
Systematic Search for SARS-CoV-2 Main Protease Inhibitors for Drug Repurposing: Ethacrynic Acid as a Potential Drug


Abstract
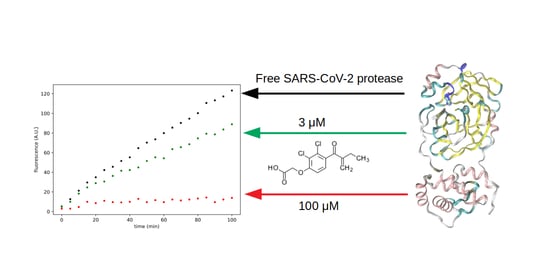
:
1. Introduction
2. Materials and Methods
3. Results
3.1. Protein Targets for Docking
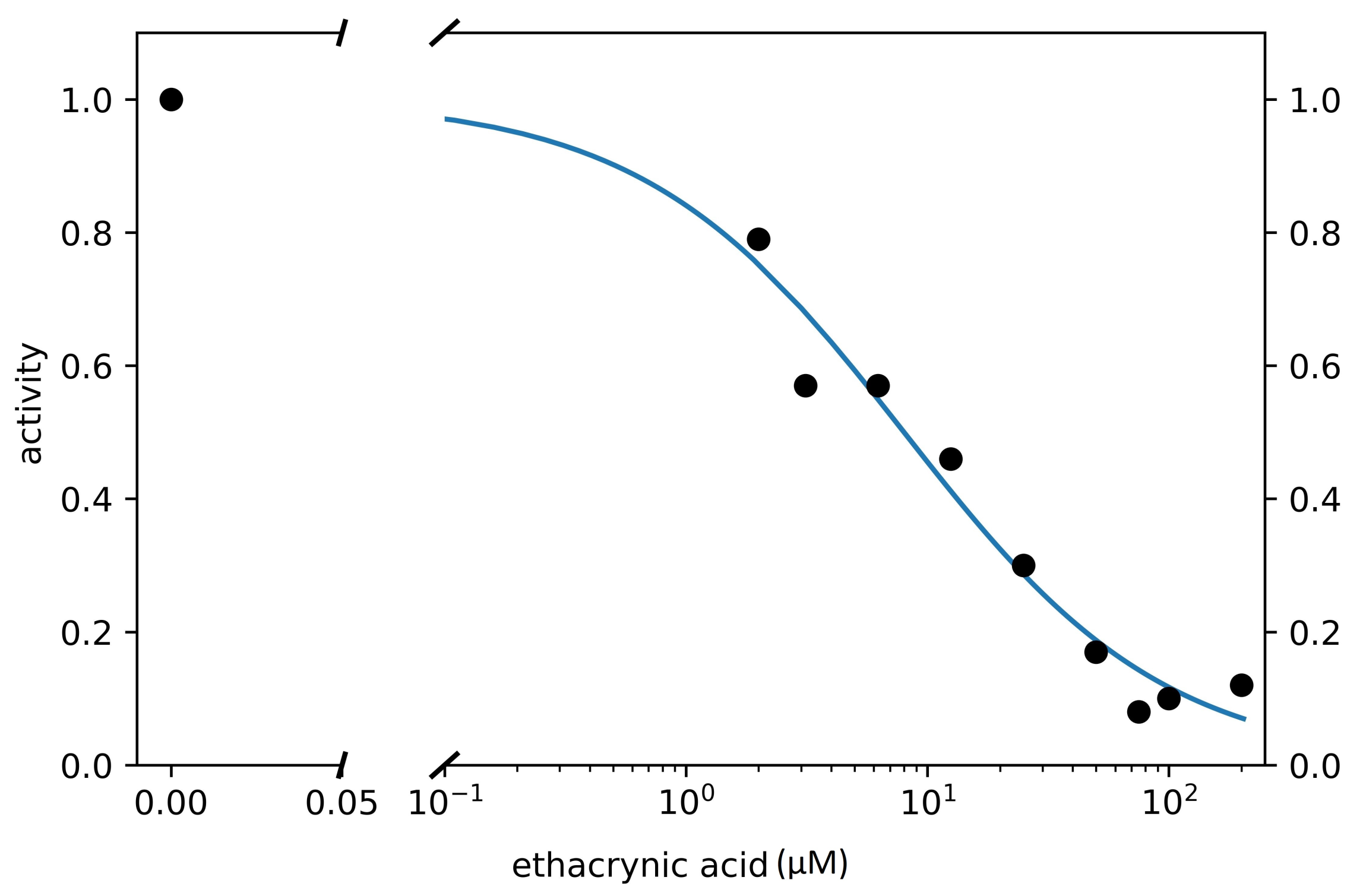
3.2. Search for Competitive Inhibitors
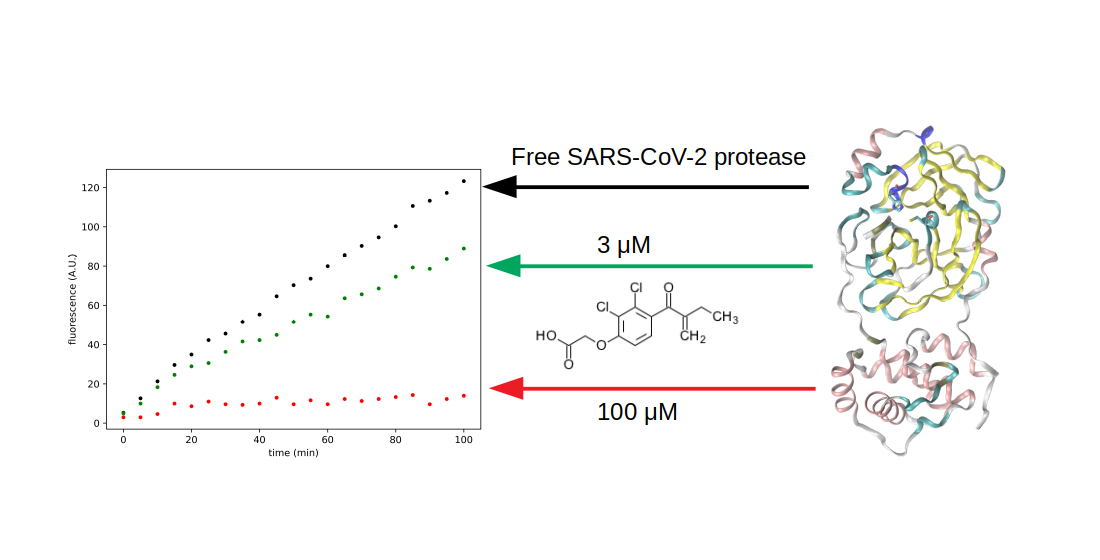
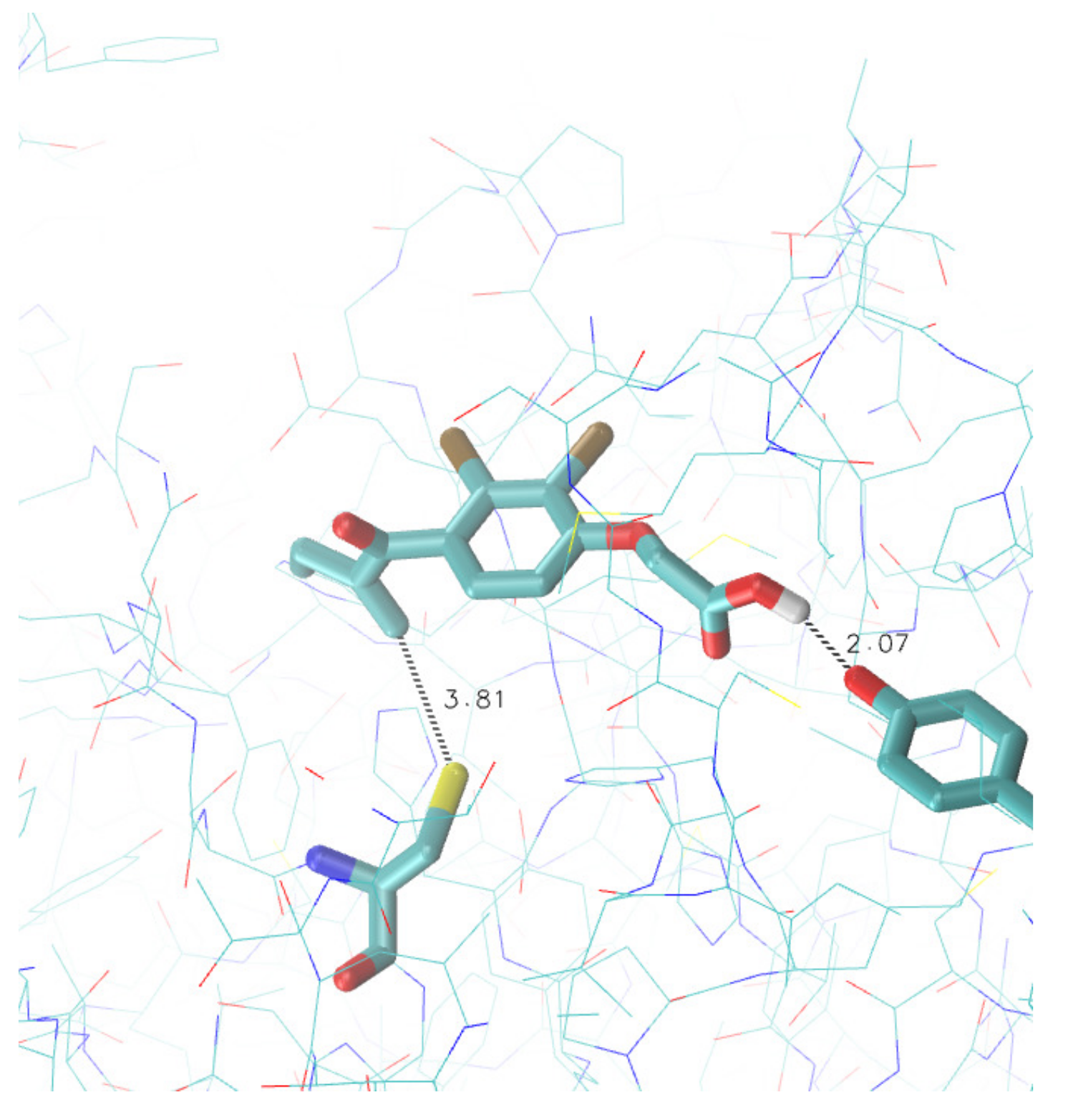
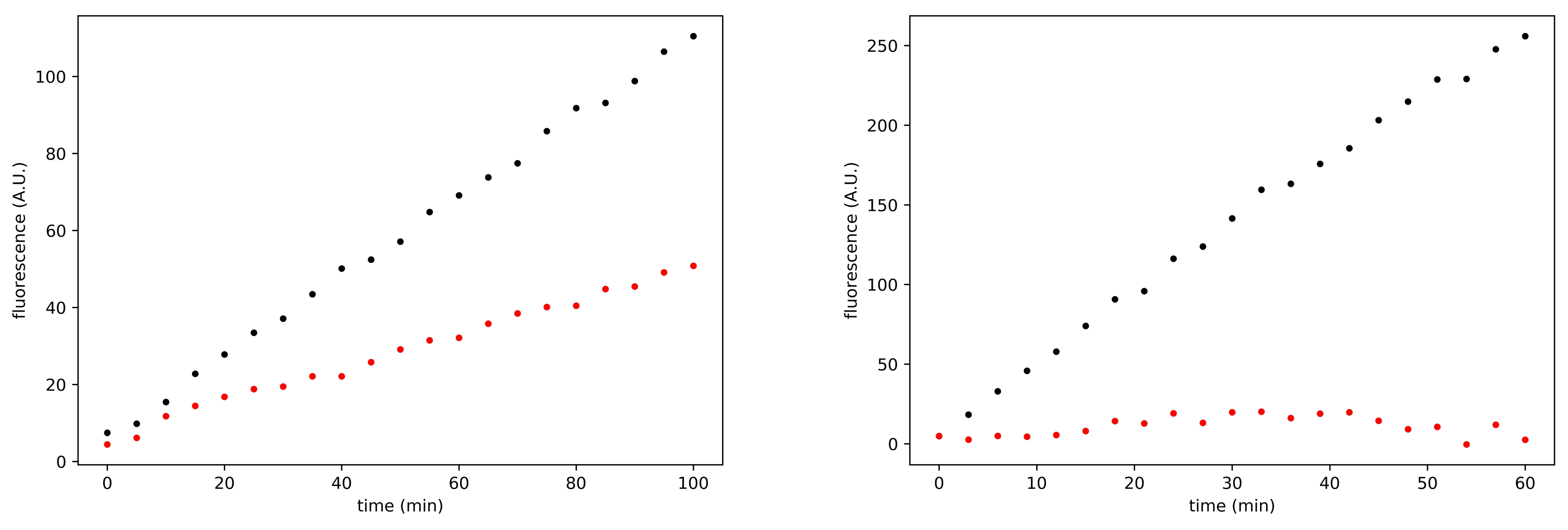
3.3. Search for Irreversible Inhibitors
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef] [Green Version]
- Gorbalenya, A.; Baker, S.; Baric, R.; de Groot, R.J. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.M.; Cavanagh, D. The molecular biology of coronaviruses. In Advances in Virus Research; Academic Press: Cambridge, MA, USA, 1997; Volume 48, pp. 1–100. [Google Scholar] [CrossRef]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Hilgenfeld, R. From SARS to MERS: Crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014, 281, 4085–4096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, K.; Palm, G.J.; Mesters, J.R.; Siddell, S.G.; Ziebuhr, J.; Hilgenfeld, R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α-helical domain. EMBO J. 2002, 21, 3213–3224. [Google Scholar] [CrossRef] [PubMed]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Chen, C.; Tan, W.; Yang, K.; Yang, H. Structure of main protease from human coronavirus NL63: Insights for wide spectrum anti-coronavirus drug design. Sci. Rep. 2016, 6, 22677. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Li, S.; Xue, F.; Zou, Y.; Chen, C.; Bartlam, M.; Rao, Z. Structure of the main protease from a global infectious human coronavirus, HCoV-HKU1. J. Virol. 2008, 82, 8647–8655. [Google Scholar] [CrossRef] [Green Version]
- Ratia, K.; Saikatendu, K.S.; Santarsiero, B.D.; Barretto, N.; Baker, S.C.; Stevens, R.C.; Mesecar, A.D. Severe acute respiratory syndrome coronavirus papain-like protease: Structure of a viral deubiquitinating enzyme. Proc. Natl. Acad. Sci. USA 2006, 103, 5717–5722. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Palese, L.L. The structural landscape of SARS-CoV-2 main protease: Hints for inhibitor search. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Kapusta, K.; Kar, S.; Collins, J.T.; Franklin, L.M.; Kolodziejczyk, W.; Leszczynski, J.; Hill, G.A. Protein reliability analysis and virtual screening of natural inhibitors for SARS-CoV-2 main protease (Mpro) through docking, molecular mechanic & dynamic, and ADMET profiling. J. Biomol. Struct. Dyn. 2020, 1–18. [Google Scholar] [CrossRef]
- Ojha, P.K.; Kar, S.; Krishna, J.G.; Roy, K.; Leszczynski, J. Therapeutics for COVID-19: From computation to practices—Where we are, where we are heading to. Mol. Divers. 2020, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, A.; Mestres-Truyol, J.; Ojeda-Montes, M.J.; Macip, G.; Saldivar-Espinoza, B.; Cereto-Massagué, A.; Pujadas, G.; Garcia-Vallvé, S. Prediction of Novel Inhibitors of the Main Protease (M-pro) of SARS-CoV-2 through Consensus Docking and Drug Reposition. Int. J. Mol. Sci. 2020, 21, 3793. [Google Scholar] [CrossRef] [PubMed]
- Muteeb, G.; Alshoaibi, A.; Aatif, M.; Rehman, M.T.; Qayyum, M.Z. Screening marine algae metabolites as high-affinity inhibitors of SARS-CoV-2 main protease (3CLpro): An in silico analysis to identify novel drug candidates to combat COVID-19 pandemic. Appl. Biol. Chem. 2020, 63, 1–12. [Google Scholar] [CrossRef]
- Li, G.; De Clercq, E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat. Rev. Drug Discov. 2020, 19, 149. [Google Scholar] [CrossRef] [Green Version]
- Low, Z.Y.; Farouk, I.A.; Lal, S.K. Drug repositioning: New approaches and future prospects for life-debilitating diseases and the COVID-19 pandemic outbreak. Viruses 2020, 12, 1058. [Google Scholar] [CrossRef]
- Martorana, A.; Gentile, C.; Lauria, A. In Silico Insights into the SARS CoV-2 Main Protease Suggest NADH Endogenous Defences in the Control of the Pandemic Coronavirus Infection. Viruses 2020, 12, 805. [Google Scholar] [CrossRef]
- Kumar, S.; Zhi, K.; Mukherji, A.; Gerth, K. Repurposing antiviral protease inhibitors using extracellular vesicles for potential therapy of COVID-19. Viruses 2020, 12, 486. [Google Scholar] [CrossRef]
