Detection of Recombinant Rousettus Bat Coronavirus GCCDC1 in Lesser Dawn Bats (Eonycteris spelaea) in Singapore

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Vertebrate Animal Care and Safety
2.2. Bat Colony Structure and Sampling Strategy
2.3. Nucleic Acid Extraction and Sequencing
2.4. Bioinformatic Analyses
2.5. Phylogenetic Analysis
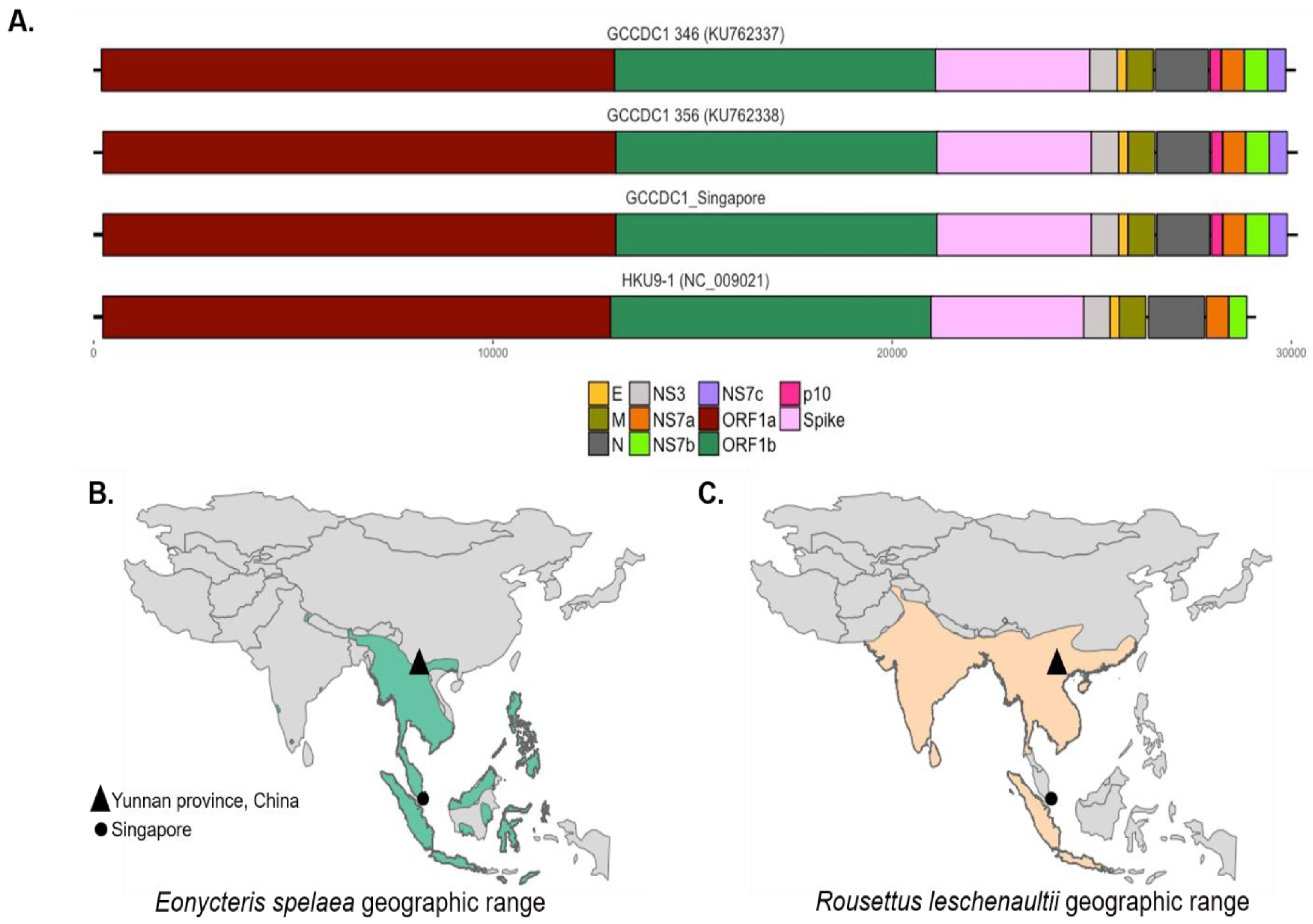
2.6. Geographic Range of Bats
3. Results
3.1. Prevalence of GCCDC1 in the Captive Colony
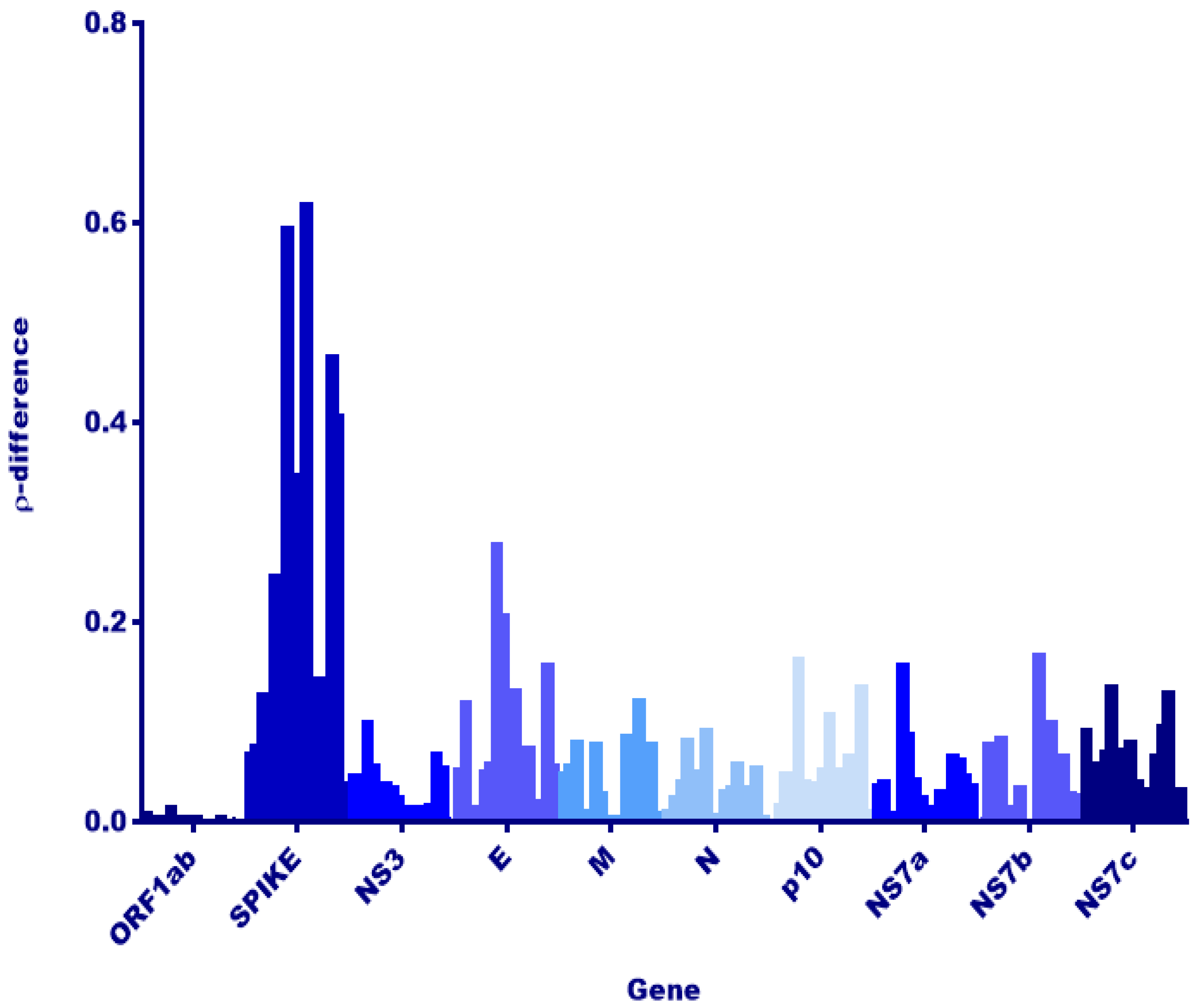
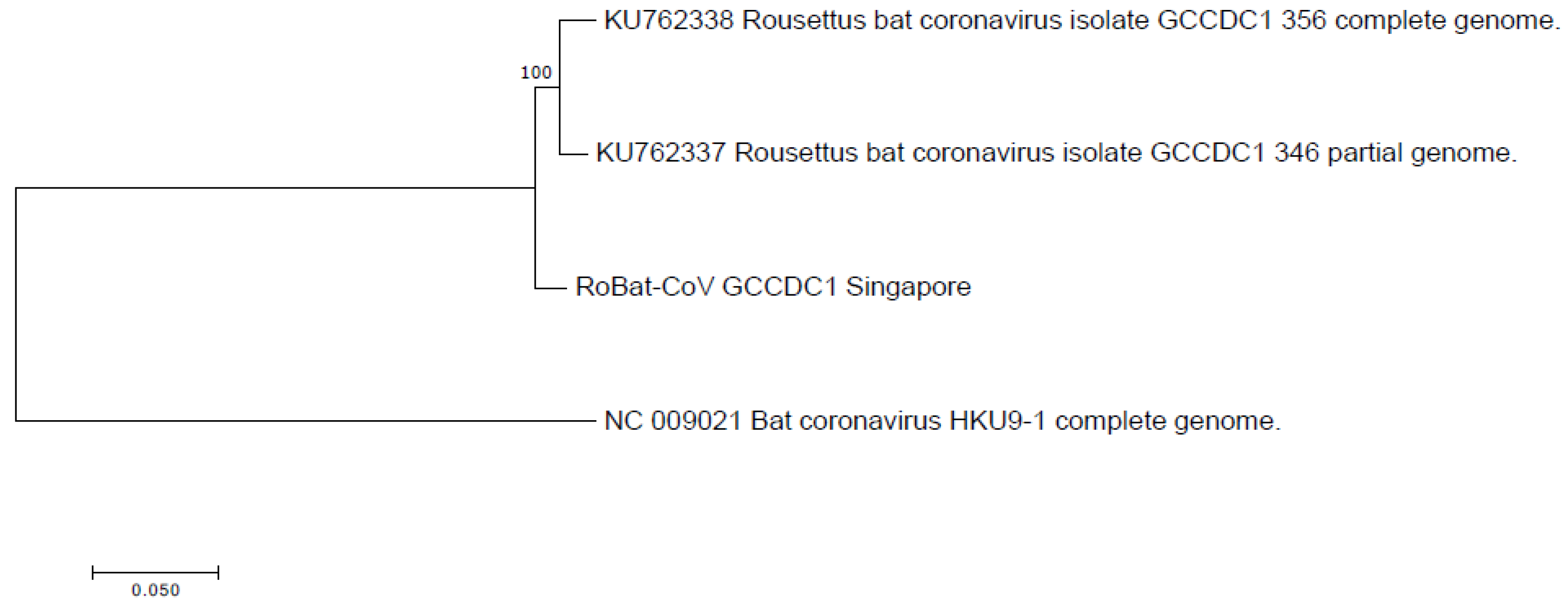
3.2. Comparison to Previously Discovered Cross-Family Recombinant Coronaviruses
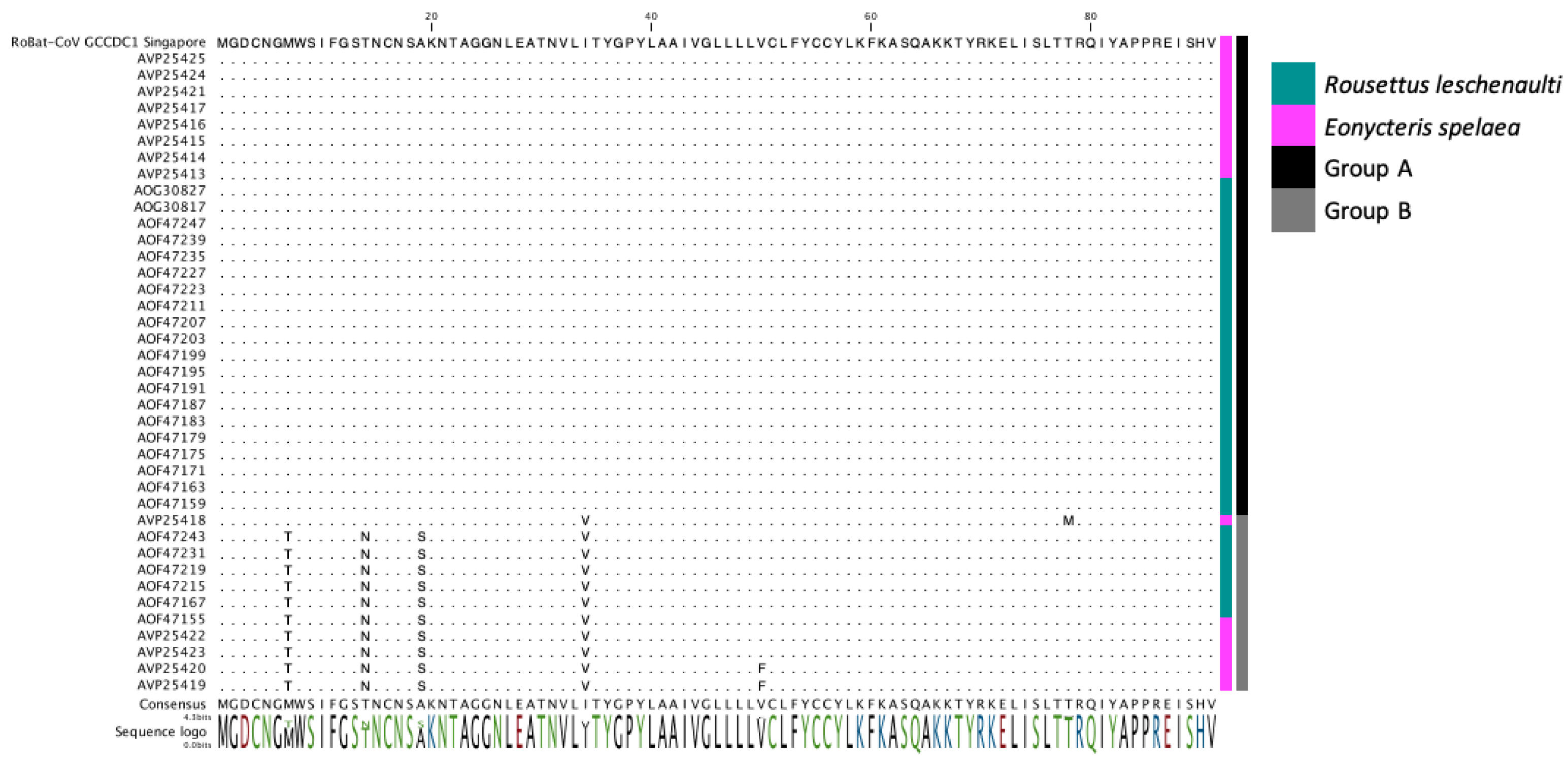
3.3. Detection of the Recombinant Genome despite Geographic and Host Differences
4. Discussion
Data Availability
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tyrrell, D.; Bynoe, M. Cultivation of Viruses from A High Proportion of Patients with Colds. Lancet 1966, 287, 76–77. [Google Scholar] [CrossRef]
- Mehand, M.S.; Al Shorbaji, F.; Millett, P.; Murgue, B. The WHO R&D Blueprint: 2018 review of emerging infectious diseases requiring urgent research and development efforts. Antivir. Res. 2018, 159, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Poon, L.L.M.; Chu, D.K.W.; Chan, K.H.; Wong, O.K.; Ellis, T.M.; Leung, Y.H.C.; Lau, S.K.P.; Woo, P.C.Y.; Suen, K.Y.; Yuen, K.-Y.; et al. Identification of a Novel Coronavirus in Bats. J. Virol. 2005, 79, 2001–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Liu, B.; Yang, J.; Jin, Q. DBatVir: The database of bat-associated viruses. Database 2014, 2014, bau021. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Li, F.; Daszak, P. