
Unpicking the Secrets of African Swine Fever Viral Replication Sites

 and
and 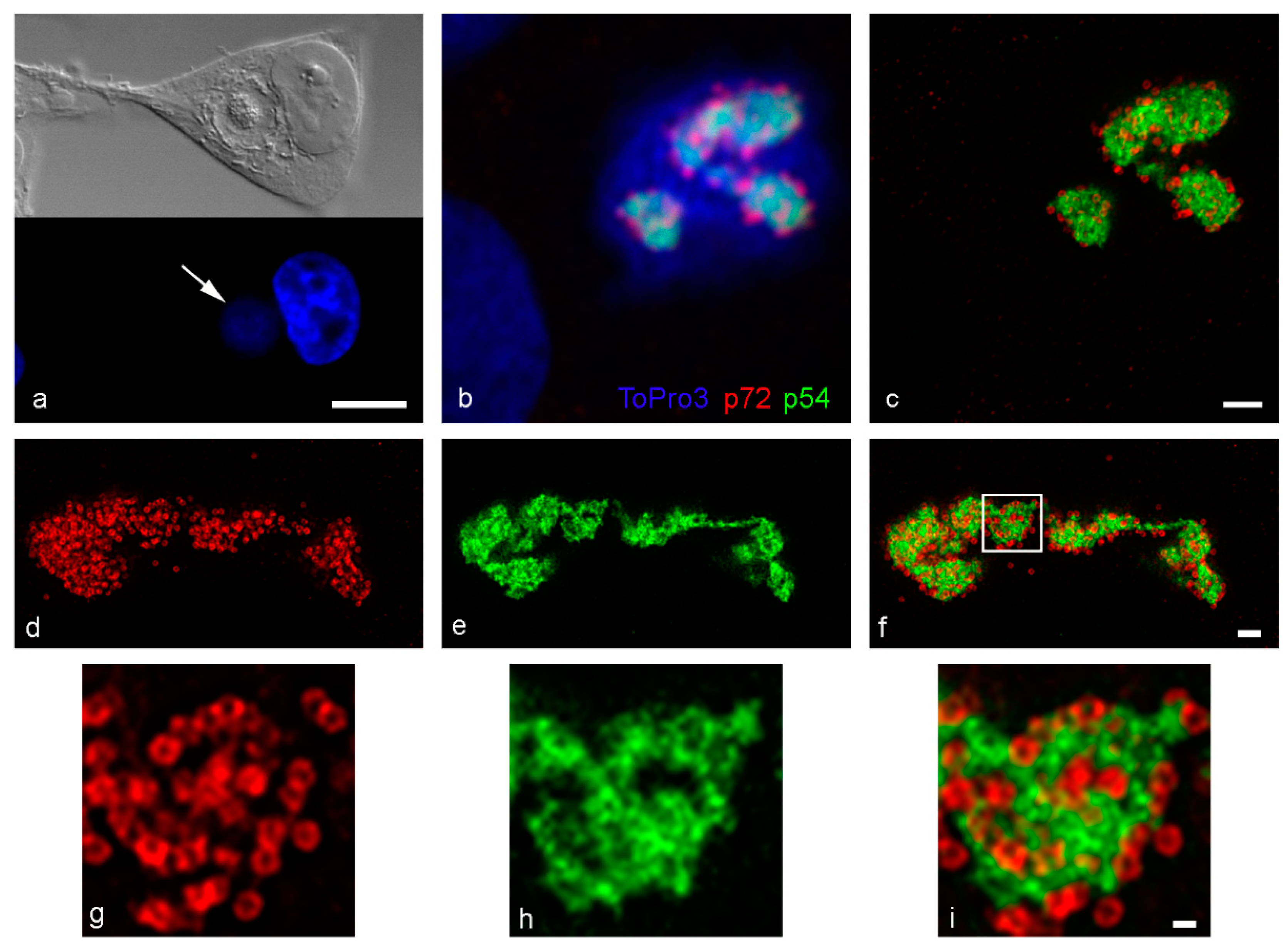{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Virus and Antibodies
2.2. Immunofluorescence and Confocal/STED Microscopy