- Palese, L.L. Conformations of the HIV-1 protease: A crystal structure data set analysis. Biochim. Biophys. Acta 2017, 1865, 1416–1422. [Google Scholar] [CrossRef]
- Palese, L.L. Analysis of the conformations of the HIV-1 protease from a large crystallographic data set. Data Brief 2017, 15, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Gnoni, A.; De Nitto, E.; Scacco, S.; Santacroce, L.; Palese, L.L. A New Look at the Structures of Old Sepsis Actors by Exploratory Data Analysis Tools. Antibiotics 2019, 8, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, X.; Yang, H.; Shen, W.; Zhao, Q.; Li, J.; Yang, K.; Chen, C.; Jin, Y.; Bartlam, M.; Rao, Z. Production of authentic SARS-CoV Mpro with enhanced activity: Application as a novel tag-cleavage endopeptidase for protein overproduction. J. Mol. Biol. 2007, 366, 965–975. [Google Scholar] [CrossRef]
- Yang, S.; Chen, S.J.; Hsu, M.F.; Wu, J.D.; Tseng, C.T.K.; Liu, Y.F.; Chen, H.C.; Kuo, C.W.; Wu, C.S.; Chang, L.W.; et al. Synthesis, Crystal Structure, Structure- Activity Relationships, and Antiviral Activity of a Potent SARS Coronavirus 3CL Protease Inhibitor. J. Med. Chem. 2006, 49, 4971–4980. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; George, S.; Schmidt, M.F.; Al-Gharabli, S.I.; Rademann, J.; Hilgenfeld, R. Peptide aldehyde inhibitors challenge the substrate specificity of the SARS-coronavirus main protease. Antiviral Res. 2011, 92, 204–212. [Google Scholar] [CrossRef]