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Genet. 2018, 17, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Van Boheemen, S.; De Graaf, M.; Lauber, C.; Bestebroer, T.M.; Raj, V.S.; Zaki, A.M.; Osterhaus, A.; Haagmans, B.L.; Gorbalenya, A.E.; Snijder, E.J.; et al. Genomic Characterization of a Newly Discovered Coronavirus Associated with Acute Respiratory Distress Syndrome in Humans. mBio 2012, 3, e00473. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Lacroix, A.; Duong, V.; Hul, V.; San, S.; Davun, H.; Omaliss, K.; Chea, S.; Hassanin, A.; Theppangna, W.; Silithammavong, S.; et al. Genetic diversity of coronaviruses in bats in Lao PDR and Cambodia. Infect. Genet. Evol. 2017, 48, 10–18. [Google Scholar] [CrossRef]
- Banerjee, A.; Kulcsar, K.; Misra, V.; Frieman, M.B.; Mossman, K. Bats and Coronaviruses. Viruses 2019, 11, 41. [Google Scholar] [CrossRef] [Green Version]
- Li, W. Bats Are Natural Reservoirs of SARS-Like Coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef]
- Corman, V.; Ithete, N.; Richards, L.R.; Schoeman, M.C.; Preiser, W.; Drosten, C.; Drexler, J.F. Rooting the Phylogenetic Tree of Middle East Respiratory Syndrome Coronavirus by Characterization of a Conspecific Virus from an African Bat. J. Virol. 2014, 88, 11297–11303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, P.C.; Huang, Y.; Lau, S.K.P.; Yuen, K.-Y. Coronavirus Genomics and Bioinformatics Analysis. Viruses 2010, 2, 1804–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, P.C.; Lau, S.K.P.; Lam, C.S.F.; Lau, C.C.Y.; Tsang, A.K.L.; Lau, J.H.N.; Bai, R.; Teng, J.L.L.; Tsang, C.C.C.; Wang, M.; et al. Discovery of Seven Novel Mammalian and Avian Coronaviruses in the Genus Deltacoronavirus Supports Bat Coronaviruses as the Gene Source of Alphacoronavirus and Betacoronavirus and Avian Coronaviruses as the Gene Source of Gammacoronavirus and Deltacoronavirus. J. Virol. 