2.3. Analysis of Protein and DNA Localization Using Click IT Chemistry
2.4. Preparation of Thawed Cryo-Sections for TEM
2.5. Immunogold Labelling of Thawed Cryo-Sections
2.6. High-Pressure Freezing and Freeze Substitution of Cell Monolayers
2.7. Chemical Fixation of Cell Monolayers for TEM
2.8. Electron Tomography (ET) of Infected Cell Monolayers
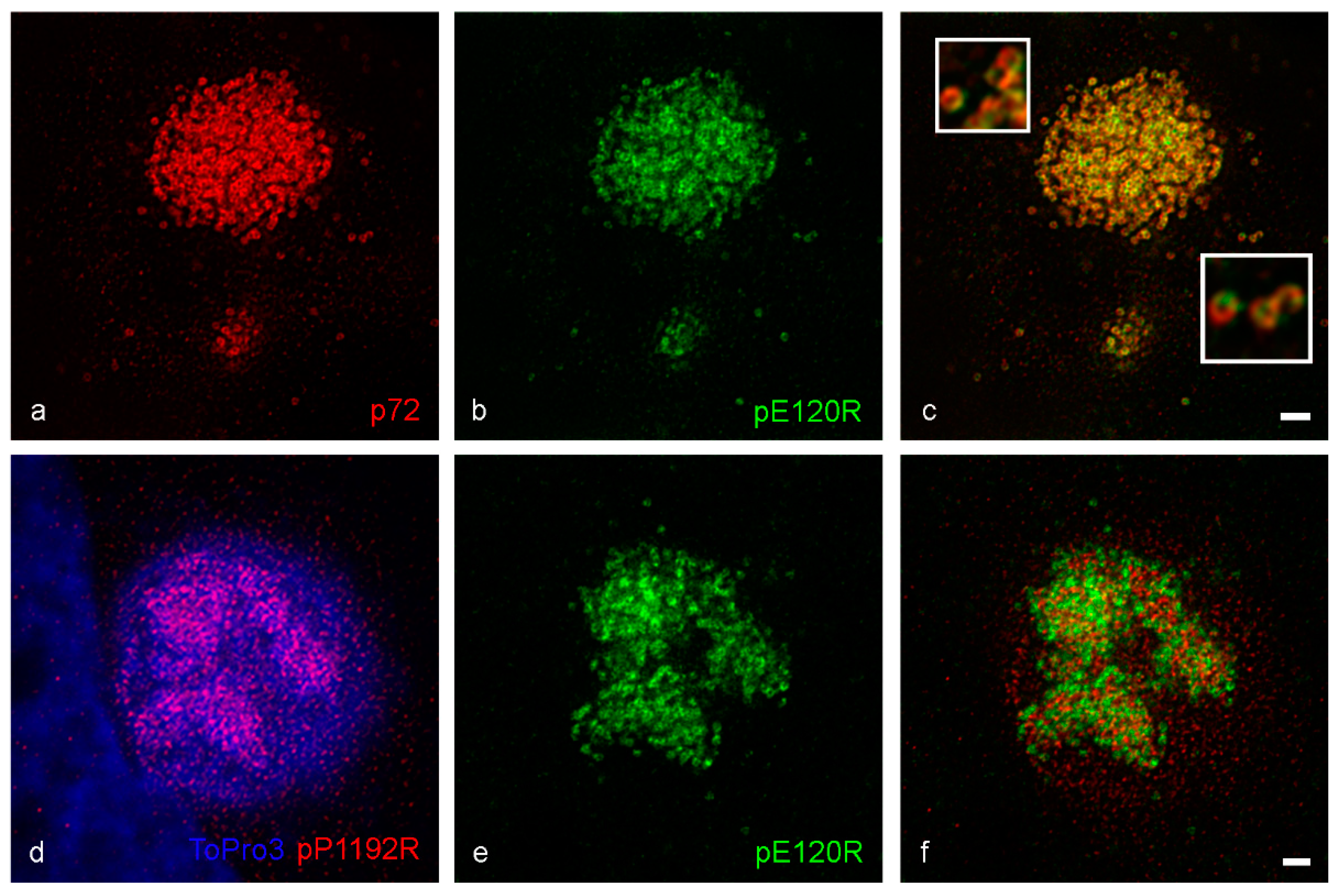
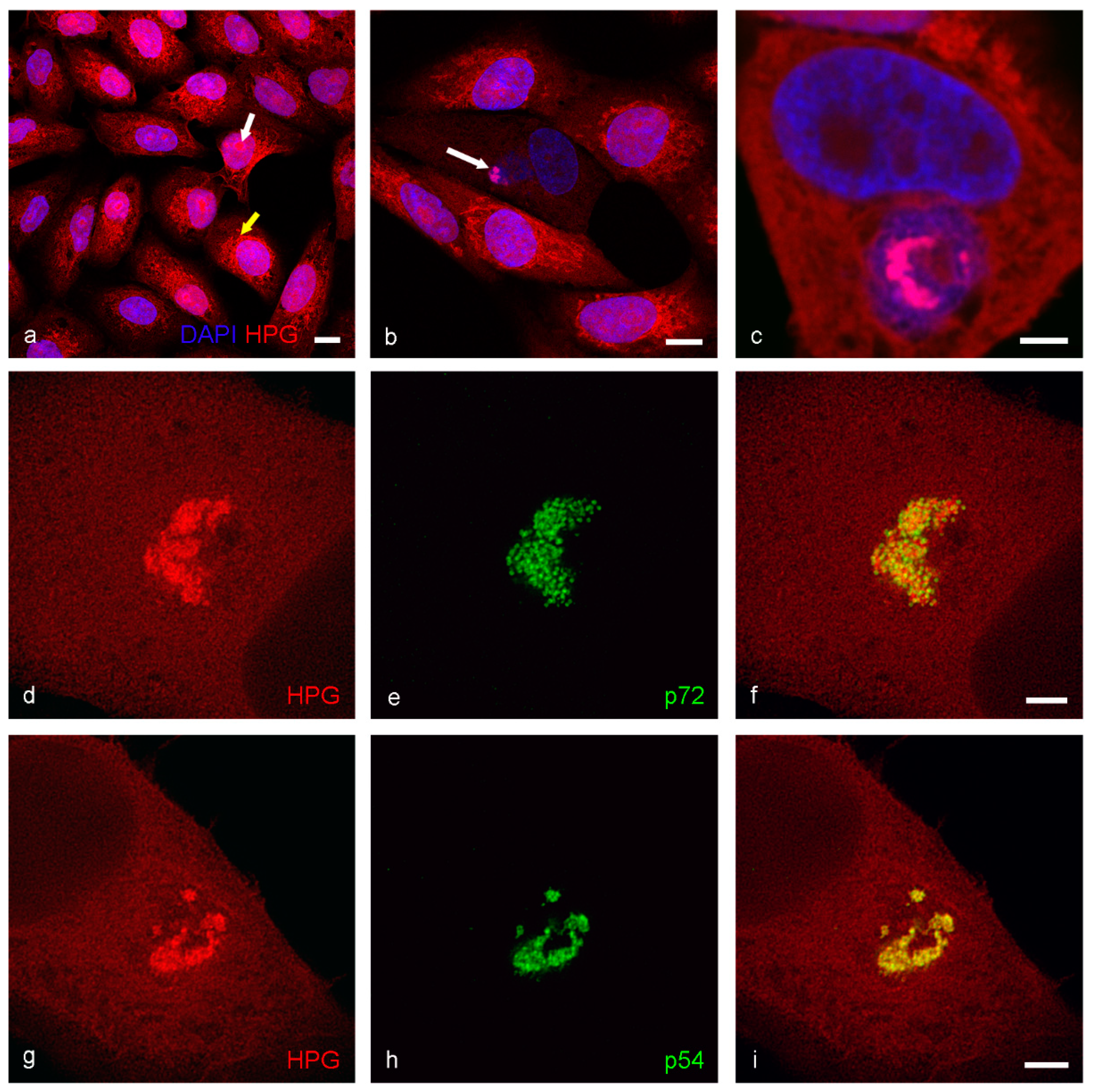
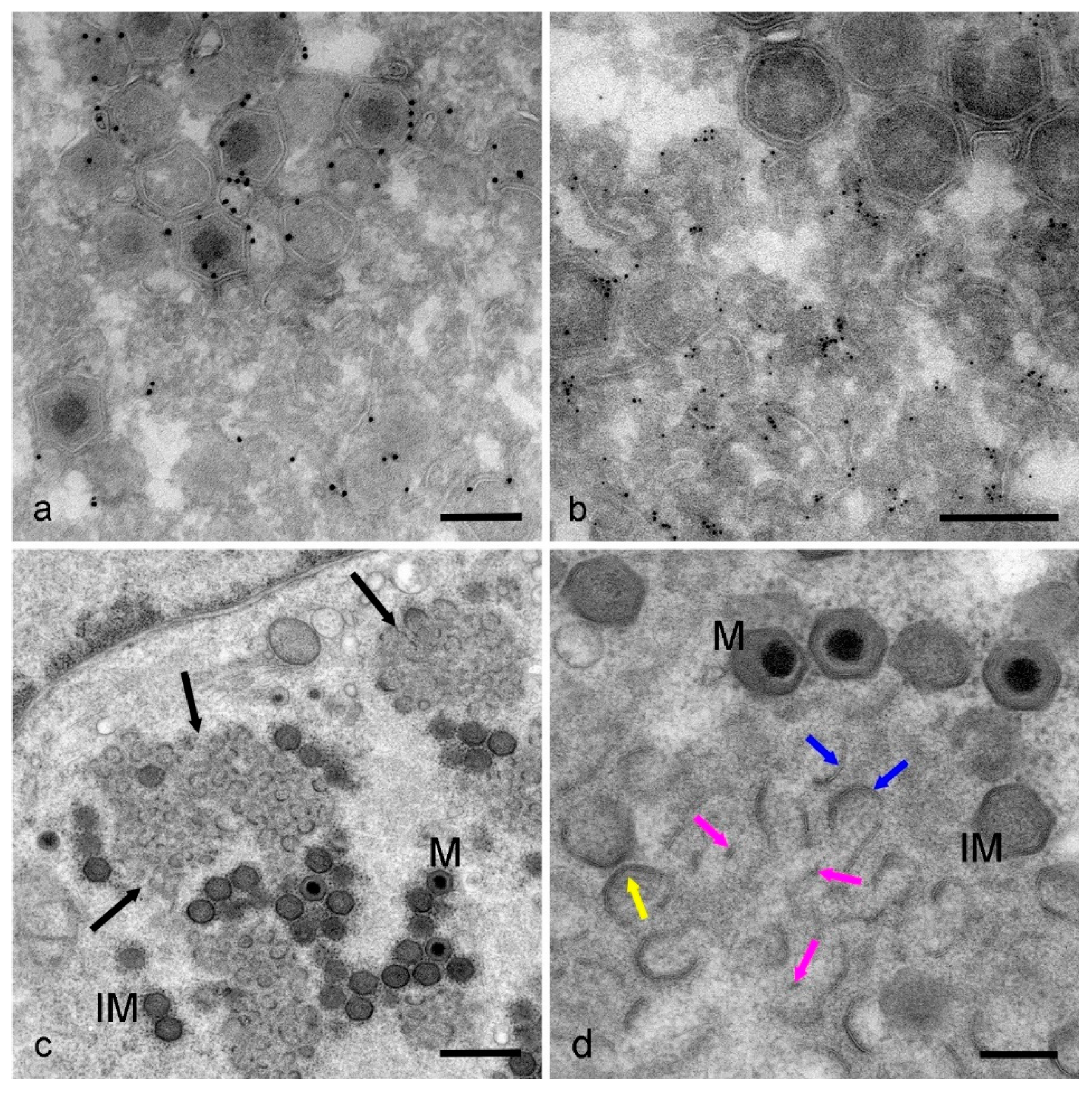
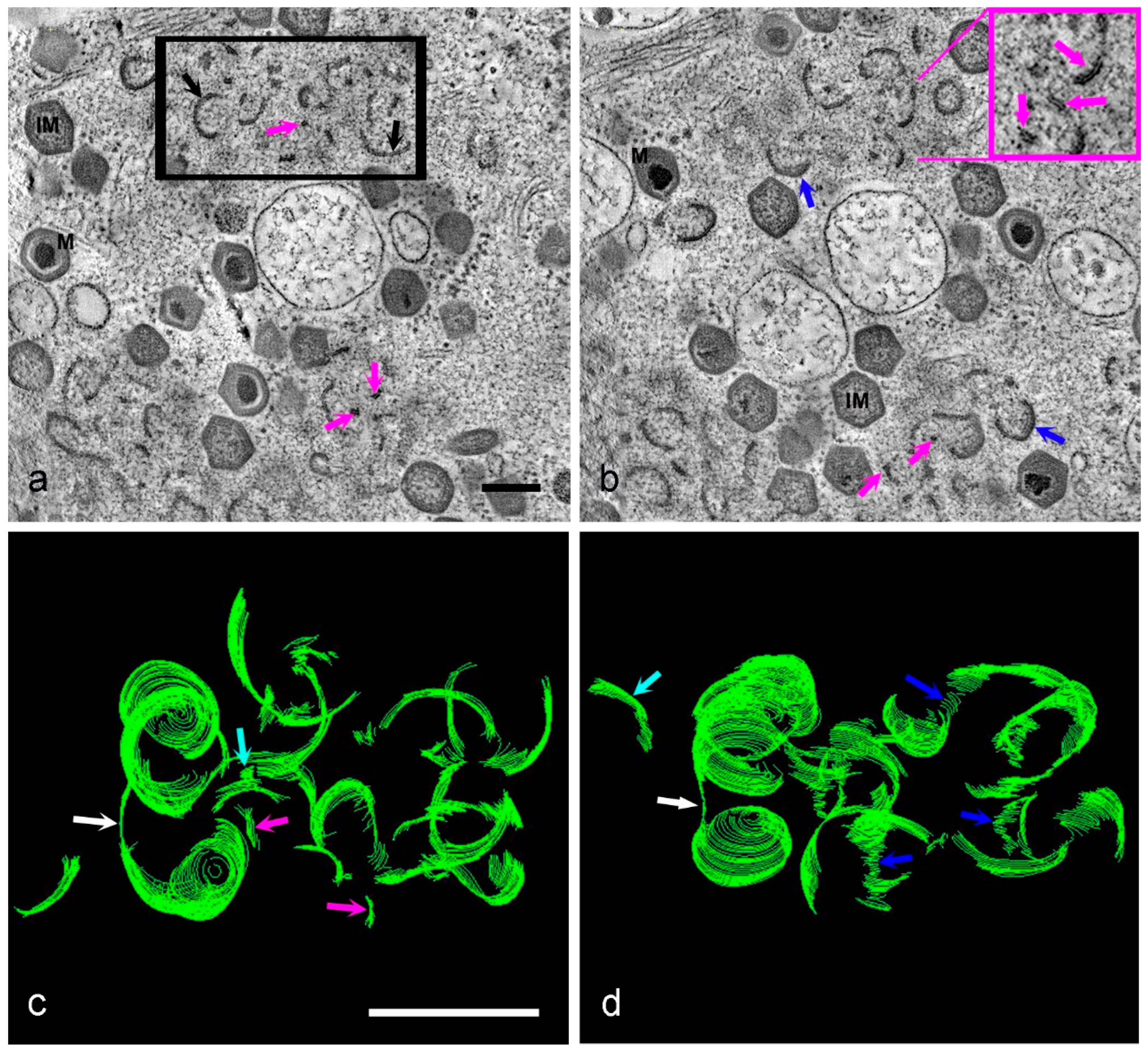