- Jacobs, J.; Grum-Tokars, V.; Zhou, Y.; Turlington, M.; Saldanha, S.A.; Chase, P.; Eggler, A.; Dawson, E.S.; Baez-Santos, Y.M.; Tomar, S.; et al. Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J. Med. Chem. 2013, 56, 534–546. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Niu, C.; Cherney, M.M.; Zhang, J.; Huitema, C.; Eltis, L.D.; Vederas, J.C.; James, M.N. A mechanistic view of enzyme inhibition and peptide hydrolysis in the active site of the SARS-CoV 3C-like peptidase. J. Mol. Biol. 2007, 371, 1060–1074. [Google Scholar] [CrossRef]
- Verschueren, K.H.; Pumpor, K.; Anemüller, S.; Chen, S.; Mesters, J.R.; Hilgenfeld, R. A structural view of the inactivation of the SARS coronavirus main proteinase by benzotriazole esters. Chem. Biol. 2008, 15, 597–606. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.C.; Kuo, C.J.; Hsu, M.F.; Liang, P.H.; Fang, J.M.; Shie, J.J.; Wang, A.H.J. Structural basis of mercury-and zinc-conjugated complexes as SARS-CoV 3C-like protease inhibitors. FEBS Lett. 2007, 581, 5454–5458. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.C.; Kuo, C.J.; Ko, T.P.; Hsu, M.F.; Tsui, Y.C.; Chang, S.C.; Yang, S.; Chen, S.J.; Chen, H.C.; Hsu, M.C.; et al. Structural basis of inhibition specificities of 3C and 