2012, 86, 3995–4008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, J.M.; Gómez-Puertas, P.; Cavanagh, D.; Gorbalenya, A.E.; Enjuanes, L. A comparative sequence analysis to revise the current taxonomy of the family Coronaviridae. Arch. Virol. 2003, 148, 2207–2235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, M.M. Recombination in Large RNA Viruses: Coronaviruses; Seminars in Virology; Elsevier: Amsterdam, The Netherlands, 1996; pp. 381–388. [Google Scholar]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.; Zhou, J.; Liu, W.; Bi, Y.; Gao, F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, I.; Silva, R.F. Creation of diversity in the animal virus world by inter-species and intra-species recombinations: Lessons learned from poultry viruses. Virus Genes 2007, 36, 1–9. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Wang, M.; Lau, S.K.P.; Xu, H.; Poon, R.W.S.; Guo, R.; Wong, B.H.L.; Gao, K.; Tsoi, H.-W.; Huang, Y.; et al. Comparative Analysis of Twelve Genomes of Three Novel Group 2c and Group 2d Coronaviruses Reveals Unique Group and Subgroup Features. J. Virol. 2006, 81, 1574–1585. [Google Scholar] [CrossRef] [Green Version]
- Amman, B.R.; Jones, M.E.B.; Sealy, T.K.; Uebelhoer, L.S.; Schuh, A.J.; Bird, B.H.; Coleman-McCray, J.D.; Martin, B.E.; Nichol, S.T.; Towner, J.S. Oral Shedding of Marburg Virus in Experimentally Infected Egyptian Fruit Bats (Rousettus Aegyptiacus). J. Wildl. Dis. 2015, 51, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Drexler, J.F.; Corman, V.; Müller, M.A.; Maganga, G.D.; Vallo, P.; Binger, T.; Gloza-Rausch, F.; Rasche, A.; Yordanov, S.; Seebens, A.; et al. Bats host major mammalian paramyxoviruses. Nat. Commun. 2012, 3, 796. [Google Scholar] [CrossRef] [Green Version]
