Tat-Based Therapies as an Adjuvant for an HIV-1 Functional Cure


Abstract
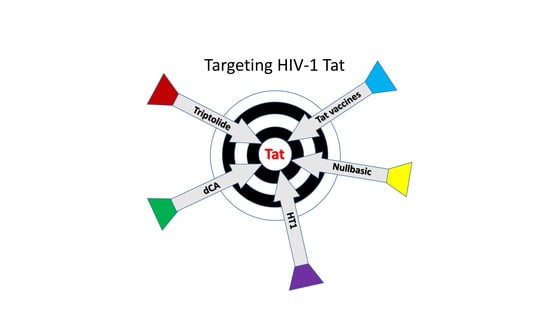
:
1. Introduction
2. Tat Transactivation of HIV Transcription
3. A Tat-Based Block-and-Lock Functional Cure Strategy
4. Tat Trans-Dominant Negative Proteins and Nullbasic
4.1. Tat Trans-Dominant Negative Proteins
4.2. Nullbasic Inhibits HIV Transcription and Reactivation from Latency
5. A HEXIM1-Tat Fusion Protein that Inhibits HIV Replication
6. Two Compounds that Inhibit Tat Activity
6.1. Didehydro-Cortistatin A (dCA)
6.2. Triptolide
7. Anti-Tat Vaccines that may Compliment cART
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alexaki, A.; Liu, Y.; Wigdahl, B. Cellular reservoirs of HIV-1 and their role in viral persistence. Curr. HIV Res. 2008, 6, 388–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araínga, M.; Edagwa, B.; Mosley, R.L.; Poluektova, L.Y.; Gorantla, S.; Gendelman, H.E. A mature macrophage is a principal HIV-1 cellular reservoir in humanized mice after treatment with long acting antiretroviral therapy. Retrovirology 2017, 14, 17. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.K.; Yukl, S.A. Tissue reservoirs of HIV. Curr. Opin. HIV AIDS 2016, 11, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Crooks, A.M.; Bateson, R.; Cope, A.B.; Dahl, N.P.; Griggs, M.K.; Kuruc, J.D.; Gay, C.L.; Eron, J.J.; Margolis, D.M.; Bosch, R.J.; et al. Precise quantitation of the latent HIV-1 reservoir: Implications for eradication strategies. J. Infect. Dis. 2015, 212, 1361–1365. [Google Scholar] [CrossRef]
- Siliciano, J.M.; Siliciano, R.F. The remarkable stability of the latent reservoir for HIV-1 in resting memory CD4+ T cells. J. Infect. Dis. 2015, 212, 1345–1347. [Google Scholar] [CrossRef] [Green Version]
- Cantero-Pérez, J.; Grau-Expósito, J.; Serra-Peinado, C.; Rosero, D.A.; Luque-Ballesteros, L.; Astorga-Gamaza, A.; Castellví, J.; Sanhueza, T.; Tapia, G.; Lloveras, B.; et al. Resident memory T cells are a cellular reservoir for HIV in the cervical mucosa. Nat. Commun. 2019, 10, 4739. [Google Scholar]
- Baxter, A.E.; Niessl, J.; Fromentin, R.; Richard, J.; Porichis, F.; Charlebois, R.; Massanella, M.; Brassard, N.; Alsahafi, N.; Delgado, G.-G.; et al. Single-cell characterization of viral translation-competent reservoirs in HIV-infected individuals. Cell Host Microbe 2016, 20, 368–380. [Google Scholar] [CrossRef] [Green Version]
- Ganor, Y.; Real, F.; Sennepin, A.; Dutertre, C.-A.; Prevedel, L.; Xu, L.; Tudor, D.; Charmeteau, B.; Couedel-Courteille, A.; Marion, S.; et al. HIV-1 reservoirs in urethral macrophages of patients under suppressive antiretroviral therapy. Nat. Microbiol. 2019, 4, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.; Kang, G.; Hattler, J.B.; Galadima, H.I.; Zhang, J.; Li, Q.; Kim, W.-K. Macrophages but not astrocytes harbor HIV DNA in the brains of HIV-1-infected aviremic individuals on suppressive antiretroviral therapy. J. Neuroimmune Pharmacol. 2019, 14, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Sigal, A.; Kim, J.T.; Balazs, A.B.; Dekel, E.; Mayo, A.; Milo, R.; Baltimore, D. Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature 2011, 477, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef]
- Colby, D.J.; Trautmann, L.; Pinyakorn, S.; Leyre, L.; Pagliuzza, A.; Kroon, E.; Rolland, M.; Takata, H.; Buranapraditkun, S.; Intasan, J.; et al. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in Fiebig I acute HIV infection. Nat. Med. 2018, 24, 923–926. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, C.; Bouchat, S.; Marcello, A. HIV-1 transcription and latency: An update. Retrovirology 2013, 10, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakre, S.; Chavez, L.; Shirakawa, K.; Verdin, E. Epigenetic regulation of HIV latency. Curr. Opin. HIV AIDS 2011, 6, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Trono, D.; Van Lint, C.; Rouzioux, C.; Verdin, E.; Barre-Sinoussi, F.; Chun, T.W.; Chomont, N. HIV persistence and the prospect of long-term drug-free remissions for HIV-infected individuals. Science 2010, 329, 174–180. [Google Scholar] [CrossRef]
- Cary, D.C.; Fujinaga, K.; Peterlin, B.M. Molecular mechanisms of HIV latency. J. Clin. Investig. 2016, 126, 448–454. [Google Scholar] [CrossRef]
- Elsheikh, M.M.; Tang, Y.; Li, D.; Jiang, G. Deep latency: A new insight into a functional HIV cure. EBioMedicine 2019, 45, 624–629. [Google Scholar] [CrossRef] [Green Version]
- Duverger, A.; Wolschendorf, F.; Zhang, M.; Wagner, F.; Hatcher, B.; Jones, J.; Cron, R.Q.; van der Sluis, R.M.; Jeeninga, R.E.; Berkhout, B.; et al. An AP-1 binding site in the enhancer/core element of the HIV-1 promoter controls the ability of HIV-1 to establish latent infection. J. Virol. 2013, 87, 2264–2277. [Google Scholar] [CrossRef] [Green Version]
- Verdin, E.; Paras, P.; Van Lint, C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation activation [published erratum appears in EMBO J. 1993, 12, 4900]. EMBO J. 1993, 12, 3249–3259. [Google Scholar] [CrossRef]
- Van Lint, C.; Emiliani, S.; Ott, M.; Verdin, E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996, 15, 1112–1120. [Google Scholar] [CrossRef] [PubMed]
- Razooky, B.S.; Pai, A.; Aull, K.; Rouzine, I.M.; Weinberger, L.S. A hardwired HIV latency program. Cell 2015, 160, 990–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Lei, X.; Ribeiro, R.M.; Perelson, A.S.; Liang, J. Probabilistic control of HIV latency and transactivation by the Tat gene circuit. Proc. Natl. Acad. Sci. USA 2018, 115, 12453. [Google Scholar] [CrossRef] [PubMed]