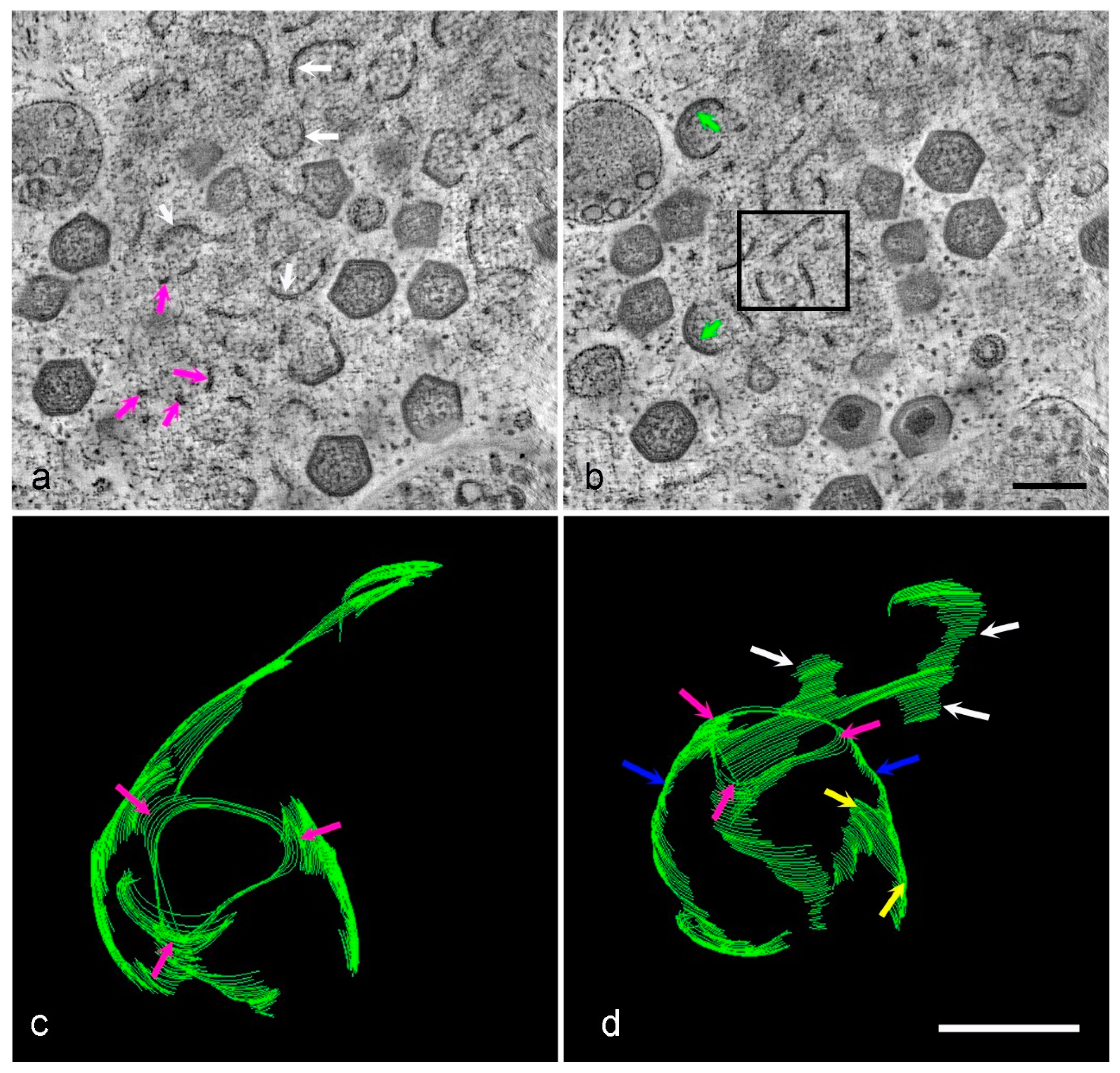
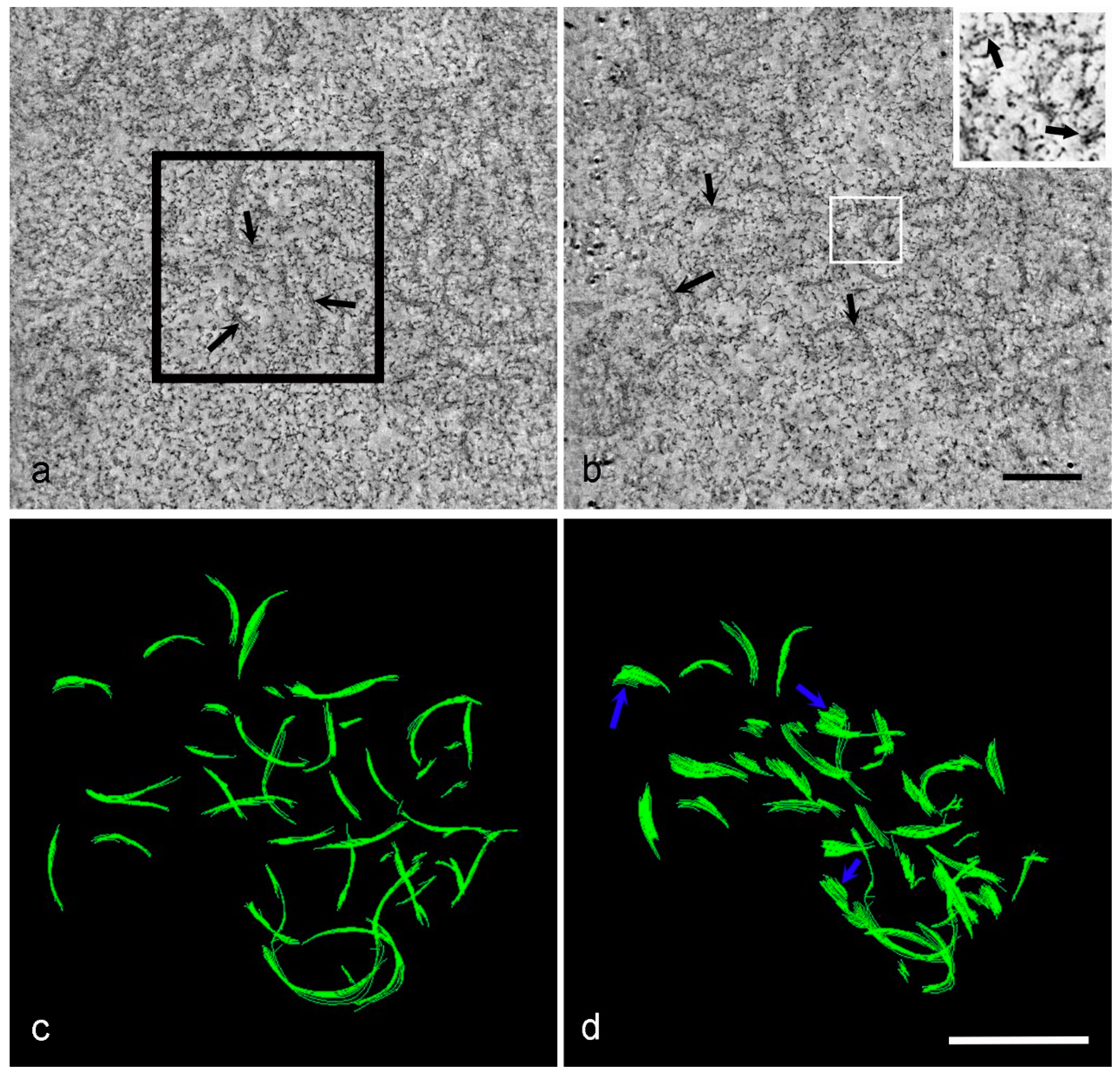
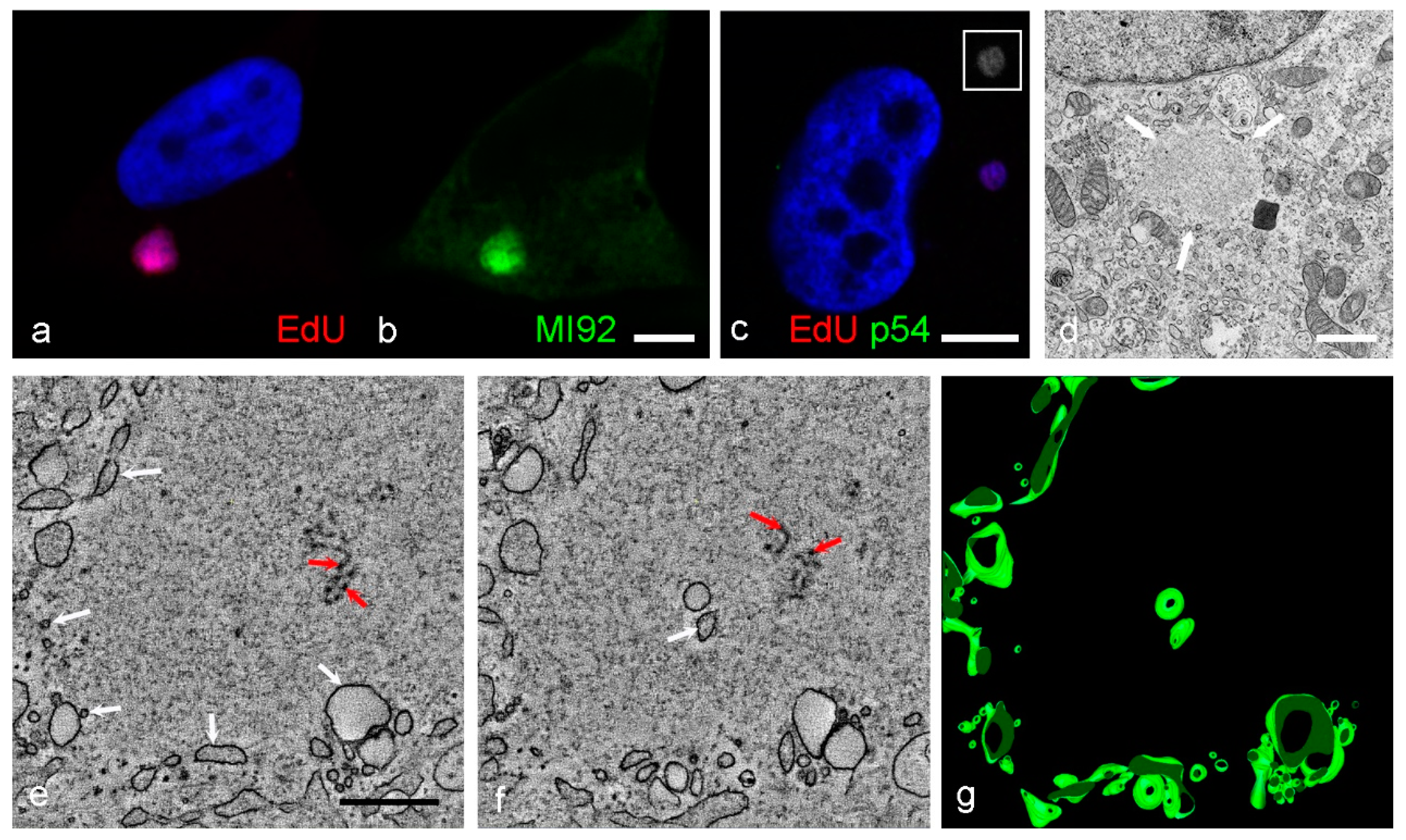
3. Results
4. Discussion
5. Conclusions
- Viral factories are complex regions with clear compartmentalization of protein and membrane components.
- Super-resolution microscopy reveals the sub-structure within virions and factory compartments with the advantage over challenging electron microscopy techniques of identifying the location of specific proteins.
- Click IT chemistry is a valuable technique for investigating the presence of newly synthesized protein.
- Different forms of membrane assembly intermediates are seen in early factories by HPF EM; membrane fragments, larger curved membranes and protein-coated membranes with the beginnings of the icosahedral profile.