- Halpin, K.; Hyatt, A.D.; Fogarty, R.; Middleton, D.; Bingham, J.; Epstein, J.H.; Rahman, S.A.; Hughes, T.; Smith, C.; Field, H.E.; et al. Pteropid Bats are Confirmed as the Reservoir Hosts of Henipaviruses: A Comprehensive Experimental Study of Virus Transmission. Am. J. Trop. Med. Hyg. 2011, 85, 946–951. [Google Scholar] [CrossRef] [Green Version]
- Olival, K.J.; Hosseini, P.R.; Zambrana-Torrelio, C.; Ross, N.; Bogich, T.; Daszak, P. Host and viral traits predict zoonotic spillover from mammals. Nature 2017, 546, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.H.; Cheng, P.K.; Lai, M.Y.; Leung, P.C.; Wong, K.K.; Lee, W.; Lim, W.W. Virulence Potential of Fusogenic Orthoreoviruses. Emerg. Infect. Dis. 2012, 18, 944–948. [Google Scholar] [CrossRef] [PubMed]
- Menachery, V.D.; Yount, B.L.; Debbink, K.; Agnihothram, S.; Gralinski, L.E.; Plante, J.; Graham, R.L.; Scobey, T.; Ge, X.-Y.; Donaldson, E.F.; et al. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat. Med. 2015, 21, 1508–1513. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Liu, W.J.; Xu, W.; Jin, T.; Zhao, Y.; Song, J.; Shi, Y.; Ji, W.; Jia, H.; Zhou, Y.; et al. A Bat-Derived Putative Cross-Family Recombinant Coronavirus with a Reovirus Gene. PLoS Pathog. 2016, 12, e1005883. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Li, B.; Jiang, R.-D.; Hu, B.-J.; Luo, D.-S.; Zhu, G.-J.; Hu, B.; Liu, H.