- Chavez, L.; Calvanese, V.; Verdin, E. HIV latency is established directly and early in both resting and activated primary CD4 T cells. PLOS Pathog. 2015, 11, e1004955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, L.; Deng, K.; Gao, H.; Xing, S.; Capoferri, A.A.; Durand, C.M.; Rabi, S.A.; Laird, G.M.; Kim, M.; Hosmane, N.N.; et al. Transcriptional reprogramming during effector-to-memory transition renders CD4+ T cells permissive for latent HIV-1 infection. Immunity 2017, 47, 766–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobrowolski, C.; Valadkhan, S.; Graham, A.C.; Shukla, M.; Ciuffi, A.; Telenti, A.; Karn, J. Entry of polarized effector cells into quiescence forces HIV latency. mBio 2019, 10, e00337-00319. [Google Scholar] [CrossRef] [Green Version]
- Bradley, T.; Ferrari, G.; Haynes, B.F.; Margolis, D.M.; Browne, E.P. Single-cell analysis of quiescent HIV infection reveals host transcriptional profiles that regulate proviral latency. Cell Rep. 2018, 25, 107–117. [Google Scholar] [CrossRef] [Green Version]
- Norton, N.J.; Mok, H.P.; Sharif, F.; Hirst, J.C.; Lever, A.M.L. HIV silencing and inducibility are heterogeneous and are affected by factors intrinsic to the virus. mBio 2019, 10, e00188-00119. [Google Scholar] [CrossRef] [Green Version]
- Clark, E.; Nava, B.; Caputi, M. Tat is a multifunctional viral protein that modulates cellular gene expression and functions. Oncotarget 2017, 8, 27569–27581. [Google Scholar] [CrossRef] [Green Version]
- Sivakumaran, H.; Cutillas, V.; Harrich, D. Revisiting transdominant-negative proteins in HIV gene therapy. Future Virol. 2013, 8, 757–768. [Google Scholar] [CrossRef]
- Kurnaeva, M.A.; Sheval, E.V.; Musinova, Y.R.; Vassetzky, Y.S. Tat basic domain: A “Swiss army knife” of HIV-1 Tat? Rev. Med. Virol. 2019, 29, e2031. [Google Scholar] [CrossRef]
- Feinberg, M.B.; Baltimore, D.; Frankel, A.D. The role of Tat in the human immunodeficiency virus life cycle indicates a primary effect on transcriptional elongation. Proc. Natl. Acad. Sci. USA 1991, 88, 4045–4049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, S.Y.; Calman, A.F.; Luciw, P.A.; Peterlin, B.M. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature 1987, 330, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Laspia, M.F.; Rice, A.P.; Mathews, M.B. HIV-1 Tat protein increases transcriptional initiation and stabilizes elongation. Cell 1989, 59, 283–292. [Google Scholar] [CrossRef]
- Marciniak, R.A.; Sharp, P.A. HIV-1 Tat protein promotes formation of more-processive elongation complexes. EMBO J. 1991, 10, 4189–4196. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.; Sharmeen, L.; Kimpton, J.; Romeo, J.M.; Garcia, J.V.; Peterlin, B.M.; Groudine, M.; Emerman, M. Cellular latency in human immunodeficiency virus-infected individuals with high CD4 levels can be detected by the presence of promoter-proximal transcripts. Proc. Natl. Acad. Sci. USA 1994, 91, 3862–3866. [Google Scholar] [CrossRef] [Green Version]
- Nabel, G.; Baltimore, D. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 1987, 326, 711–713. [Google Scholar] [CrossRef]
- Weinberger, L.S.; Burnett, J.C.; Toettcher, J.E.; Arkin, A.P.; Schaffer, D.V. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell 2005, 122, 169–182. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, M.; Chen, L.F.; Chen, R. P-tefb: Finding its ways to release promoter-proximally paused RNA polymerase II. Transcription 2018, 9, 88–94. [Google Scholar] [CrossRef]
- Rice, A.P. The HIV-1 Tat protein: Mechanism of action and target for HIV-1 cure strategies. Curr. Pharm. Des. 2017, 23, 4098–4102. [Google Scholar] [CrossRef] [Green Version]
- Dahabieh, M.S.; Battivelli, E.; Verdin, E. Understanding HIV latency: The road to an HIV cure. Annu. Rev. Med. 2015, 66, 407–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planelles, V. An ounce of Tat prevention is worth a pound of functional cure. mBio 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vranckx, L.S.; Demeulemeester, J.; Saleh, S.; Boll, A.; Vansant, G.; Schrijvers, R.; Weydert, C.; Battivelli, E.; Verdin, E.; Cereseto, A.; et al. Ledgin-mediated inhibition of integrase-ledgf/p75 interaction reduces reactivation of residual latent HIV. EBioMedicine 2016, 8, 248–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darcis, G.; Van Driessche, B.; Van Lint, C. HIV latency: Should we shock or lock? Trends Immunol. 2017, 38, 217–228. [Google Scholar] [CrossRef]
- Mousseau, G.; Mediouni, S.; Valente, S.T. Targeting HIV transcription: The quest for a functional cure. Curr Top. Microbiol. Immunol. 2015, 389, 121–145. [Google Scholar]
- Mousseau, G.; Kessing, C.F.; Fromentin, R.; Trautmann, L.; Chomont, N.; Valente, S.T. The Tat inhibitor didehydro-cortistatin A prevents HIV-1 reactivation from latency. MBio 2015, 6, e00465. [Google Scholar] [CrossRef] [Green Version]
- Jean, M.J.; Hayashi, T.; Huang, H.; Brennan, J.; Simpson, S.; Purmal, A.; Gurova, K.; Keefer, M.C.; Kobie, J.J.; Santoso, N.G.; et al. Curaxin CBL0100 blocks HIV-1 replication and reactivation through inhibition of viral transcriptional elongation. Front. Microbiol. 2017, 8, 2007. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Choi, M.S.; Inn, K.S.; Kim, B.J. Inhibition of HIV-1 reactivation by a telomerase-derived peptide in a HSP90-dependent manner. Sci. Rep. 2016, 6, 28896. [Google Scholar] [CrossRef] [Green Version]