- It is still not known where the membrane fragments come from or how they travel into the factory.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koonin, E.V.; Yutin, N. Evolution of the Large Nucleocytoplasmic DNA Viruses of Eukaryotes and Convergent Origins of Viral Gigantism. In Advances in Virus Research; Academic Press Inc.: Cambridge, MA, USA, 2019; Volume 103, pp. 167–202. [Google Scholar]
- Andreani, J.; Khalil, J.Y.B.; Sevvana, M.; Benamar, S.; Di Pinto, F.; Bitam, I.; Colson, P.; Klose, T.; Rossmann, M.G.; Raoult, D.; et al. Pacmanvirus, a New Giant Icosahedral Virus at the Crossroads between Asfarviridae and Faustoviruses. J. Virol. 2017, 91, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heath, C.M.; Windsor, M.; Wileman, T. Aggresomes Resemble Sites Specialized for Virus Assembly. J. Cell Biol. 2001, 153, 449–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, L.K.; Chapman, D.A.G.; Netherton, C.L.; Upton, C. African swine fever virus replication and genomics. Virus Res. 2013, 173, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Chinchar, V.G.; Waltzek, T.B.; Subramaniam, K. Ranaviruses and other members of the family Iridoviridae: Their place in the virosphere. Virology 2017, 511, 259–271. [Google Scholar] [CrossRef]
- Andrés, G.; García-Escudero, R.; Viñuela, E.; Salas, M.L.; Rodríguez, J.M. African Swine Fever Virus Structural Protein pE120R Is Essential for Virus Transport from Assembly Sites to Plasma Membrane but Not for Infectivity. J. Virol. 2001, 75, 6758–6768. [Google Scholar] [CrossRef] [Green Version]
- Alejo, A.; Matamoros, T.; Guerra, M.; Andrés, G. A Proteomic Atlas of the African Swine Fever Virus Particle. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E.V.; Yutin, N. Origin and evolution of eukaryotic large nucleo-cytoplasmic DNA viruses. Intervirology 2010, 53, 284–292. [Google Scholar] [CrossRef] [Green Version]
- Suzan-Monti, M.; la Scola, B.; Barrassi, L.; Espinosa, L.; Raoult, D. Ultrastructural characterization of the giant volcano-like virus factory of Acanthamoeba polyphaga Mimivirus. PLoS ONE 2007, 2. [Google Scholar] [CrossRef]
- Heaton, N.S.; Perera, R.; Berger, K.L.; Khadka, S.; Lacoount, D.L.; Kuhn, J.K.; Randall, G. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 17345–17350. [Google Scholar] [CrossRef] [Green Version]
- Martín-Acebes, M.A.; Blázquez, A.B.; de Oya, N.J.; Escribano-Romero, E.; Saiz, J.C. West nile virus replication requires fatty acid synthesis but is independent on phosphatidylinositol-4-phosphate lipids. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [Green Version]
- Yan, B.; Chu, H.; Yang, D.; Sze, K.H.; Lai, P.M.; Yuan, S.; Shuai, H.; Wang, Y.; Kao, R.Y.-T.; Chan, J.-F.-W.; et al. Characterization of the lipidomic profile of human coronavirus-infected cells: Implications for lipid metabolism remodeling upon coronavirus replication. Viruses 2019, 11, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, J.M.; Salas, M.L. African swine fever virus transcription. Virus Res. 2013, 173, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Suarez, C.; Andres, G.; Kolovou, A.; Hoppe, S.; Salas, M.L.; Walther, P.; Locker, J.K. African swine fever virus assembles a single membrane derived from rupture of the endoplasmic reticulum. Cell. Microbiol. 2015, 17, 1683–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrés, G.; García-Escudero, R.; Simón-Mateo, C.; Viñuela, E. African Swine Fever Virus Is Enveloped by a Two-Membraned Collapsed Cisterna Derived from the Endoplasmic Reticulum. J. Virol. 1998, 72, 8988–9001. [Google Scholar] [CrossRef] [PubMed]
- Salas, M.L.; Andrés, G. African swine fever virus morphogenesis. Virus Res. 2013, 173, 29–41. [Google Scholar] [CrossRef]
- Jouvenet, N.; Monaghan, P.; Way, M.; Wileman, T. Transport of African Swine Fever Virus from Assembly Sites to the Plasma Membrane Is Dependent on Microtubules and Conventional Kinesin. J. Virol. 2004, 78, 7990–8001. [Google Scholar] [CrossRef] [Green Version]
- Castro, I.F.; Volonté, L.; Risco, C. Virus factories: Biogenesis and structural design. Cell. Microbiol. 2013, 15, 24–34. [Google Scholar] [CrossRef]
- Katsafanas, G.C.; Moss, B. Colocalization of Transcription and Translation within Cytoplasmic Poxvirus Factories Coordinates Viral Expression and Subjugates Host Functions. Cell Host Microbe 2007, 2, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Castelló, A.; Quintas, A.; Sanchez, E.G.; Sabina, P.; Nogal, M.; Carrasco, L.; Revilla, Y. Regulation of host translational machinery by African swine fever virus. PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef] [Green Version]
- Andrés, G.; Charro, D.; Matamoros, T.; Dillard, R.S.; Abrescia, N.G.A. The cryo-EM structure of African swine fever virus unravels a unique architecture comprising two icosahedral protein capsids and two lipoprotein membranes. J. Biol. Chem. 2020, 295, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Meng, Y.; Chai, Y.; Li, L.; Sun, H.; Gaoa, G.F.; Tan, S.; Jianxun, Q. Cryo-EM Structure of the African Swine Fever Virus. Cell Host Microbe 2019, 26, 836–843.e3. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhao, D.; Wang, J.; Zhang, Y.; Wang, M.; Gao, Y.; Li, F.; Wang, J.; Bu, B.; Rao, Z.; et al. Architecture of African swine fever virus and implications for viral assembly. Science 2019, 366, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Rouiller, I.; Brookes, S.M.; Hyatt, A.D.; Windsor, M.; Wileman, T. African Swine Fever Virus Is Wrapped by the Endoplasmic Reticulum. J. Virol. 1998, 72, 2373–2387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locker, J.K.; Chlanda, P.; Sachsenheimer, T.; Brügger, B. Poxvirus membrane biogenesis: Rupture not disruption. Cellular Microbiol. 2013, 15, 190–199. [Google Scholar] [CrossRef] [Green Version]