-Z.; Zhang, Y.; Yang, X.; et al. Longitudinal Surveillance of Betacoronaviruses in Fruit Bats in Yunnan Province, China During 2009–2016. Virol. Sin. 2018, 33, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obameso, J.O.; Li, H.; Jia, H.; Han, M.; Zhu, S.; Huang, C.; Zhao, Y.; Zhao, M.; Bai, Y.; Yuan, F.; et al. The persistent prevalence and evolution of cross-family recombinant coronavirus GCCDC1 among a bat population: A two-year follow-up. Sci. China Life Sci. 2017, 60, 1357–1363. [Google Scholar] [CrossRef] [Green Version]
- Geoghegan, J.; Duchene, S.; Holmes, E.C. Comparative analysis estimates the relative frequencies of co-divergence and cross-species transmission within viral families. PLoS Pathog. 2017, 13, e1006215. [Google Scholar] [CrossRef]
- Paskey, A.C.; Ng, J.H.J.; Rice, G.K.; Ni Chia, W.; Philipson, C.W.; Foo, R.J.H.; Cer, R.Z.; Long, K.A.; Lueder, M.R.; Frey, K.G.; et al. The temporal RNA virome patterns of a lesser dawn bat (Eonycteris spelaea) colony revealed by deep sequencing. Virus Evol. 2020, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Mendenhall, I.H.; Borthwick, S.; Neves, E.S.; Low, D.; Linster, M.; Liang, B.; Skiles, M.; Jayakumar, J.; Han, H.; Gunalan, V.; et al. Identification of a Lineage D Betacoronavirus in Cave Nectar Bats (Eonycteris spelaea) in Singapore and an Overview of Lineage D Reservoir Ecology in SE Asian Bats. Transbound. Emerg. Dis. 2016, 64, 1790–1800. [Google Scholar] [CrossRef]
- Paskey, A.C.; Frey, K.; Schroth, G.P.; Gross, S.; Hamilton, T.; Bishop-Lilly, K.A. Enrichment post-library preparation enhances the sensitivity of high-throughput sequencing-based detection and characterization of viruses from complex samples. BMC Genom. 2019, 20, 155. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner; Lawrence Berkeley National Lab.: Berkeley, CA, USA, 2014. [Google Scholar]
