
The Not-So-Good, the Bad and the Ugly: HPV E5, E6 and E7 Oncoproteins in the Orchestration of Carcinogenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Carcinogenic Orchestration by E5, E6 and E7
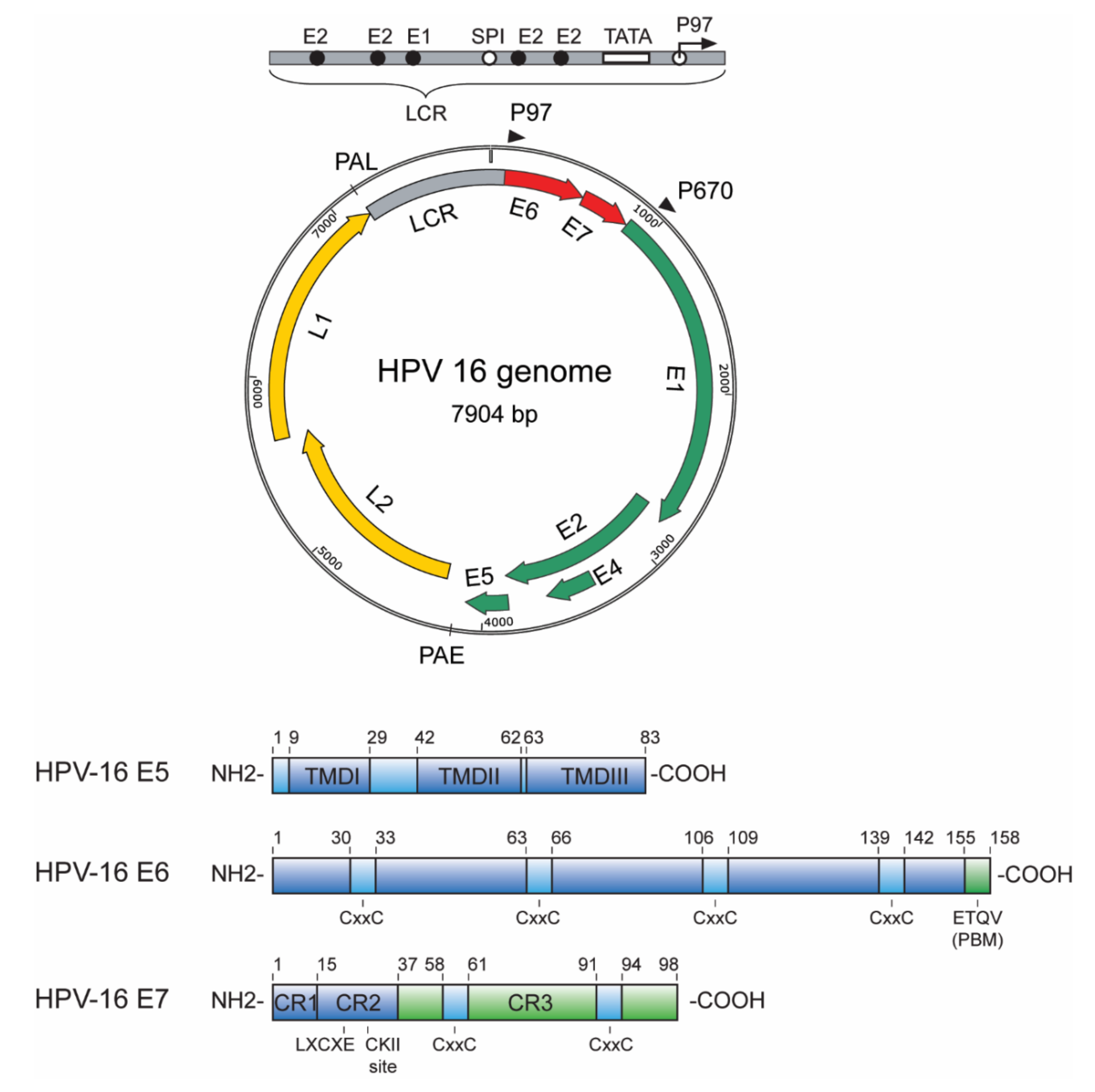
1.1. E5, E6 and E7 Oncoproteins—An Overview
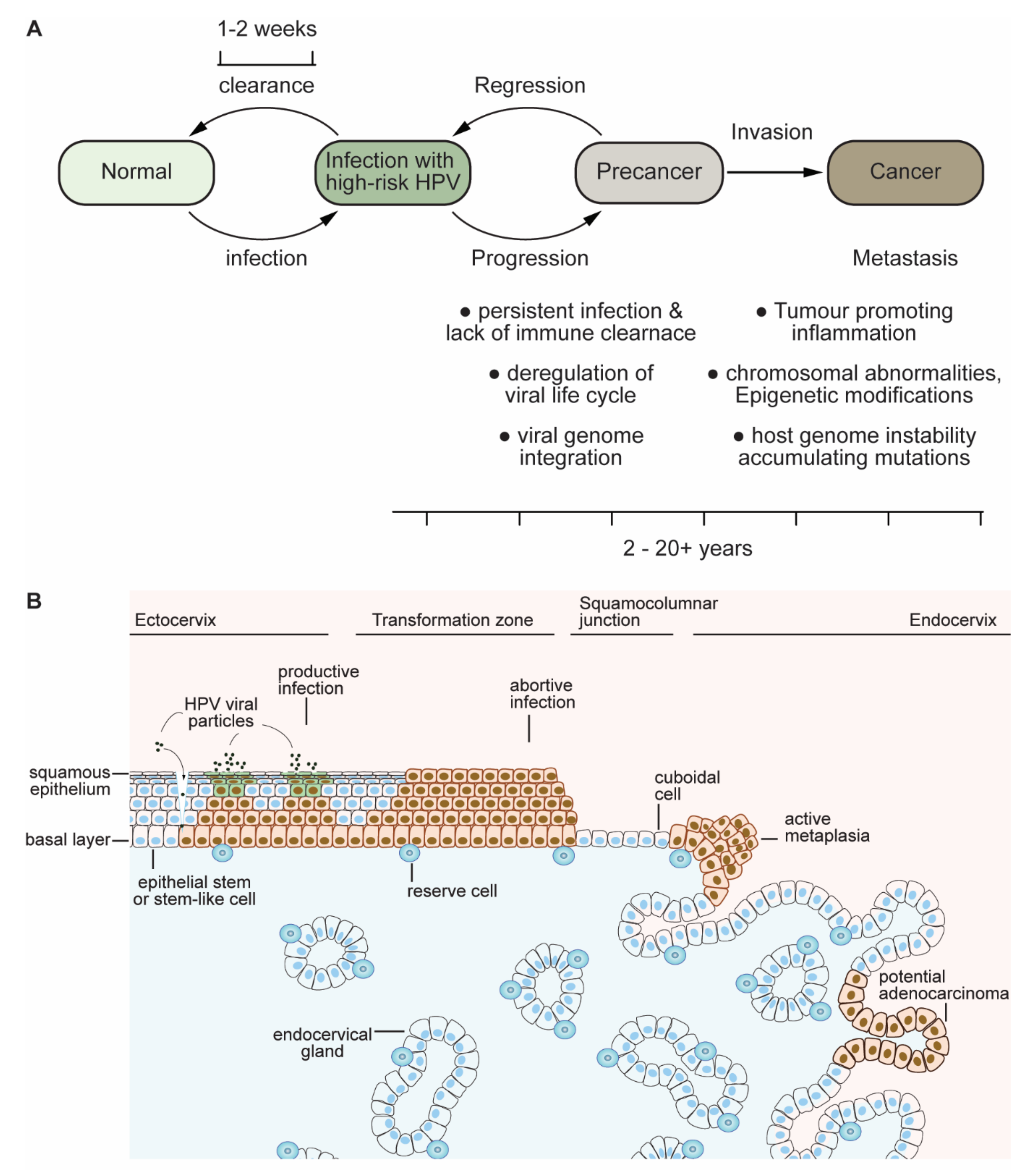
1.2. Clues and Cues to Transformation and Cancer by HPV-Oncoproteins
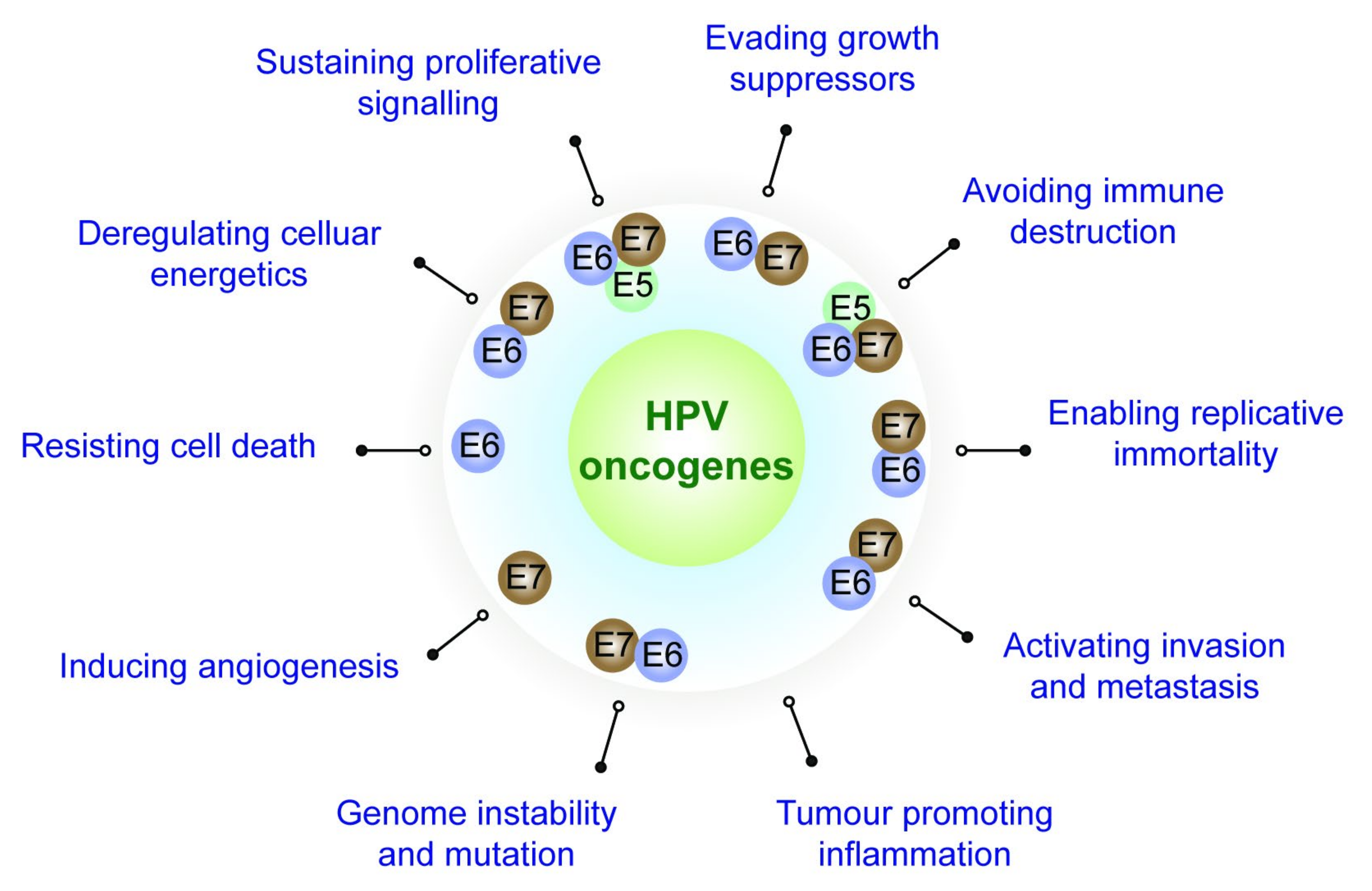
2. Role Played by the HPV Oncoproteins towards Attaining the Cancer Hallmarks
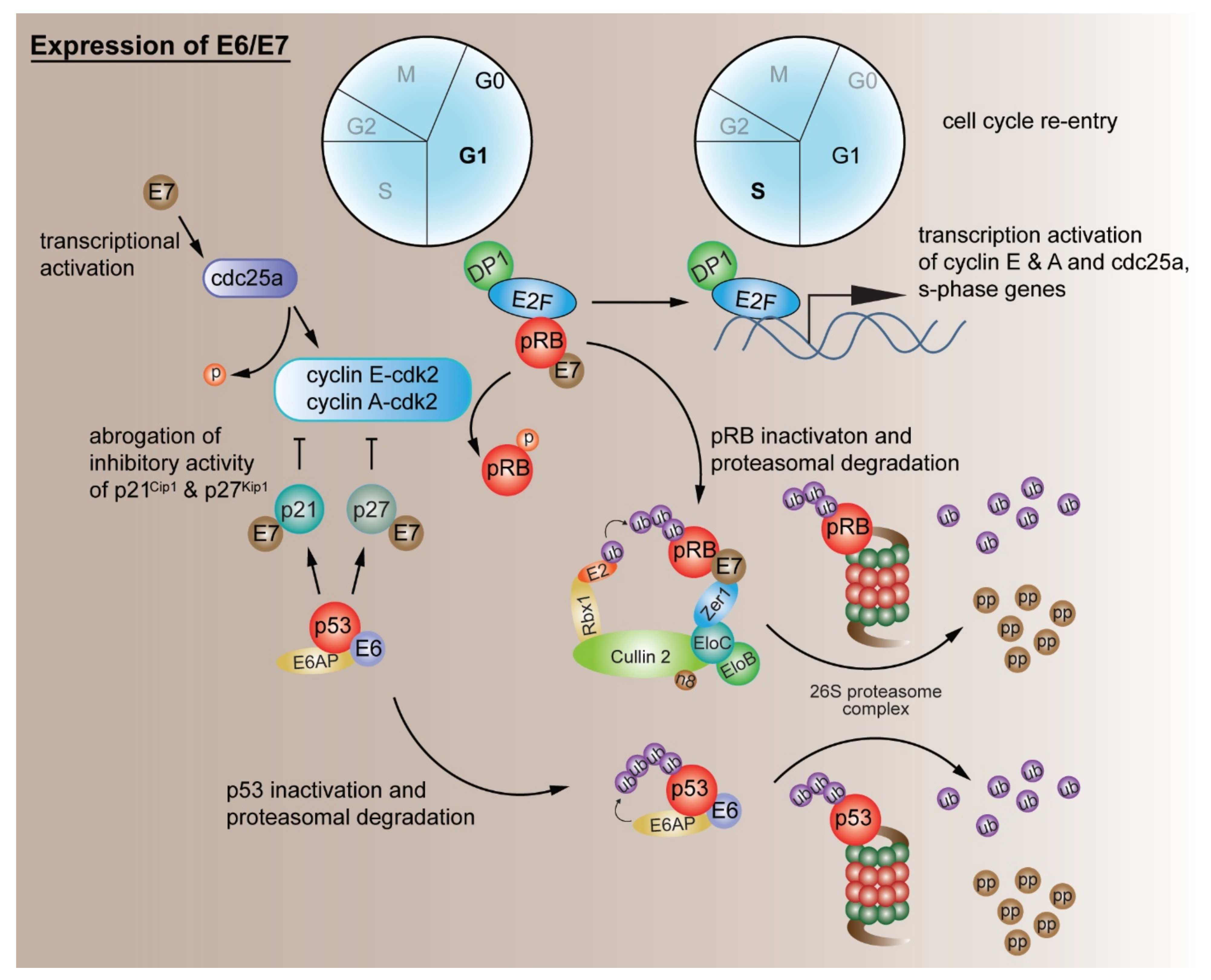
2.1. Evading Growth Suppressors
G1/S Cell Cycle Checkpoint Deregulation by Other Mechanisms
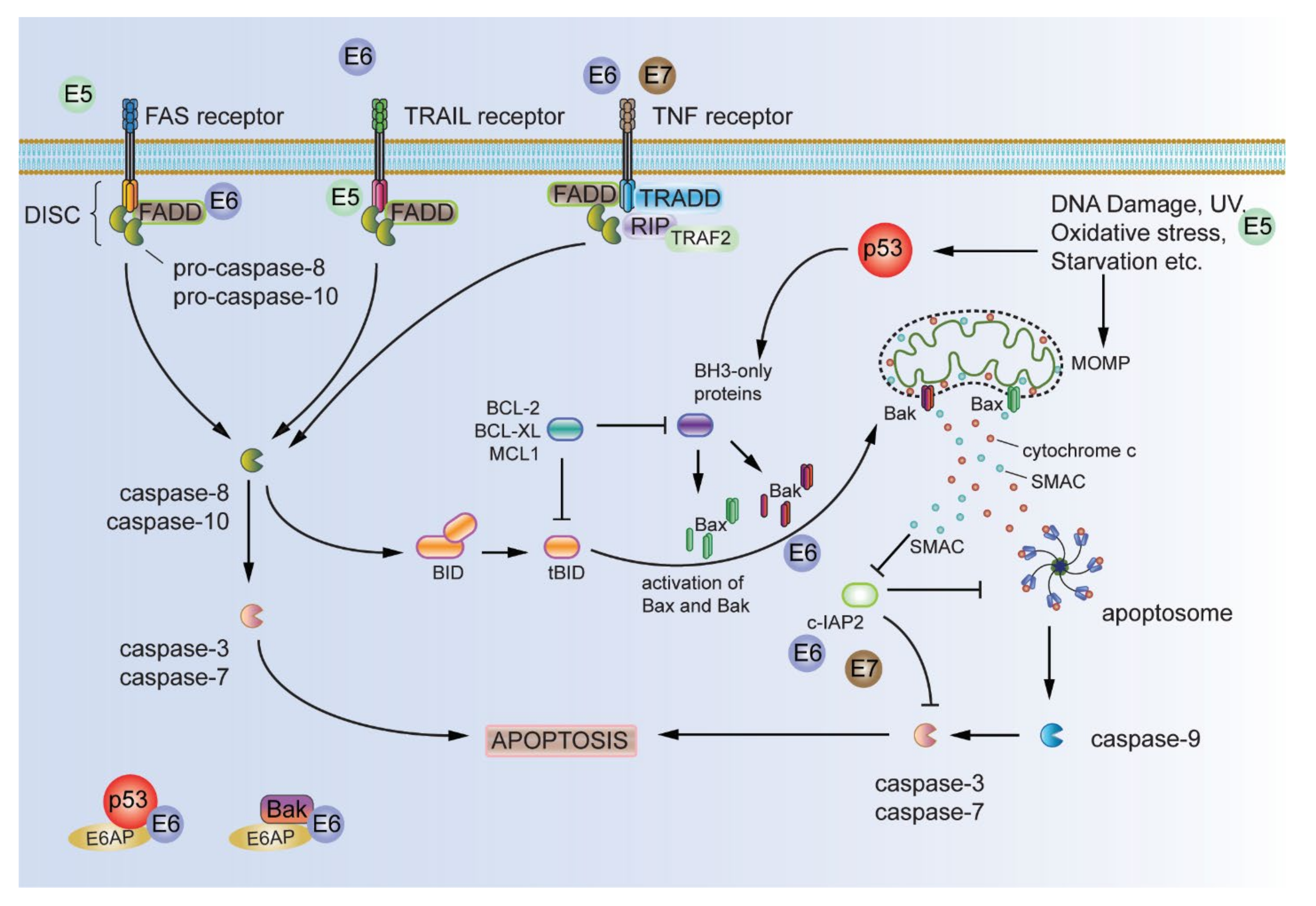
2.2. Resisting Cell Death
Resistance to Anoikis and Anchorage Independence
2.3. Sustaining Proliferative Signalling
Modulation of Cellular Signalling Pathways
2.4. Enabling Replicative Immortality
2.5. Activating Invasion and Metastasis
Role of E6 PDZ Binding Motif (PBM) and Polarity Deregulation
2.6. Inducing Angiogenesis
2.7. Deregulating Cellular Energetics and Metabolism
2.8. Genome Instability and the Consequent Mutation of Hallmark-Enabling Genes
2.8.1. HPV E7 and Epigenetic Reprograming
2.8.2. Modulation of MicroRNAs by HPV E7
2.9. Avoiding Immune Destruction