3C-like proteases by zinc-coordinating and peptidomimetic compounds. J. Biol. Chem. 2009, 284, 7646–7655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, I.L.; Mahindroo, N.; Liang, P.H.; Peng, Y.H.; Kuo, C.J.; Tsai, K.C.; Hsieh, H.P.; Chao, Y.S.; Wu, S.Y. Structure-based drug design and structural biology study of novel nonpeptide inhibitors of severe acute respiratory syndrome coronavirus main protease. J. Med. Chem. 2006, 49, 5154–5161. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, T.; Takemoto, C.; Kim, Y.T.; Wang, H.; Nishii, W.; Terada, T.; Shirouzu, M.; Yokoyama, S. SARS-CoV 3CL protease cleaves its C-terminal autoprocessing site by novel subsite cooperativity. Proc. Natl. Acad. Sci. USA 2016, 113, 12997–13002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.W.; Cherney, M.M.; Huitema, C.; Liu, J.; James, K.E.; Powers, J.C.; Eltis, L.D.; James, M.N. Crystal structures of the main peptidase from the SARS coronavirus inhibited by a substrate-like aza-peptide epoxide. J. Mol. Biol. 2005, 353, 1137–1151. [Google Scholar] [CrossRef] [PubMed]
- Fearon, D.; Owen, C.; Douangamath, A.; Lukacik, P.; Powell, A.; Strain-Damerell, C.; Resnick, E.; Krojer, T.; Gehrtz, P.; Wild, C. PanDDA Analysis Group Deposition of SARS-CoV-2 Main Protease Fragment Screen. 2020. Available online: https://doi.org/https://doi.org/10.2210/pdb5RET/pdb (accessed on 13 January 2021).