- Anderson, I.; Low, J.S.; Weston, S.; Weinberger, M.; Zhyvoloup, A.; Labokha, A.A.; Corazza, G.; Kitson, R.A.; Moody, C.J.; Marcello, A.; et al. Heat shock protein 90 controls HIV-1 reactivation from latency. Proc. Natl. Acad. Sci. USA 2014, 111, E1528–E1537. [Google Scholar] [CrossRef] [Green Version]
- Besnard, E.; Hakre, S.; Kampmann, M.; Lim, H.W.; Hosmane, N.N.; Martin, A.; Bassik, M.C.; Verschueren, E.; Battivelli, E.; Chan, J.; et al. The mTOR complex controls HIV latency. Cell Host Microbe 2016, 20, 785–797. [Google Scholar] [CrossRef] [Green Version]
- Margolis, D.M.; Garcia, J.V.; Hazuda, D.J.; Haynes, B.F. Latency reversal and viral clearance to cure HIV-1. Science 2016, 353, aaf6517. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Anderson, J.L.; Lewin, S.R. Getting the “kill” into “shock and kill”: Strategies to eliminate latent HIV. Cell Host Microbe 2018, 23, 14–26. [Google Scholar] [CrossRef]
- Aguilera, L.U.; Rodriguez-Gonzalez, J. Modeling the effect of Tat inhibitors on HIV latency. J. Theor. Biol. 2019, 473, 20–27. [Google Scholar] [CrossRef]
- Tabarrini, O.; Desantis, J.; Massari, S. Recent advances in the identification of Tat-mediated transactivation inhibitors: Progressing toward a functional cure of HIV. Future Med. Chem. 2016, 8, 421–442. [Google Scholar] [CrossRef]
- Pearson, L.; Garcia, J.; Wu, F.; Modesti, N.; Nelson, J.; Gaynor, R. A transdominant Tat mutant that inhibits Tat-induced gene expression from the human immunodeficiency virus long terminal repeat. Proc. Natl. Acad. Sci. USA 1990, 87, 5079–5083. [Google Scholar] [CrossRef] [Green Version]
- Modesti, N.; Garcia, J.; Debouck, C.; Peterlin, M.; Gaynor, R. Trans-dominant Tat mutants with alterations in the basic domain inhibit HIV-1 gene expression. New Biol. 1991, 3, 759–768. [Google Scholar]
- Kishi, M.; Nishino, Y.; Sumiya, M.; Ohki, K.; Kimura, T.; Goto, T.; Nakai, M.; Kakinuma, M.; Ikuta, K. Cells surviving infection by human immunodeficiency virus type 1: Vif or Vpu mutants produce non-infectious or markedly less cytopathic viruses. J. Gen. Virol. 1992, 73, 77–87. [Google Scholar] [CrossRef]
- Bevec, D.; Dobrovnik, M.; Hauber, J.; Bohnlein, E. Inhibition of human immunodeficiency virus type 1 replication in human T cells by retroviral-mediated gene transfer of a dominant-negative Rev trans-activator. Proc. Natl. Acad. Sci. USA 1992, 89, 9870–9874. [Google Scholar] [CrossRef] [Green Version]
- Malim, M.H.; Freimuth, W.W.; Liu, J.; Boyle, T.J.; Lyerly, H.K.; Cullen, B.R.; Nabel, G.J. Stable expression of transdominant Rev protein in human T cells inhibits human immunodeficiency virus replication. J. Exp. Med. 1992, 176, 1197–1201. [Google Scholar] [CrossRef] [Green Version]
- Ulich, C.; Harrich, D.; Estes, P.; Gaynor, R.B. Inhibition of human immunodeficiency virus type 1 replication is enhanced by a combination of transdominant Tat and Rev proteins. J. Virol. 1996, 70, 4871–4876. [Google Scholar] [CrossRef] [Green Version]
- Harrich, D.; Ulich, C.; Garcia-Martinez, L.F.; Gaynor, R.B. Tat is required for efficient HIV-1 reverse transcription. EMBO J. 1997, 16, 1224–1235. [Google Scholar] [CrossRef] [PubMed]
- Apolloni, A.; Meredith, L.W.; Suhrbier, A.; Kiernan, R.; Harrich, D. The HIV-1 Tat protein stimulates reverse transcription in vitro. Curr. HIV Res. 2007, 5, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Boudier, C.; Humbert, N.; Chaminade, F.; Chen, Y.; de Rocquigny, H.; Godet, J.; Mauffret, O.; Fossé, P.; Mély, Y. Dynamic interactions of the HIV-1 Tat with nucleic acids are critical for Tat activity in reverse transcription. Nucleic Acids Res. 2014, 42, 1065–1078. [Google Scholar] [CrossRef] [Green Version]
- Meredith, L.W.; Sivakumaran, H.; Major, L.; Suhrbier, A.; Harrich, D. Potent inhibition of HIV-1 replication by a Tat mutant. PLoS ONE 2009, 4, e7769. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.H.; Apolloni, A.; Cutillas, V.; Sivakumaran, H.; Martin, S.; Li, D.; Wei, T.; Wang, R.; Jin, H.; Spann, K.; et al. A mutant Tat protein inhibits HIV-1 reverse transcription by targeting the reverse transcription complex. J. Virol. 2015, 89, 4827–4836. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.H.; Sivakumaran, H.; Jones, A.; Li, D.; Harper, C.; Wei, T.; Jin, H.; Rustanti, L.; Meunier, F.A.; Spann, K.; et al. A HIV-1 Tat mutant protein disrupts HIV-1 Rev function by targeting the DEAD-box RNA helicase DDX1. Retrovirology 2014, 11, 121. [Google Scholar] [CrossRef]
- Lin, M.H.; Sivakumaran, H.; Apolloni, A.; Wei, T.; Jans, D.A.; Harrich, D. Nullbasic, a potent anti-HIV Tat mutant, induces CRM1-dependent disruption of HIV Rev trafficking. PLoS ONE 2012, 7, e51466. [Google Scholar] [CrossRef] [Green Version]
- Apolloni, A.; Lin, M.H.; Sivakumaran, H.; Li, D.; Kershaw, M.H.; Harrich, D. A mutant Tat protein provides strong protection from HIV-1 infection in human CD4+ T cells. Hum. Gene Ther. 2013, 24, 270–282. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Li, D.; Sivakumaran, H.; Lor, M.; Rustanti, L.; Cloonan, N.; Wani, S.; Harrich, D. Shutdown of HIV-1 transcription in T cells by Nullbasic, a mutant Tat protein. MBio 2016, 7, e00518-16. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Sun, Y.; Li, D.; Lin, M.-H.; Lor, M.; Rustanti, L.; Harrich, D. Strong in vivo inhibition of HIV-1 replication by Nullbasic, a Tat mutant. mBio 2019, 10, e01769-01719. [Google Scholar] [CrossRef] [Green Version]
- Fraldi, A.; Varrone, F.; Napolitano, G.; Michels, A.A.; Majello, B.; Bensaude, O.; Lania, L. Inhibition of Tat activity by the HEXIM1 protein. Retrovirology 2005, 2, 42. [Google Scholar] [CrossRef] [Green Version]
