucleotide sequence of porcine circovirus associated with postweaning multisystemic wasting syndrome in pigs. J. Virol. 1998, 72, 5262–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allan, G.M.; Kennedy, S.; McNeilly, F.; Foster, J.C.; Ellis, J.A.; Krakowka, S.J.; Meehan, B.M.; Adair, B.M. Experimental reproduction of severe wasting disease by co-infection of pigs with porcine circovirus and porcine parvovirus. J. Comp. Pathol. 1999, 121, 1–11. [Google Scholar] [CrossRef]
- Sanchez, R.E., Jr.; Nauwynck, H.J.; McNeilly, F.; Allan, G.M.; Pensaert, M.B. Porcine circovirus 2 infection in swine foetuses inoculated at different stages of gestation. Vet. Microbiol. 2001, 83, 169–176. [Google Scholar] [CrossRef]
- Nawagitgul, P.; Morozov, I.; Bolin, S.R.; Harms, P.A.; Sorden, S.D.; Paul, P.S. Open reading frame 2 of porcine circovirus type 2 encodes a major capsid protein. J. Gen. Virol. 2000, 81, 2281–2287. [Google Scholar] [CrossRef]
- Misinzo, G.; Delputte, P.L.; Meerts, P.; Lefebvre, D.J.; Nauwynck, H.J. Porcine circovirus 2 uses heparan sulfate and chondroitin sulfate B glycosaminoglycans as receptors for its attachment to host cells. J. Virol. 2006, 80, 3487–3494. [Google Scholar] [CrossRef] [Green Version]
- Rostand, K.S.; Esko, J.D. Microbial adherence to and invasion through proteoglycans. Infect. Immun. 1997, 65, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cagno, V.; Tseligka, E.D.; Jones, S.T.; Tapparel, C. Heparan Sulfate Proteoglycans and Viral Attachment: True Receptors or Adaptation Bias? Viruses 2019, 11, 596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prydz, K.; Dalen, K.T. Synthesis and sorting of proteoglycans. J. Cell Sci. 2000, 113, 193–205. [Google Scholar] [PubMed]
- Xu, Y.; Martinez, P.; Seron, K.; Luo, G.; Allain, F.; Dubuisson, J.; Belouzard, S. Characterization of hepatitis C virus interaction with heparan sulfate proteoglycans. J. Virol. 2015, 89, 3846–3858. [Google Scholar] [CrossRef] [Green Version]
- Shieh, M.T.; WuDunn, D.; Montgomery, R.I.; Esko, J.D.; Spear, P.G. Cell surface receptors for herpes simplex virus are heparan sulfate proteoglycans. J. Cell Biol. 1992, 116, 1273–1281. [Google Scholar] [CrossRef]
- Tan, C.W.; Poh, C.L.; Sam, I.C.; Chan, Y.F. Enterovirus 71 uses cell surface heparan sulfate glycosaminoglycan as an attachment receptor. J. Virol. 2013, 87, 611–620. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, M.; Anindita, P.D.; Ito, N.; Sugiyama, M.; Carr, M.; Fukuhara, H.; Ose, T.; Maenaka, K.; Takada, A.; Hall, W.W.; et al. The Role of Heparan Sulfate Proteoglycans as an Attachment Factor for Rabies Virus Entry and Infection. J. Infect. Dis. 2018, 217, 1740–1749. [Google Scholar] [CrossRef] [Green Version]
- Dhindwal, S.; Avila, B.; Feng, S.; Khayat, R. Porcine Circovirus 2 Uses a Multitude of weak binding sites to interact with heparan sulfate, and the interactions do not follow the symmetry of the Capsid. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.T.; Chen, T.Y.; Lin, S.C.; Chung, C.Y.; Lin, T.C.; Wang, G.H.; Anderson, R.; Lin, C.C.; Richardson, C.D. Broad-spectrum antiviral activity of chebulagic acid and punicalagin against viruses that use glycosaminoglycans for entry. BMC Microbiol. 2013, 13, 187. [Google Scholar] [CrossRef] [Green Version]
- Narotzki, B.; Reznick, A.Z.; Aizenbud, D.; Levy, Y. Green tea: A promising natural product in oral health. Arch. Oral Biol. 2012, 57, 429–435. [Google Scholar] [CrossRef]