- Charif, D.; Thioulouse, J.; Lobry, J.R.; Perrière, G. Online synonymous codon usage analyses with the ade4 and seqinR packages. Bioinformatics 2004, 21, 545–547. [Google Scholar] [CrossRef] [PubMed]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, C.; Rosell-Ambal, G.; Tabaranza, B.; Carino, P.; Helgen, K.; Molur, S.; Srinivasulu, C. The IUCN Red List of Threatened Species 2008. 2008. Available online: https://dx.doi.org/10.2305/IUCN.UK.2008.RLTS.T7787A12850087.en (accessed on 26 November 2019).
- Tennekes, M. Tmap: Thematic Maps in R. J. Stat. Softw. 2018, 84, 1–39. [Google Scholar] [CrossRef] [Green Version]
- Behura, S.; Severson, D.W. Codon usage bias: Causative factors, quantification methods and genome-wide patterns: With emphasis on insect genomes. Boil. Rev. 2012, 88, 49–61. [Google Scholar] [CrossRef]
- Subudhi, S.; Rapin, N.; Misra, V. Immune System Modulation and Viral Persistence in Bats: Understanding Viral Spillover. Viruses 2019, 11, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, P.C.Y.; Lau, S.K.P.; Lam, C.S.F.; Lai, K.K.Y.; Huang, Y.; Lee, P.; Luk, G.S.M.; Dyrting, K.C.; Chan, K.-H.; Yuen, K.-Y. Comparative Analysis of Complete Genome Sequences of Three Avian Coronaviruses Reveals a Novel Group 3c Coronavirus. J. Virol. 2008, 83, 908–917. [Google Scholar] [CrossRef] [Green Version]
- Woo, P.C.; Lau, S.K.P.; Li, K.S.; Tsang, A.K.; Yuen, K.-Y. Genetic relatedness of the novel human group C betacoronavirus to Tylonycteris bat coronavirus HKU4 and Pipistrellus bat coronavirus HKU5. Emerg. Microbes Infect. 2012, 1, e35. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.P.; Poon, R.W.S.; Wong, B.H.L.; Wang, M.; Huang, Y.; Xu, H.; Guo, R.; Li, K.S.M.; Gao, K.; Chan, K.-H.; et al. Coexistence of Different Genotypes in the Same Bat and Serological Characterization of Rousettus Bat Coronavirus HKU9 Belonging to a Novel Betacoronavirus Subgroup. J. Virol. 2010, 84, 11385–11394. [Google Scholar] [CrossRef] [Green Version]
- Plowright, R.K.; Eby, P.; Hudson, P.J.; Smith, I.L.; Westcott, D.; Bryden, W.L.; Middleton, D.; Reid, P.A.; McFarlane, R.; Martin, G.; et al. Ecological dynamics of emerging bat virus spillover. Proc. R. Soc. B Boil. Sci. 2015, 282, 20142124. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Zeng, L.-P.; Yang, X.; Ge, X.-Y.; Zhang, W.; Li, B.; Xie, J.-Z.; Shen, X.-R.; Zhang, Y.-Z.; Wang, N.; et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017, 13, e1006698. [Google Scholar] [CrossRef]
- Laing, E.D.; Mendenhall, I.H.; Linster, M.; Low, D.H.W.; Chen, Y.; Yan, L.; Sterling, S.L.; Borthwick, S.; Neves, E.S.; Lim, J.S.L.; et al. Serologic Evidence of Fruit Bat Exposure to Filoviruses, Singapore, 2011–2016. Emerg. Infect. Dis. 2018, 24, 122–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dovih, P.; Laing, E.D.; Chen, Y.; Low, D.H.W.; Ansil, B.R.; Yang, X.; Shi, Z.; Broder, C.C.; Smith, G.J.D.; Linster, M.; et al. Filovirus-reactive antibodies in humans and bats in Northeast India imply zoonotic spillover. PLoS Negl. Trop. Dis. 2019, 13, e0007733. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Date | # Swabs Sequenced | # Swabs Positive for GCCDC1 (%) | # Bats Sampled | # Bats Positive for GCCDC1 (%) |
|---|---|---|---|---|
| April 2016 | 41 | 11 (26.8%) | 18 | 8 (44.4%) |