- Leoz, M.; Kukanja, P.; Luo, Z.; Huang, F.; Cary, D.C.; Peterlin, B.M.; Fujinaga, K. HEXIM1-Tat chimera inhibits HIV-1 replication. PLOS Pathogens 2018, 14, e1007402. [Google Scholar] [CrossRef]
- Mousseau, G.; Clementz, M.A.; Bakeman, W.N.; Nagarsheth, N.; Cameron, M.; Shi, J.; Baran, P.; Fromentin, R.; Chomont, N.; Valente, S.T. An analog of the natural steroidal alkaloid cortistatin A potently suppresses Tat-dependent HIV transcription. Cell Host Microbe 2012, 12, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Mediouni, S.; Chinthalapudi, K.; Ekka, M.K.; Usui, I.; Jablonski, J.A.; Clementz, M.A.; Mousseau, G.; Nowak, J.; Macherla, V.R.; Beverage, J.N.; et al. Didehydro-cortistatin A inhibits HIV-1 by specifically binding to the unstructured basic region of Tat. MBio 2019, 10, e02662-18. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Mousseau, G.; Valente, S.T. Tat inhibition by didehydro-cortistatin A promotes heterochromatin formation at the HIV-1 long terminal repeat. Epigenetics Chromatin 2019, 12, 23. [Google Scholar] [CrossRef]
- Mediouni, S.; Kessing, C.F.; Jablonski, J.A.; Thenin-Houssier, S.; Clementz, M.; Kovach, M.D.; Mousseau, G.; de Vera, I.M.S.; Li, C.; Kojetin, D.J.; et al. The Tat inhibitor didehydro-cortistatin A suppresses SIV replication and reactivation. FASEB J. 2019, 33, 8280–8293. [Google Scholar] [CrossRef]
- Wan, Z.; Chen, X. Triptolide inhibits human immunodeficiency virus type 1 replication by promoting proteasomal degradation of Tat protein. Retrovirology 2014, 11, 88. [Google Scholar] [CrossRef]
- Ensoli, B.; Fiorelli, V.; Ensoli, F.; Lazzarin, A.; Visintini, R.; Narciso, P.; Di Carlo, A.; Tripiciano, A.; Longo, O.; Bellino, S.; et al. The preventive phase I trial with the HIV-1 Tat-based vaccine. Vaccine 2009, 28, 371–378. [Google Scholar] [CrossRef]
- Ensoli, B.; Nchabeleng, M.; Ensoli, F.; Tripiciano, A.; Bellino, S.; Picconi, O.; Sgadari, C.; Longo, O.; Tavoschi, L.; Joffe, D.; et al. HIV-Tat immunization induces cross-clade neutralizing antibodies and CD4+ T cell increases in antiretroviral-treated South African volunteers: A randomized phase II clinical trial. Retrovirology 2016, 13, 34. [Google Scholar] [CrossRef] [Green Version]
- Cafaro, A.; Caputo, A.; Fracasso, C.; Maggiorella, M.T.; Goletti, D.; Baroncelli, S.; Pace, M.; Sernicola, L.; Koanga-Mogtomo, M.L.; Betti, M.; et al. Control of SHIV-89.6p-infection of cynomolgus monkeys by HIV-1 Tat protein vaccine. Nat. Med. 1999, 5, 643–650. [Google Scholar] [CrossRef]
- Maggiorella, M.T.; Baroncelli, S.; Michelini, Z.; Fanales-Belasio, E.; Moretti, S.; Sernicola, L.; Cara, A.; Negri, D.R.; Butto, S.; Fiorelli, V.; et al. Long-term protection against SHIV89.6p replication in HIV-1 Tat vaccinated cynomolgus monkeys. Vaccine 2004, 22, 3258–3269. [Google Scholar] [CrossRef]
- Ensoli, F.; Cafaro, A.; Casabianca, A.; Tripiciano, A.; Bellino, S.; Longo, O.; Francavilla, V.; Picconi, O.; Sgadari, C.; Moretti, S.; et al. HIV-1 Tat immunization restores immune homeostasis and attacks the HAART-resistant blood HIV DNA: Results of a randomized phase II exploratory clinical trial. Retrovirology 2015, 12, 33. [Google Scholar] [CrossRef] [Green Version]
- Sgadari, C.; Monini, P.; Tripiciano, A.; Picconi, O.; Casabianca, A.; Orlandi, C.; Moretti, S.; Francavilla, V.; Arancio, A.; Paniccia, G.; et al. Continued decay of HIV proviral DNA upon vaccination with HIV-1 Tat of subjects on long-term ART: An 8-year follow-up study. Front. Immunol. 2019, 10, 233. [Google Scholar] [CrossRef]
- Ensoli, B.; Bellino, S.; Tripiciano, A.; Longo, O.; Francavilla, V.; Marcotullio, S.; Cafaro, A.; Picconi, O.; Paniccia, G.; Scoglio, A.; et al. Therapeutic immunization with HIV-1 Tat reduces immune activation and loss of regulatory T-cells and improves immune function in subjects on HAART. PLoS ONE 2010, 5, e13540. [Google Scholar] [CrossRef] [Green Version]
- Opi, S.; Peloponese, J.M.; Esquieu, D.; Campbell, G.; de Mareuil, J.; Walburger, A.; Solomiac, M.; Gregoire, C.; Bouveret, E.; Yirrell, D.L.; et al. Tat HIV-1 primary and tertiary structures critical to immune response against non-homologous variants. J. Biol. Chem. 2002, 277, 35915–35919. [Google Scholar] [CrossRef] [Green Version]
- Loret, E.P.; Darque, A.; Jouve, E.; Loret, E.A.; Nicolino-Brunet, C.; Morange, S.; Castanier, E.; Casanova, J.; Caloustian, C.; Bornet, C.; et al. Intradermal injection of a Tat Oyi-based therapeutic HIV vaccine reduces of 1.5 log copies/mL the HIV RNA rebound median and no HIV DNA rebound following cart interruption in a phase I/II randomized controlled clinical trial. Retrovirology 2016, 13, 21. [Google Scholar] [CrossRef] [Green Version]
- Watkins, J.D.; Lancelot, S.; Campbell, G.R.; Esquieu, D.; de Mareuil, J.; Opi, S.; Annappa, S.; Salles, J.P.; Loret, E.P. Reservoir cells no longer detectable after a heterologous SHIV challenge with the synthetic HIV-1 Tat Oyi vaccine. Retrovirology 2006, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Spina, C.A.; Anderson, J.; Archin, N.M.; Bosque, A.; Chan, J.; Famiglietti, M.; Greene, W.C.; Kashuba, A.; Lewin, S.R.; Margolis, D.M.; et al. An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS Pathog. 2013, 9, e1003834. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, C.; Archin, N.; Michaels, D.; Belkina, A.C.; Denis, G.V.; Bradner, J.; Sebastiani, P.; Margolis, D.M.; Montano, M. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J. Leukoc Biol. 2012, 92, 1147–1154. [Google Scholar] [CrossRef]