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Halko, N.; Martinsson, P.G.; Tropp, J.A. Finding structure with randomness: Probabilistic algorithms for constructing approximate matrix decompositions. SIAM Rev. 2011, 53, 217–288. [Google Scholar] [CrossRef]
- Palese, L.L. A random version of principal component analysis in data clustering. Comput. Biol. Chem. 2018, 73, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Shityakov, S.; Förster, C. In silico predictive model to determine vector-mediated transport properties for the blood–brain barrier choline transporter. Adv. Appl. Bioinform. Chem. 2014, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterling, T.; Irwin, J.J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Ye, F.; Feng, Y.; Yu, F.; Wang, Q.; Wu, Y.; Zhao, C.; Sun, H.; Huang, B.; Niu, P.; et al. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020, 11, 4417. [Google Scholar] [CrossRef] [PubMed]
- Vuong, W.; Khan, M.B.; Fischer, C.; Arutyunova, E.; Lamer, T.; Shields, J.; Saffran, H.A.; McKay, R.T.; van Belkum, M.J.; Joyce, M.; et al. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2020, 11, 4282. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Sellner, M.; Neranjan, S.; Smieško, M.; Lill, M.A. Potential Inhibitors for Novel Coronavirus Protease Identified by Virtual Screening of 606 Million Compounds. Int. J. Mol. Sci. 2020, 21, 3626. [Google Scholar] [CrossRef]
- Bzówka, M.; Mitusińska, K.; Raczyńska, A.; Samol, A.; Tuszyński, J.A.; Góra, A. Structural and Evolutionary Analysis Indicate That the SARS-CoV-2 Mpro Is a Challenging Target for Small-Molecule Inhibitor Design. Int. J. Mol. Sci. 2020, 21, 3099. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Liu, W.; Wacker, D.; Gati, C.; Han, G.W.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; Katritch, V.; Barty, A.; et al. Serial femtosecond crystallography of G protein–coupled receptors. Science 2013, 342, 1521–1524. [Google Scholar] [CrossRef] [Green Version]
- Weisberg, E.; Manley, P.W.; Breitenstein, W.; Brüggen, J.; Cowan-Jacob, S.W.; Ray, A.; Huntly, B.; Fabbro, D.; Fendrich, G.; Hall-Meyers, E.; et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 2005, 7, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Tazikeh-Lemeski, E.; Moradi, S.; Raoufi, R.; Shahlaei, M.; Janlou, M.A.M.; Zolghadri, S. Targeting SARS-COV-2 non-structural protein 16: A virtual drug repurposing study. J. Biomol. Struct. Dyn. 2020, 1–14. [Google Scholar] [CrossRef]
- Bauer, R.A. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 2015, 20, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Melvin, K.E.W.; Farrelly, R.O.; North, J.D.K. Ethacrynic Acid: A New Oral Diuretic. Br. Med. J. 1963, 1, 1521–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molnar, J.; Somberg, J.C. The clinical pharmacology of ethacrynic acid. Am. J. Ther. 2009, 16, 86–92. [Google Scholar] [CrossRef]
- Kaeppler, U.; Stiefl, N.; Schiller, M.; Vicik, R.; Breuning, A.; Schmitz, W.; Rupprecht, D.; Schmuck, C.; Baumann, K.; Ziebuhr, J.; et al. A new lead for nonpeptidic active-site-directed inhibitors of the severe acute respiratory syndrome coronavirus main protease discovered by a combination of screening and docking methods. J. Med. Chem. 2005, 48, 6832–6842. [Google Scholar] [CrossRef]