- Yang, C.S.; Wang, H. Cancer Preventive Activities of Tea Catechins. Molecules 2016, 21, 1679. [Google Scholar] [CrossRef]
- Gonzalez, Y.; Torres-Mendoza, D.; Jones, G.E.; Fernandez, P.L. Marine Diterpenoids as Potential Anti-Inflammatory Agents. Mediat. Inflamm. 2015, 2015, 263543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chacko, S.M.; Thambi, P.T.; Kuttan, R.; Nishigaki, I. Beneficial effects of green tea: A literature review. Chin. Med. 2010, 5, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kono, K.; Tatara, I.; Takeda, S.; Arakawa, K.; Hara, Y. Antibacterial activity of epigallocatechin gallate against methicillin-resistant Staphylococcus aureus. Kansenshogaku zasshi. J. Jpn. Assoc. Infect. Dis. 1994, 68, 1518–1522. [Google Scholar] [CrossRef] [Green Version]
- Song, J.M.; Lee, K.H.; Seong, B.L. Antiviral effect of catechins in green tea on influenza virus. Antivir. Res. 2005, 68, 66–74. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Honda, M.; Ikigai, H.; Hara, Y.; Shimamura, T. Inhibitory effects of (-)-epigallocatechin gallate on the life cycle of human immunodeficiency virus type 1 (HIV-1). Antivir. Res. 2002, 53, 19–34. [Google Scholar] [CrossRef]
- Nakayama, M.; Suzuki, K.; Toda, M.; Okubo, S.; Hara, Y.; Shimamura, T. Inhibition of the infectivity of influenza virus by tea polyphenols. Antivir. Res. 1993, 21, 289–299. [Google Scholar] [CrossRef]
- Carneiro, B.M.; Batista, M.N.; Ana, C.B.; Nogueira, M.L.; Rahal, P. The green tea molecule EGCG inhibits Zika virus entry. Virology 2016, 496, 215–218. [Google Scholar] [CrossRef]
- Isaacs, C.E.; Wen, G.Y.; Xu, W.; Jia, J.H.; Rohan, L.; Corbo, C.; Di Maggio, V.; Jenkins, E.C., Jr.; Hillier, S. Epigallocatechin gallate inactivates clinical isolates of herpes simplex virus. Antimicrob. Agents Chemother. 2008, 52, 962–970. [Google Scholar] [CrossRef] [Green Version]
- Ciesek, S.; von Hahn, T.; Colpitts, C.C.; Schang, L.M.; Friesland, M.; Steinmann, J.; Manns, M.P.; Ott, M.; Wedemeyer, H.; Meuleman, P.; et al. The green tea polyphenol, epigallocatechin-3-gallate, inhibits hepatitis C virus entry. Hepatology 2011, 54, 1947–1955. [Google Scholar] [CrossRef]
- Kawai, K.; Tsuno, N.H.; Kitayama, J.; Okaji, Y.; Yazawa, K.; Asakage, M.; Hori, N.; Watanabe, T.; Takahashi, K.; Nagawa, H. Epigallocatechin gallate, the main component of tea polyphenol, binds to CD4 and interferes with gp120 binding. J. Allergy Clin. Immunol. 2003, 112, 951–957. [Google Scholar] [CrossRef]
- O’Donnell, C.D.; Kovacs, M.; Akhtar, J.; Valyi-Nagy, T.; Shukla, D. Expanding the role of 3-O sulfated heparan sulfate in herpes simplex virus type-1 entry. Virology 2010, 397, 389–398. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.Y.; Chen, Q.X.; Ye, J.X.; Shen, H.G.; Chen, T.F.; Shang, S.B. Serological investigation and genomic characterization of PCV2 isolates from different geographic regions of Zhejiang province in China. Vet. Res. Commun. 2006, 30, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.Y.; Shang, S.B.; Gong, H.; Chen, Q.X.; Wu, J.X.; Shen, H.G.; Chen, T.F.; Guo, J.Q. In vitro expression, monoclonal antibody and bioactivity for capsid protein of porcine circovirus type II without nuclear localization signal. J. Biotechnol. 2005, 118, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Hjulsager, C.K.; Grau-Roma, L.; Sibila, M.; Enoe, C.; Larsen, L.; Segales, J. Inter-laboratory and inter-assay comparison on two real-time PCR techniques for quantification of PCV2 nucleic acid extracted from field samples. Vet. Microbiol. 2009, 133, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xu, Z.; Zheng, W. A Review of the Antiviral Role of Green Tea Catechins. Molecules 2017, 22, 1337. [Google Scholar] [CrossRef] [Green Version]