2.10. Tumour-Promoting Immune Cell Infiltration (Inflammation)
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Biological Hallmarks of Cancer. In Holland—Frei Cancer Medicine; Wiley-Blackwell Publishers: Hoboken, NJ, USA, 2016; pp. 1–10. [Google Scholar]
- Plummer, M.; de Martel, C.; Vignat, J.; Ferlay, J.; Bray, F.; Franceschi, S. Global burden of cancers attributable to infections in 2012: A synthetic analysis. Lancet Glob. Health 2016, 4, e609–e616. [Google Scholar] [CrossRef] [Green Version]
- De Martel, C.; Plummer, M.; Vignat, J.; Franceschi, S. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int. J. Cancer 2017, 141, 664–670. [Google Scholar] [CrossRef] [Green Version]
- Kombe Kombe, A.J.; Li, B.; Zahid, A.; Mengist, H.M.; Bounda, G.A.; Zhou, Y.; Jin, T. Epidemiology and Burden of Human Papillomavirus and Related Diseases, Molecular Pathogenesis, and Vaccine Evaluation. Front. Public Health 2020, 8, 552028. [Google Scholar] [CrossRef]
- Wild, C.P.; Weiderpass, E.; Stewart, B.W. (Eds.) World Cancer Report: Cancer Research for Cancer Prevention; Licence: CC BY-NC-ND 3.0 IGO; International Agency for Research on Cancer: Lyon, France, 2020; Available online: http://publications.iarc.fr/586 (accessed on 15 May 2021).