| July 2016 | 28 | 4 (14.3%) | 19 | 4 (21.1%) |
| October 2016 | 75 | 55 (73.3%) | 20 | 20 (100%) |
| January 2017 | 14 | 1 (7.1%) | 11 | 1 (9.1%) |
| May 2017 | 21 | 0 (0%) | 15 | 0 (0%) |
| September 2017 | 27 | 1 (3.7%) | 13 | 1 (7.7%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paskey, A.C.; Ng, J.H.J.; Rice, G.K.; Chia, W.N.; Philipson, C.W.; Foo, R.J.H.; Cer, R.Z.; Long, K.A.; Lueder, M.R.; Lim, X.F.; et al. Detection of Recombinant Rousettus Bat Coronavirus GCCDC1 in Lesser Dawn Bats (Eonycteris spelaea) in Singapore. Viruses 2020, 12, 539. https://doi.org/10.3390/v12050539
Paskey AC, Ng JHJ, Rice GK, Chia WN, Philipson CW, Foo RJH, Cer RZ, Long KA, Lueder MR, Lim XF, et al. Detection of Recombinant Rousettus Bat Coronavirus GCCDC1 in Lesser Dawn Bats (Eonycteris spelaea) in Singapore. Viruses. 2020; 12(5):539. https://doi.org/10.3390/v12050539
Chicago/Turabian StylePaskey, Adrian C., Justin H. J. Ng, Gregory K. Rice, Wan Ni Chia, Casandra W. Philipson, Randy J.H. Foo, Regina Z. Cer, Kyle A. Long, Matthew R. Lueder, Xiao Fang Lim, and et al. 2020. "Detection of Recombinant Rousettus Bat Coronavirus GCCDC1 in Lesser Dawn Bats (Eonycteris spelaea) in Singapore" Viruses 12, no. 5: 539. https://doi.org/10.3390/v12050539
APA StylePaskey, A. C., Ng, J. H. J., Rice, G. K., Chia, W. N., Philipson, C. W., Foo, R. J. H., Cer, R. Z., Long, K. A., Lueder, M. R., Lim, X. F., Frey, K. G., Hamilton, T., Anderson, D. E., Laing, E. D., Mendenhall, I. H., Smith, G. J., Wang, L. -F., & Bishop-Lilly, K. A. (2020). Detection of Recombinant Rousettus Bat Coronavirus GCCDC1 in Lesser Dawn Bats (Eonycteris spelaea) in Singapore. Viruses, 12(5), 539. https://doi.org/10.3390/v12050539