- Kreuger, J.; Kjellen, L. Heparan sulfate biosynthesis: Regulation and variability. J. Histochem. Cytochem. 2012, 60, 898–907. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Esko, J.D. Demystifying heparan sulfate-protein interactions. Annu. Rev. Biochem. 2014, 83, 129–157. [Google Scholar] [CrossRef]
- Billings, P.C.; Pacifici, M. Interactions of signaling proteins, growth factors and other proteins with heparan sulfate: Mechanisms and mysteries. Connect. Tissue Res. 2015, 56, 272–280. [Google Scholar] [CrossRef] [Green Version]
- Cardin, A.D.; Weintraub, H.J. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 1989, 9, 21–32. [Google Scholar] [CrossRef]
- Shriver, Z.; Capila, I.; Venkataraman, G.; Sasisekharan, R. Heparin and heparan sulfate: Analyzing structure and microheterogeneity. Handb. Exp. Pharmacol. 2012, 159–176. [Google Scholar] [CrossRef] [Green Version]
- Zulueta, M.M.; Lin, S.Y.; Hu, Y.P.; Hung, S.C. Synthetic heparin and heparan sulfate oligosaccharides and their protein interactions. Curr. Opin. Chem. Biol. 2013, 17, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.; Xiao, Y.; Chen, H.; Luo, F.; Du, G.; Zeng, F. Multiple antiviral approaches of (-)-epigallocatechin-3-gallate (EGCG) against porcine reproductive and respiratory syndrome virus infection in vitro. Antivir. Res. 2018, 158, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Hour, M.J.; Lai, H.C.; Chen, C.H.; Chang, P.J.; Huang, S.H.; Lin, C.W. Epigallocatechin-3-gallate inhibits the early stages of Japanese encephalitis virus infection. Virus Res. 2018, 253, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Liu, S.; Li, C.; Yang, L.; Zu, Y. In vitro evaluation of the antiviral activity of the synthetic epigallocatechin gallate analog-epigallocatechin gallate (EGCG) palmitate against porcine reproductive and respiratory syndrome virus. Viruses 2014, 6, 938–950. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Song, D.; Wang, S.; Dai, Y.; Zhou, J.; Gu, J. Antiviral Effect of Epigallocatechin Gallate via Impairing Porcine Circovirus Type 2 Attachment to Host Cell Receptor. Viruses 2020, 12, 176. https://doi.org/10.3390/v12020176
Li J, Song D, Wang S, Dai Y, Zhou J, Gu J. Antiviral Effect of Epigallocatechin Gallate via Impairing Porcine Circovirus Type 2 Attachment to Host Cell Receptor. Viruses. 2020; 12(2):176. https://doi.org/10.3390/v12020176
Chicago/Turabian StyleLi, Jiarong, Dongfeng Song, Shengnan Wang, Yadong Dai, Jiyong Zhou, and Jinyan Gu. 2020. "Antiviral Effect of Epigallocatechin Gallate via Impairing Porcine Circovirus Type 2 Attachment to Host Cell Receptor" Viruses 12, no. 2: 176. https://doi.org/10.3390/v12020176
APA StyleLi, J., Song, D., Wang, S., Dai, Y., Zhou, J., & Gu, J. (2020). Antiviral Effect of Epigallocatechin Gallate via Impairing Porcine Circovirus Type 2 Attachment to Host Cell Receptor. Viruses, 12(2), 176. https://doi.org/10.3390/v12020176