- McBride, A.A.; Warburton, A. The role of integration in oncogenic progression of HPV-associated cancers. PLoS Pathog. 2017, 13, e1006211. [Google Scholar] [CrossRef] [Green Version]
- McBride, A.A. Mechanisms and strategies of papillomavirus replication. Biol. Chem. 2017, 398, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Della Fera, A.N.; Warburton, A.; Coursey, T.L.; Khurana, S.; McBride, A.A. Persistent Human Papillomavirus Infection. Viruses 2021, 13, 321. [Google Scholar] [CrossRef]
- Gao, G.; Johnson, S.H.; Vasmatzis, G.; Pauley, C.E.; Tombers, N.M.; Kasperbauer, J.L.; Smith, D.I. Common fragile sites (CFS) and extremely large CFS genes are targets for human papillomavirus integrations and chromosome rearrangements in oropharyngeal squamous cell carcinoma. Genes Chromosomes Cancer 2017, 56, 59–74. [Google Scholar] [CrossRef]
- Thorland, E.C.; Myers, S.L.; Persing, D.H.; Sarkar, G.; McGovern, R.M.; Gostout, B.S.; Smith, D.I. Human papillomavirus type 16 integrations in cervical tumors frequently occur in common fragile sites. Cancer Res. 2000, 60, 5916–5921. [Google Scholar] [PubMed]
- Durst, M.; Croce, C.M.; Gissmann, L.; Schwarz, E.; Huebner, K. Papillomavirus sequences integrate near cellular oncogenes in some cervical carcinomas. Proc. Natl. Acad. Sci. USA 1987, 84, 1070–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burk, R.D.; Chen, Z.; Saller, C.; Tarvin, K.; Carvalho, A.L.; Scapulatempo-Neto, C.; Silveira, H.C.; Fregnani, J.H.; Creighton, C.J.; Anderson, M.L.; et al. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar] [CrossRef]
- Cullen, A.P.; Reid, R.; Campion, M.; Lorincz, A.T. Analysis of the physical state of different human papillomavirus DNAs in intraepithelial and invasive cervical neoplasm. J. Virol. 1991, 65, 606–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, I.M.; DiNardo, L.J.; Windle, B. Integration of Human Papillomavirus Genomes in Head and Neck Cancer: Is It Time to Consider a Paradigm Shift? Viruses 2017, 9, 208. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.; Baldwin, A.; Edwards, K.M.; Hayakawa, H.; Nguyen, C.L.; Owens, M.; Grace, M.; Huh, K. Mechanisms of human papillomavirus-induced oncogenesis. J. Virol. 2004, 78, 11451–11460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butz, K.; Ristriani, T.; Hengstermann, A.; Denk, C.; Scheffner, M.; Hoppe-Seyler, F. siRNA targeting of the viral E6 oncogene efficiently kills human papillomavirus-positive cancer cells. Oncogene 2003, 22, 5938–5945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamato, K.; Yamada, T.; Kizaki, M.; Ui-Tei, K.; Natori, Y.; Fujino, M.; Nishihara, T.; Ikeda, Y.; Nasu, Y.; Saigo, K.; et al. New highly potent and specific E6 and E7 siRNAs for treatment of HPV16 positive cervical cancer. Cancer Gene Ther. 2008, 15, 140–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshinouchi, M.; Yamada, T.; Kizaki, M.; Fen, J.; Koseki, T.; Ikeda, Y.; Nishihara, T.; Yamato, K. In vitro and in vivo growth suppression of human papillomavirus 16-positive cervical cancer cells by E6 siRNA. Mol. Ther. J. Am. Soc. Gene Ther. 2003, 8, 762–768. [Google Scholar] [CrossRef]
- Ren, S.; Gaykalova, D.A.; Guo, T.; Favorov, A.V.; Fertig, E.J.; Tamayo, P.; Callejas-Valera, J.L.; Allevato, M.; Gilardi, M.; Santos, J.; et al. HPV E2, E4, E5 drive alternative carcinogenic pathways in HPV positive cancers. Oncogene 2020, 39, 6327–6339. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Schiffman, M.; Doorbar, J.; Wentzensen, N.; de Sanjose, S.; Fakhry, C.; Monk, B.J.; Stanley, M.A.; Franceschi, S. Carcinogenic human papillomavirus infection. Nat. Rev. Dis. Primers 2016, 2, 16086. [Google Scholar] [CrossRef]
- Doorbar, J.; Griffin, H. Refining our understanding of cervical neoplasia and its cellular origins. Papillomavirus Res. 2019, 7, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Gieswein, C.E.; Sharom, F.J.; Wildeman, A.G. Oligomerization of the E5 protein of human papillomavirus type 16 occurs through multiple hydrophobic regions. Virology 2003, 313, 415–426. [Google Scholar] [CrossRef]