- Chlanda, P.; Carbajal, M.A.; Cyrklaff, M.; Griffiths, G.; Krijnse-Locker, J. Membrane Rupture Generates Single Open Membrane Sheets during Vaccinia Virus Assembly. Cell Host Microbe 2009, 6, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Mutsafi, Y.; Shimoni, E.; Shimon, A.; Minsky, A. Membrane Assembly during the Infection Cycle of the Giant Mimivirus. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef]
- Enjuanes, L.; Carrascosa, A.L.; Moreno, M.A.; Vinuela, E. Titration of African swine fever (ASF) virus. J. Gen. Virol. 1976, 32, 471–477. [Google Scholar] [CrossRef]
- Sanz, A.; Garcia-Barreno, B.; Nogal, M.L.; Vinuela, E.; Enjuanes, L. Monoclonal antibodies specific for African swine fever virus proteins. J. Virol. 1985, 54, 199–206. [Google Scholar] [CrossRef] [Green Version]
- Cobbold, C.; Whittle, J.T.; Wileman, T. Involvement of the endoplasmic reticulum in the assembly and envelopment of African swine fever virus. J. Virol. 1996, 70, 8382–8390. [Google Scholar] [CrossRef] [Green Version]
- Netherton, C.L.; Simpson, J.; Haller, O.; Wileman, T.E.; Takamatsu, H.-H.; Monaghan, P.; Taylor, G. Inhibition of a Large Double-Stranded DNA Virus by MxA Protein. J. Virol. 2009, 83, 2310–2320. [Google Scholar] [CrossRef] [Green Version]
- Coelho, J.; Martins, C.; Ferreira, F.; Leitão, A. African swine fever virus ORF P1192R codes for a functional type II DNA topoisomerase. Virology 2015, 474, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, C.; Teo, H.; Serwa, R.A.; O’hare, P. Spatial and Temporal Resolution of Global Protein Synthesis during HSV Infection Using Bioorthogonal Precursors and Click Chemistry. PLoS Pathog. 2016, 12. [Google Scholar] [CrossRef] [Green Version]
- Hawes, P.; Netherton, C.L.; Mueller, M.; Wileman, T.; Monaghan, P. Rapid freeze-substitution preserves membranes in high-pressure frozen tissue culture cells. J. Microsc. 2007, 226, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Hawes, P.C.; Netherton, C.L.; Wileman, T.E.; Monaghan, P. The Envelope of Intracellular African Swine Fever Virus Is Composed of a Single Lipid Bilayer. J. Virol. 2008, 82, 7905–7912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kremer, J.R.; Mastronarde, D.N.; McIntosh, J.R. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 1996, 116, 71–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojo, G.; García-Beato, R.; Viñuela, E.; Salas, M.L.; Salas, J. Replication of African swine fever virus DNA in infected cells. Virology 1999, 257, 524–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez, C.; Welsch, S.; Chlanda, P.; Hagen, W.; Hoppe, S.; Kolovou, A.; Pagnier, I.; Raoult, D.; Locker, J.K. Open membranes are the precursors for assembly of large DNA viruses. Cell. Microbiol. 2013, 15, 1883–1895. [Google Scholar] [CrossRef] [Green Version]
- Quemin, E.R.; Corroyer-Dulmont, S.; Baskaran, A.; Penard, E.; Gazi, A.D.; Christo-Foroux, E.; Walther, P.; Abergel, C.; Locker, J.K. Complex Membrane Remodeling during Virion Assembly of the 30,000-Year-Old Mollivirus Sibericum. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Cackett, G.; Matelska, D.; Sýkora, M.; Portugal, R.; Malecki, M.; Bähler, J.; Dixon, L.; Werner, F. The African Swine Fever Virus Transcriptome. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [Green Version]
- Keßler, C.; Forth, J.H.; Keil, G.M.; Mettenleiter, T.C.; Blome, S.; Karger, A. The intracellular proteome of African swine fever virus. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Reis, A.L.; Parkhouse, R.M.E.; Penedos, A.R.; Martins, C.; Leitáo, A. Systematic analysis of longitudinal serological responses of pigs infected experimentally with African swine fever virus. J. Gen. Virol. 2007, 88, 2426–2434. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, A.S.; Cohen, S. Lipid Droplet and Peroxisome Biogenesis: Do They Go Hand-in-Hand? Front. Cell Dev. Biol. 2019, 7, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, J.M.; García-Escudero, R.; Salas, M.L.; Andrés, G. African Swine Fever Virus Structural Protein p54 Is Essential for the Recruitment of Envelope Precursors to Assembly Sites. J. Virol. 2004, 78, 4299–4313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, A.S.; Zhang, H.; Prinz, W.A. Organelle biogenesis in the endoplasmic reticulum. Nat. Cell Biol. 2017, 19, 876–882. [Google Scholar] [CrossRef]
- Hyman, A.A.; Weber, C.A.; Jülicher, F. Liquid-liquid phase separation in biology. In Annual Review of Cell and Developmental Biology; Annual Reviews: Palo Alto, CA, USA, 2014; Volume 30, pp. 39–58. [Google Scholar] [CrossRef] [Green Version]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357, 6357. [Google Scholar] [CrossRef] [Green Version]
- Nasheri, N.; Joyce, M.; Rouleau, Y.; Yang, P.; Yao, S.; Tyrrell, D.L.; Pezacky, J.P. Modulation of fatty acid synthase enzyme activity and expression during hepatitis C virus replication. Chem. Biol. 2013, 20, 570–582. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aicher, S.-M.; Monaghan, P.; Netherton, C.L.; Hawes, P.C. Unpicking the Secrets of African Swine Fever Viral Replication Sites. Viruses 2021, 13, 77. https://doi.org/10.3390/v13010077
Aicher S-M, Monaghan P, Netherton CL, Hawes PC. Unpicking the Secrets of African Swine Fever Viral Replication Sites. Viruses. 2021; 13(1):77. https://doi.org/10.3390/v13010077
Chicago/Turabian StyleAicher, Sophie-Marie, Paul Monaghan, Christopher L. Netherton, and Philippa C. Hawes. 2021. "Unpicking the Secrets of African Swine Fever Viral Replication Sites" Viruses 13, no. 1: 77. https://doi.org/10.3390/v13010077
APA StyleAicher, S. -M., Monaghan, P., Netherton, C. L., & Hawes, P. C. (2021). Unpicking the Secrets of African Swine Fever Viral Replication Sites. Viruses, 13(1), 77. https://doi.org/10.3390/v13010077