- Ylisastigui, L.; Archin, N.M.; Lehrman, G.; Bosch, R.J.; Margolis, D.M. Coaxing HIV-1 from resting CD4 T cells: Histone deacetylase inhibition allows latent viral expression. AIDS 2004, 18, 1101–1108. [Google Scholar] [CrossRef]
- Archin, N.M.; Espeseth, A.; Parker, D.; Cheema, M.; Hazuda, D.; Margolis, D.M. Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS Res. Hum. Retroviruses 2009, 25, 207–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras, X.; Schweneker, M.; Chen, C.S.; McCune, J.M.; Deeks, S.G.; Martin, J.; Peterlin, B.M. Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells. J. Biol. Chem. 2009, 284, 6782–6789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.F.; Strain, M.C.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rustanti, L.; Jin, H.; Lor, M.; Lin, M.H.; Rawle, D.J.; Harrich, D. A mutant Tat protein inhibits infection of human cells by strains from diverse HIV-1 subtypes. Virol. J. 2017, 14, 52. [Google Scholar] [CrossRef] [PubMed]
- AJ, C.Q.; Bugai, A.; Barboric, M. Cracking the control of RNA polymerase II elongation by 7SK snRNP and P-TEFb. Nucleic Acids Res. 2016, 44, 7527–7539. [Google Scholar]
- Yik, J.H.; Chen, R.; Pezda, A.C.; Samford, C.S.; Zhou, Q. A human immunodeficiency virus type 1 Tat-like arginine-rich RNA-binding domain is essential for HEXIM1 to inhibit RNA polymerase II transcription through 7SK snRNA-mediated inactivation of P-TEFb. Mol. Cell Biol. 2004, 24, 5094–5105. [Google Scholar] [CrossRef] [Green Version]
- Michels, A.A.; Fraldi, A.; Li, Q.; Adamson, T.E.; Bonnet, F.; Nguyen, V.T.; Sedore, S.C.; Price, J.P.; Price, D.H.; Lania, L.; et al. Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. EMBO J. 2004, 23, 2608–2619. [Google Scholar] [CrossRef] [Green Version]
- Yik, J.H.; Chen, R.; Nishimura, R.; Jennings, J.L.; Link, A.J.; Zhou, Q. Inhibition of P-TEFb (CDK9/cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol. Cell 2003, 12, 971–982. [Google Scholar] [CrossRef]
- Tahirov, T.H.; Babayeva, N.D.; Varzavand, K.; Cooper, J.J.; Sedore, S.C.; Price, D.H. Crystal structure of HIV-1 Tat complexed with human P-TEFb. Nature 2010, 465, 747–751. [Google Scholar] [CrossRef]
- Shi, J.; Manolikakes, G.; Yeh, C.H.; Guerrero, C.A.; Shenvi, R.A.; Shigehisa, H.; Baran, P.S. Scalable synthesis of cortistatin A and related structures. J. Am. Chem. Soc. 2011, 133, 8014–8027. [Google Scholar] [CrossRef] [Green Version]
- Pelish, H.E.; Liau, B.B.; Nitulescu, I.I.; Tangpeerachaikul, A.; Poss, Z.C.; Da Silva, D.H.; Caruso, B.T.; Arefolov, A.; Fadeyi, O.; Christie, A.L.; et al. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature 2015, 526, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Cee, V.J.; Chen, D.Y.; Lee, M.R.; Nicolaou, K.C. Cortistatin A is a high-affinity ligand of protein kinases ROCK, CDK8, and CDK11. Angew. Chem. Int. Ed. Engl. 2009, 48, 8952–8957. [Google Scholar] [CrossRef] [PubMed]
- Cary, D.C.; Rheinberger, M.; Rojc, A.; Peterlin, B.M. HIV transcription is independent of mediator kinases. AIDS Res. Hum. Retroviruses 2019, 35, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Mediouni, S.; Jablonski, J.; Paris, J.J.; Clementz, M.A.; Thenin-Houssier, S.; McLaughlin, J.P.; Valente, S.T. Didehydro-cortistatin A inhibits HIV-1 Tat mediated neuroinflammation and prevents potentiation of cocaine reward in Tat transgenic mice. Curr. HIV Res. 2015, 13, 64–79. [Google Scholar] [CrossRef] [Green Version]
- Turner, A.W.; Margolis, D.M. Chromatin regulation and the histone code in HIV latency. Yale J. Biol. Med. 2017, 90, 229–243. [Google Scholar]
- Easley, R.; Carpio, L.; Dannenberg, L.; Choi, S.; Alani, D.; Van Duyne, R.; Guendel, I.; Klase, Z.; Agbottah, E.; Kehn-Hall, K.; et al. Transcription through the HIV-1 nucleosomes: Effects of the PBAF complex in Tat activated transcription. Virology 2010, 405, 322–333. [Google Scholar] [CrossRef] [Green Version]
- Kessing, C.F.; Nixon, C.C.; Li, C.; Tsai, P.; Takata, H.; Mousseau, G.; Ho, P.T.; Honeycutt, J.B.; Fallahi, M.; Trautmann, L.; et al. In vivo suppression of HIV rebound by didehydro-cortistatin A, a “block-and-lock” strategy for HIV-1 treatment. Cell Rep. 2017, 21, 600–611. [Google Scholar] [CrossRef] [Green Version]
- Rice, A.P. Unexpected mutations in HIV-1 that confer resistance to the Tat inhibitor didehydro-cortistatin A. MBio 2019, 10, e01547-19. [Google Scholar] [CrossRef] [Green Version]
- Mousseau, G.; Aneja, R.; Clementz, M.A.; Mediouni, S.; Lima, N.S.; Haregot, A.; Kessing, C.F.; Jablonski, J.A.; Thenin-Houssier, S.; Nagarsheth, N.; et al. Resistance to the Tat inhibitor didehydro-cortistatin A is mediated by heightened basal HIV-1 transcription. MBio 2019, 10, e01750-18. [Google Scholar] [CrossRef] [Green Version]
- Bao, J.; Dai, S.-M. A chinese herb Tripterygium wilfordii hook F in the treatment of rheumatoid arthritis: Mechanism, efficacy, and safety. Rheumatol. Int. 2011, 31, 1123–1129. [Google Scholar] [CrossRef]
- Tao, X.; Younger, J.; Fan, F.Z.; Wang, B.; Lipsky, P.E. Benefit of an extract of Tripterygium wilfordii hook F in patients with rheumatoid arthritis: A double-blind, placebo-controlled study. Arthritis Rheum 2002, 46, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-R.; Dai, Y.; Zhao, J.; Lin, L.; Wang, Y.; Wang, Y. A mechanistic overview of triptolide and celastrol, natural products from Tripterygium wilfordii hook F. Front. Pharmacol. 2018, 9, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.T.; Kim, S.Y.; Lee, J.; Kim, K.; Park, S.H.; Park, Y.S.; Lim, H.Y.; Kang, W.K.; Park, J.O. Triptolide as a novel agent in pancreatic cancer: The validation using patient derived pancreatic tumor cell line. BMC Cancer 2018, 18, 1103. [Google Scholar] [CrossRef] [PubMed]