- Yang, D.H.; Wildeman, A.G.; Sharom, F.J. Overexpression, purification, and structural analysis of the hydrophobic E5 protein from human papillomavirus type 16. Protein Expr. Purif. 2003, 30, 1–10. [Google Scholar] [CrossRef]
- Disbrow, G.L.; Sunitha, I.; Baker, C.C.; Hanover, J.; Schlegel, R. Codon optimization of the HPV-16 E5 gene enhances protein expression. Virology 2003, 311, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Bravo, I.G.; Alonso, A. Mucosal human papillomaviruses encode four different E5 proteins whose chemistry and phylogeny correlate with malignant or benign growth. J. Virol. 2004, 78, 13613–13626. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Ceresa, B.P. Characterization of the plasma membrane localization and orientation of HPV16 E5 for cell-cell fusion. Virology 2009, 393, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Disbrow, G.L.; Hanover, J.A.; Schlegel, R. Endoplasmic reticulum-localized human papillomavirus type 16 E5 protein alters endosomal pH but not trans-Golgi pH. J. Virol. 2005, 79, 5839–5846. [Google Scholar] [CrossRef] [Green Version]
- Schiffman, M.; Herrero, R.; Desalle, R.; Hildesheim, A.; Wacholder, S.; Rodriguez, A.C.; Bratti, M.C.; Sherman, M.E.; Morales, J.; Guillen, D.; et al. The carcinogenicity of human papillomavirus types reflects viral evolution. Virology 2005, 337, 76–84. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, M.S.; Lowy, D.R.; Schiller, J.T. Papillomavirus polypeptides E6 and E7 are zinc-binding proteins. J. Virol. 1989, 63, 1404–1407. [Google Scholar] [CrossRef] [Green Version]
- Grossman, S.R.; Laimins, L.A. E6 protein of human papillomavirus type 18 binds zinc. Oncogene 1989, 4, 1089–1093. [Google Scholar]
- Tang, S.; Tao, M.; McCoy, J.P., Jr.; Zheng, Z.M. The E7 oncoprotein is translated from spliced E6*I transcripts in high-risk human papillomavirus type 16- or type 18-positive cervical cancer cell lines via translation reinitiation. J. Virol. 2006, 80, 4249–4263. [Google Scholar] [CrossRef] [Green Version]
- Doorbar, J.; Parton, A.; Hartley, K.; Banks, L.; Crook, T.; Stanley, M.; Crawford, L. Detection of novel splicing patterns in a HPV16-containing keratinocyte cell line. Virology 1990, 178, 254–262. [Google Scholar] [CrossRef]