- Smee, D.F.; Hurst, B.L.; Wong, M.H. Lack of efficacy of aurintricarboxylic acid and ethacrynic acid against vaccinia virus respiratory infections in mice. Antivir. Chem. Chemother. 2010, 20, 201–205. [Google Scholar] [CrossRef]
- Lacreta, F.P.; Brennan, J.M.; Nash, S.L.; Comis, R.L.; Tew, K.D.; O’Dwyer, P.J. Pharmakokinetics and bioavailability study of ethacrynic acid as a modulator of drug resistance in patients with cancer. J. Pharmacol. Exp. Ther. 1994, 270, 1186–1191. [Google Scholar]
- Soltys, B.J.; Gupta, R.S. Changes in mitochondrial shape and distribution induced by ethacrynic acid and the transient formation of a mitochondrial reticulum. J. Cell. Physiol. 1994, 159, 281–294. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 5R7Y | 5R7Z | 5R80 | 5R81 | 5R82 | 5R83 | 5R84 | 5RE4 | 5RE5 | 5RE6 |
| 5RE7 | 5RE8 | 5RE9 | 5REA | 5REB | 5REC | 5RED | 5REE | 5REF | 5REG |
| 5REH | 5REI | 5REJ | 5REK | 5REL | 5REM | 5REN | 5REO | 5REP | 5RER |
| 5RES | 5RET | 5REU | 5REV | 5REW | 5REX | 5REY | 5REZ | 5RF0 | 5RF1 |
| 5RF2 | 5RF3 | 5RF4 | 5RF5 | 5RF6 | 5RF7 | 5RF8 | 5RF9 | 5RFB | 5RFC |
| 5RFD | 5RFE | 5RFF | 5RFG | 5RFH | 5RFI | 5RFJ | 5RFK | 5RFL | 5RFM |
| 5RFN | 5RFO | 5RFP | 5RFQ | 5RFR | 5RFS | 5RFT | 5RFU | 5RFV | 5RFW |
| 5RFX | 5RFY | 5RFZ | 5RG0 | 6LU7 | 6M03 | 6W63 | 6Y84 |
| Drug | ZINC ID | Binding Affinity (kcal mol) |
|---|---|---|
| Dutasteride | ZINC3932831 | −9.45 |
| Ergotamine | ZINC52955754 | −9.20 |
| Dihydroergotamine | ZINC3978005 | −9.15 |
| Trametinib | ZINC43100709 | −9.10 |
| Nilotinib | ZINC6716957 | −8.95 |
| Bromocriptine | ZINC53683151 | −8.85 |
| Idarubicin | ZINC3920266 | −8.80 |
| Epirubicin | ZINC3938704 | −8.80 |
| Irinotecan | ZINC1612996 | −8.80 |
| Delafloxacin | ZINC3827556 | −8.80 |
| Naldemedine | ZINC100378061 | −8.80 |
| Lumacaftor | ZINC64033452 | −8.75 |
| Olaparib | ZINC40430143 | −8.75 |
| Daunorubicin | ZINC3917708 | −8.75 |
| Pimozide | ZINC4175630 | −8.75 |
| Palbociclib | ZINC3938686 | −8.75 |
| Teniposide | ZINC4099008 | −8.75 |
| Raltegravir | ZINC13831130 | −8.70 |
| Pazopanib | ZINC11617039 | −8.65 |
| Netupitant | ZINC11681563 | −8.65 |
| Saquinavir | ZINC26664090 | −8.60 |
| Eltrombopag | ZINC11679756 | −8.60 |
| Ciclesonide | ZINC3915154 | −8.60 |
| Midostaurin | ZINC100013130 | −8.55 |
| Azilsartan | ZINC14210642 | −8.55 |
| Tadalafil | ZINC3993855 | −8.55 |
| Vemurafenib | ZINC52509366 | −8.50 |
| Enasidenib | ZINC222731806 | −8.50 |
| Deferasirox | ZINC1481815 | −8.50 |
| Rolapitant | ZINC3816514 | −8.50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Isgrò, C.; Sardanelli, A.M.; Palese, L.L. Systematic Search for SARS-CoV-2 Main Protease Inhibitors for Drug Repurposing: Ethacrynic Acid as a Potential Drug. Viruses 2021, 13, 106. https://doi.org/10.3390/v13010106
Isgrò C, Sardanelli AM, Palese LL. Systematic Search for SARS-CoV-2 Main Protease Inhibitors for Drug Repurposing: Ethacrynic Acid as a Potential Drug. Viruses. 2021; 13(1):106. https://doi.org/10.3390/v13010106
Chicago/Turabian StyleIsgrò, Camilla, Anna Maria Sardanelli, and Luigi Leonardo Palese. 2021. "Systematic Search for SARS-CoV-2 Main Protease Inhibitors for Drug Repurposing: Ethacrynic Acid as a Potential Drug" Viruses 13, no. 1: 106. https://doi.org/10.3390/v13010106
APA StyleIsgrò, C., Sardanelli, A. M., & Palese, L. L. (2021). Systematic Search for SARS-CoV-2 Main Protease Inhibitors for Drug Repurposing: Ethacrynic Acid as a Potential Drug. Viruses, 13(1), 106. https://doi.org/10.3390/v13010106