- Ziaei, S.; Halaby, R. Immunosuppressive, anti-inflammatory and anti-cancer properties of triptolide: A mini review. Avicenna J. Phytomed. 2016, 6, 149–164. [Google Scholar]
- Qiu, D.; Zhao, G.; Aoki, Y.; Shi, L.; Uyei, A.; Nazarian, S.; Ng, J.C.H.; Kao, P.N. Immunosuppressant PG490 (triptolide) inhibits T-cell interleukin-2 expression at the level of purine-box/nuclear factor of activated T-cells and NF-κB transcriptional activation. J. Biol. Chem. 1999, 274, 13443–13450. [Google Scholar] [CrossRef] [Green Version]
- Vispé, S.; DeVries, L.; Créancier, L.; Besse, J.; Bréand, S.; Hobson, D.J.; Svejstrup, J.Q.; Annereau, J.-P.; Cussac, D.; Dumontet, C.; et al. Triptolide is an inhibitor of RNA polymerase I and II–dependent transcription leading predominantly to down-regulation of short-lived mRNA. Mol. Cancer Ther. 2009, 8, 2780. [Google Scholar]
- Wang, Y.; Lu, J.-j.; He, L.; Yu, Q. Triptolide (TPL) inhibits global transcription by inducing proteasome-dependent degradation of RNA polymerase II (Pol II). PLoS ONE 2011, 6, e23993. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.X.; Smith, E.R.; Shilatifard, A. Born to run: Control of transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2018, 19, 464–478. [Google Scholar] [CrossRef]
- Liang, X.; Xie, R.; Su, J.; Ye, B.; Wei, S.; Liang, Z.; Bai, R.; Chen, Z.; Li, Z.; Gao, X. Inhibition of RNA polymerase III transcription by triptolide attenuates colorectal tumorigenesis. J. Exp. Clin. Cancer Res. 2019, 38, 217. [Google Scholar] [CrossRef]
- Titov, D.V.; Gilman, B.; He, Q.-L.; Bhat, S.; Low, W.-K.; Dang, Y.; Smeaton, M.; Demain, A.L.; Miller, P.S.; Kugel, J.F.; et al. XPB, a subunit of TFIIH, is a target of the natural product triptolide. Nat. Chem. Biol. 2011, 7, 182–188. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Gao, X.; Shilatifard, A. Stably paused genes revealed through inhibition of transcription initiation by the TFIIH inhibitor triptolide. Genes Dev. 2015, 29, 39–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derdeyn, C.A.; Decker, J.M.; Sfakianos, J.N.; Wu, X.; O’Brien, W.A.; Ratner, L.; Kappes, J.C.; Shaw, G.M.; Hunter, E. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J. Virol. 2000, 74, 8358–8367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platt, E.J.; Wehrly, K.; Kuhmann, S.E.; Chesebro, B.; Kabat, D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol. 1998, 72, 2855–2864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Decker, J.M.; Liu, H.; Zhang, Z.; Arani, R.B.; Kilby, J.M.; Saag, M.S.; Wu, X.; Shaw, G.M.; Kappes, J.C. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother. 2002, 46, 1896–1905. [Google Scholar] [CrossRef] [Green Version]
- Sivakumaran, H.; Lin, M.-H.; Apolloni, A.; Cutillas, V.; Jin, H.; Li, D.; Wei, T.; Harrich, D. Overexpression of PRMT6 does not suppress HIV-1 Tat transactivation in cells naturally lacking PRMT6. Virol. J. 2013, 10, 207. [Google Scholar] [CrossRef] [Green Version]
- Fan, D.; Guo, Q.; Shen, J.; Zheng, K.; Lu, C.; Zhang, G.; Lu, A.; He, X. The effect of triptolide in rheumatoid arthritis: From basic research towards clinical translation. Int. J. Mol. Sci. 2018, 19, 376. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, G. HIV-1 Tat protein as a potential AIDS vaccine. Nat. Med. 1996, 2, 960–964. [Google Scholar] [CrossRef]
- Singh, A.; Weinberger, L.S. Stochastic gene expression as a molecular switch for viral latency. Curr. Opin. Microbiol. 2009, 12, 460–466. [Google Scholar] [CrossRef] [Green Version]
- Ensoli, B.; Barillari, G.; Salahuddin, S.Z.; Gallo, R.C.; Wong-Staal, F. Tat protein of HIV-1 stimulates growth of cells derived from Kaposi’s sarcoma lesions of AIDS patients. Nature 1990, 345, 84–86. [Google Scholar] [CrossRef]
- Westendorp, M.O.; Frank, R.; Ochsenbauer, C.; Stricker, K.; Dhein, J.; Walczak, H.; Debatin, K.M.; Krammer, P.H. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature 1995, 375, 497–500. [Google Scholar] [CrossRef]
- Xiao, H.; Neuveut, C.; Tiffany, H.L.; Benkirane, M.; Rich, E.A.; Murphy, P.M.; Jeang, K.T. Selective CXCR4 antagonism by Tat: Implications for in vivo expansion of coreceptor use by HIV-1. Proc. Natl. Acad. Sci. USA 2000, 97, 11466–11471. [Google Scholar] [CrossRef] [Green Version]
- Rayne, F.; Debaisieux, S.; Bonhoure, A.; Beaumelle, B. HIV-1 Tat is unconventionally secreted through the plasma membrane. Cell Biol. Int. 2010, 34, 409–413. [Google Scholar] [CrossRef]
- Mele, A.R.; Marino, J.; Chen, K.; Pirrone, V.; Janetopoulos, C.; Wigdahl, B.; Klase, Z.; Nonnemacher, M.R. Defining the molecular mechanisms of HIV-1 Tat secretion: Ptdins(4,5)p2 at the epicenter. Traffic 2018, 19, 655–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.J.; Ueda, Y.; Shi, B.; Borodyansky, L.; Huang, L.; Li, Y.Z.; Pardee, A.B. Tat protein induces self-perpetuating permissivity for productive HIV-1 infection. Proc. Natl. Acad. Sci. USA 1997, 94, 8116–8120. [Google Scholar] [CrossRef] [Green Version]
- Li, C.J.; Friedman, D.J.; Wang, C.; Metelev, V.; Pardee, A.B. Induction of apoptosis in uninfected lymphocytes by HIV-1 Tat protein. Science 1995, 268, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Howcroft, T.K.; Strebel, K.; Martin, M.A.; Singer, D.S. Repression of MHC class I gene promoter activity by two-exon tat of HIV. Science 1993, 260, 1320–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, W.A.; Robinson, S.M.; Nath, A. Permeability of the blood-brain barrier to HIV-1 Tat. Exp. Neurol. 2005, 193, 218–227. [Google Scholar] [CrossRef]
- Price, T.O.; Ercal, N.; Nakaoke, R.; Banks, W.A. HIV-1 viral proteins gp120 and Tat induce oxidative stress in brain endothelial cells. Brain Res. 2005, 1045, 57–63. [Google Scholar] [CrossRef]
- Rojas-Celis, V.; Valiente-Echeverria, F.; Soto-Rifo, R.; Toro-Ascuy, D. New challenges of HIV-1 infection: How HIV-1 attacks and resides in the central nervous system. Cells 2019, 8, 1245. [Google Scholar] [CrossRef] [Green Version]
- Brake, D.A.; Goudsmit, J.; Krone, W.J.; Schammel, P.; Appleby, N.; Meloen, R.H.; Debouck, C. Characterization of murine monoclonal antibodies to the Tat protein from human immunodeficiency virus type 1. J. Virol. 1990, 64, 962–965. [Google Scholar] [CrossRef] [Green Version]
- Krone, W.J.; Debouck, C.; Epstein, L.G.; Heutink, P.; Meloen, R.; Goudsmit, J. Natural antibodies to HIV-Tat epitopes and expression of HIV-1 genes in vivo. J. Med. Virol. 1988, 26, 261–270. [Google Scholar] [CrossRef]
- Reiss, P.; Lange, J.M.; de Ronde, A.; de Wolf, F.; Dekker, J.; Debouck, C.; Goudsmit, J. Speed of progression to AIDS and degree of antibody response to accessory gene products of HIV-1. J. Med. Virol. 1990, 30, 163–168. [Google Scholar] [CrossRef]
- Re, M.C.; Furlini, G.; Vignoli, M.; Ramazzotti, E.; Roderigo, G.; De Rosa, V.; Zauli, G.; Lolli, S.; Capitani, S.; La Placa, M. Effect of antibody to HIV-1 Tat protein on viral replication in vitro and progression of HIV-1 disease in vivo. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1995, 10, 408–416. [Google Scholar] [CrossRef]
- Zagury, J.F.; Sill, A.; Blattner, W.; Lachgar, A.; Le Buanec, H.; Richardson, M.; Rappaport, J.; Hendel, H.; Bizzini, B.; Gringeri, A.; et al. Antibodies to the HIV-1 Tat protein correlated with nonprogression to AIDS: A rationale for the use of Tat toxoid as an HIV-1 vaccine. J. Hum. Virol. 1998, 1, 282–292. [Google Scholar]
- Re, M.C.; Vignoli, M.; Furlini, G.; Gibellini, D.; Colangeli, V.; Vitone, F.; La Placa, M. Antibodies against full-length Tat protein and some low-molecular-weight Tat-peptides correlate with low or undetectable viral load in HIV-1 seropositive patients. J. Clin. Virol. 2001, 21, 81–89. [Google Scholar] [CrossRef]
- Robertson, D.L.; Anderson, J.P.; Bradac, J.A.; Carr, J.K.; Foley, B.; Funkhouser, R.K.; Gao, F.; Hahn, B.H.; Kalish, M.L.; Kuiken, C.; et al. HIV-1 nomenclature proposal. Science 2000, 288, 55–56. [Google Scholar] [CrossRef]
- Ratner, L.; Haseltine, W.; Patarca, R.; Livak, K.J.; Starcich, B.; Josephs, S.F.; Doran, E.R.; Rafalski, J.A.; Whitehorn, E.A.; Baumeister, K.; et al. Complete nucleotide sequence of the AIDS virus, HTLV-III. Nature 1985, 313, 277–284. [Google Scholar] [CrossRef]
- Huet, T.; Dazza, M.C.; Brun-Vezinet, F.; Roelants, G.E.; Wain-Hobson, S. A highly defective HIV-1 strain isolated from a healthy Gabonese individual presenting an atypical western blot. AIDS 1989, 3, 707–715. [Google Scholar] [CrossRef]
- Garcia, J.A.; Harrich, D.; Pearson, L.; Mitsuyasu, R.; Gaynor, R.B. Functional domains required for Tat-induced transcriptional activation of the HIV-1 long terminal repeat. EMBO J. 1988, 7, 3143–3147. [Google Scholar] [CrossRef]
- Foley, B.; Leitner, T.; Apetrei, C.; Hahn, B.; Mizrachi, I.; Mullins, J.; Rambaut, A.; Wolinsky, S.; Korber, B. HIV Sequence Compendium 2018; Theoretical Biology and Biophysics Group, Los Alamos National Laboratory: LA-UR, NM, USA, 2018; Volume 18, p. 25673. [Google Scholar]
- Pauza, C.D.; Trivedi, P.; Wallace, M.; Ruckwardt, T.J.; Le Buanec, H.; Lu, W.; Bizzini, B.; Burny, A.; Zagury, D.; Gallo, R.C. Vaccination with Tat toxoid attenuates disease in simian/HIV-challenged macaques. Proc. Natl. Acad. Sci. USA 2000, 97, 3515–3519. [Google Scholar] [CrossRef]
- Stittelaar, K.J.; Gruters, R.A.; Schutten, M.; van Baalen, C.A.; van Amerongen, G.; Cranage, M.; Liljestrom, P.; Sutter, G.; Osterhaus, A.D. Comparison of the efficacy of early versus late viral proteins in vaccination against SIV. Vaccine 2002, 20, 2921–2927. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Casimiro, D.R.; Schleif, W.A.; Wang, F.; Davies, M.E.; Zhang, Z.Q.; Fu, T.M.; Finnefrock, A.C.; Handt, L.; Citron, M.P.; et al. Vectored Gag and Env but not Tat show efficacy against simian-human immunodeficiency virus 89.6p challenge in mamu-a*01-negative rhesus monkeys. J. Virol. 2005, 79, 12321–12331. [Google Scholar] [CrossRef] [Green Version]
- Cafaro, A.; Tripiciano, A.; Picconi, O.; Sgadari, C.; Moretti, S.; Buttò, S.; Monini, P.; Ensoli, B. Anti-Tat immunity in HIV-1 infection: Effects of naturally occurring and vaccine-induced antibodies against Tat on the course of the disease. Vaccines 2019, 7, 99. [Google Scholar] [CrossRef] [Green Version]
- Bellino, S.; Tripiciano, A.; Picconi, O.; Francavilla, V.; Longo, O.; Sgadari, C.; Paniccia, G.; Arancio, A.; Angarano, G.; Ladisa, N.; et al. The presence of anti-Tat antibodies in HIV-infected individuals is associated with containment of CD4+ T-cell decay and viral load, and with delay of disease progression: Results of a 3-year cohort study. Retrovirology 2014, 11, 49. [Google Scholar] [CrossRef] [Green Version]
- Demirhan, I.; Chandra, A.; Mueller, F.; Mueller, H.; Biberfeld, P.; Hasselmayer, O.; Chandra, P. Antibody spectrum against the viral transactivator protein in patients with human immunodeficiency virus type 1 infection and Kaposi’s sarcoma. J. Hum. Virol. 2000, 3, 137–143. [Google Scholar]
- Chen, Q.; Li, L.; Liao, W.; Zhang, H.; Wang, J.; Sheng, B.; Zhang, H.; Huang, X.; Ding, Y.; Zhang, T.; et al. Characterization of Tat antibody responses in Chinese individuals infected with HIV-1. PLoS ONE 2013, 8, e60825. [Google Scholar] [CrossRef] [Green Version]
- Butto, S.; Fiorelli, V.; Tripiciano, A.; Ruiz-Alvarez, M.J.; Scoglio, A.; Ensoli, F.; Ciccozzi, M.; Collacchi, B.; Sabbatucci, M.; Cafaro, A.; et al. Sequence conservation and antibody cross-recognition of clade B human immunodeficiency virus (HIV) type 1 Tat protein in HIV-1-infected italians, ugandans, and south africans. J. Infect. Dis. 2003, 188, 1171–1180. [Google Scholar] [CrossRef]
- Rezza, G.; Fiorelli, V.; Dorrucci, M.; Ciccozzi, M.; Tripiciano, A.; Scoglio, A.; Collacchi, B.; Ruiz-Alvarez, M.; Giannetto, C.; Caputo, A.; et al. The presence of anti-Tat antibodies is predictive of long-term nonprogression to AIDS or severe immunodeficiency: Findings in a cohort of HIV-1 seroconverters. J. Infect. Dis 2005, 191, 1321–1324. [Google Scholar] [CrossRef] [Green Version]
- Viglianti, G.A.; Mullins, J.I. Functional comparison of transactivation by simian immunodeficiency virus from rhesus macaques and human immunodeficiency virus type 1. J. Virol. 1988, 62, 4523–4532. [Google Scholar] [CrossRef] [Green Version]
- Kamori, D.; Ueno, T. HIV-1 Tat and viral latency: What we can learn from naturally occurring sequence variations. Front. Microbiol. 2017, 8, 80. [Google Scholar] [CrossRef] [Green Version]
- Rodman, T.C.; To, S.E.; Hashish, H.; Manchester, K. Epitopes for natural antibodies of human immunodeficiency virus (HIV)-negative (normal) and HIV-positive sera are coincident with two key functional sequences of HIV Tat protein. Proc. Natl. Acad. Sci. USA 1993, 90, 7719. [Google Scholar] [CrossRef] [Green Version]
- Shojania, S.; O’Neil J, D. HIV-1 Tat is a natively unfolded protein: The solution conformation and dynamics of reduced HIV-1 Tat1-72 by NMR spectroscopy. J. Biol. Chem. 2006, 281, 8347–8356. [Google Scholar] [CrossRef] [Green Version]
- Riddell, J.T.; Amico, K.R.; Mayer, K.H. HIV pre-exposure prophylaxis: A review. JAMA 2018, 319, 1261–1268. [Google Scholar] [CrossRef]
- Hong, F.F.; Mellors, J.W. Changes in HIV reservoirs during long-term antiretroviral therapy. Curr. Opin. HIV AIDS 2015, 10, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Ghose, R.; Liou, L.Y.; Herrmann, C.H.; Rice, A.P. Induction of TAK (cyclin T1/P-TEFb) in purified resting CD4(+) T lymphocytes by combination of cytokines. J. Virol. 2001, 75, 11336–11343. [Google Scholar] [CrossRef] [Green Version]
- Sung, T.L.; Rice, A.P. Effects of prostratin on cyclin T1/P-TEFb function and the gene expression profile in primary resting CD4+ T cells. Retrovirology 2006, 3, 66. [Google Scholar] [CrossRef] [Green Version]
- Morgan, R.A.; Walker, R.; Carter, C.S.; Natarajan, V.; Tavel, J.A.; Bechtel, C.; Herpin, B.; Muul, L.; Zheng, Z.; Jagannatha, S.; et al. Preferential survival of CD4+ T lymphocytes engineered with anti-human immunodeficiency virus (HIV) genes in HIV-infected individuals. Hum. Gene Ther. 2005, 16, 1065–1074. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Anti-Tat Agent | Type | Inhibits; Proposed Mechanism(s) | Reference(s) |
|---|---|---|---|
| Nullbasic | Mutant Tat protein | 1.Tat transactivation; binds to P-TEFb | [64,68,69,70] |
| 2. Reverse transcription; binds to reverse transcriptase | [64,65] | ||
| 3. Rev; binds to DDX1 | [64,66,67] | ||
| HT1 | HEXIM1-Tat fusion | Tat transactivation; binds to P-TEFb and inactivates CDK9 | [71,72] |
| dCA | Small compound | 1. Tat transactivation; binds Tat basic domain and prevents interaction with TAR RNA | [46,73,74] |
| 2. HIV transcription levels; heterochromatin formation on HIV LTR promoter | [75,76] | ||
| Triptolide | Small compound | 1. Decreases Tat steady state levels; mechanism unclear | [77] |
| Tat-BH10 | Vaccine | 1. Tat; Tat neutralizing antibodies | [78,79] |
| 2. Virus production; anti-Tat cellular responses | [78,80,81] | ||
| 3. Provirus DNA load in blood lymphocytes; anti-Tat humoral/cellular responses | [82,83] | ||
| 4. Reduced immune activation; possibly due to reduced viral burden | [82,84] | ||
| Tat-Oyi | Vaccine | 1. Tat; Tat neutralizing antibodies | [85,86] |
| 2. Virus production, mechanism unclear | [86] | ||
| 3. Viral rebound after treatment interruption; mechanism unclear | [86] | ||
| 3. Provirus DNA; mechanism unclear | [86,87] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, H.; Li, D.; Lin, M.-H.; Li, L.; Harrich, D. Tat-Based Therapies as an Adjuvant for an HIV-1 Functional Cure. Viruses 2020, 12, 415. https://doi.org/10.3390/v12040415
Jin H, Li D, Lin M-H, Li L, Harrich D. Tat-Based Therapies as an Adjuvant for an HIV-1 Functional Cure. Viruses. 2020; 12(4):415. https://doi.org/10.3390/v12040415
Chicago/Turabian StyleJin, Hongping, Dongsheng Li, Min-Hsuan Lin, Li Li, and David Harrich. 2020. "Tat-Based Therapies as an Adjuvant for an HIV-1 Functional Cure" Viruses 12, no. 4: 415. https://doi.org/10.3390/v12040415
APA StyleJin, H., Li, D., Lin, M. -H., Li, L., & Harrich, D. (2020). Tat-Based Therapies as an Adjuvant for an HIV-1 Functional Cure. Viruses, 12(4), 415. https://doi.org/10.3390/v12040415