- Smotkin, D.; Wettstein, F.O. Transcription of human papillomavirus type 16 early genes in a cervical cancer and a cancer-derived cell line and identification of the E7 protein. Proc. Natl. Acad. Sci. USA 1986, 83, 4680–4684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajiro, M.; Zheng, Z.-M. E6^E7, a Novel Splice Isoform Protein of Human Papillomavirus 16, Stabilizes Viral E6 and E7 Oncoproteins via HSP90 and GRP78. mBio 2015, 6, e02068-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pim, D.; Banks, L. HPV-18 E6*I protein modulates the E6-directed degradation of p53 by binding to full-length HPV-18 E6. Oncogene 1999, 18, 7403–7408. [Google Scholar] [CrossRef] [Green Version]
- Pim, D.; Massimi, P.; Banks, L. Alternatively spliced HPV-18 E6* protein inhibits E6 mediated degradation of p53 and suppresses transformed cell growth. Oncogene 1997, 15, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Ajiro, M.; Tang, S.; Doorbar, J.; Zheng, Z.-M. Serine/Arginine-Rich Splicing Factor 3 and Heterogeneous Nuclear Ribonucleoprotein A1 Regulate Alternative RNA Splicing and Gene Expression of Human Papillomavirus 18 through Two Functionally Distinguishable cis Elements. J. Virol. 2016, 90, 9138–9152. [Google Scholar] [CrossRef] [Green Version]
- Nomine, Y.; Masson, M.; Charbonnier, S.; Zanier, K.; Ristriani, T.; Deryckere, F.; Sibler, A.P.; Desplancq, D.; Atkinson, R.A.; Weiss, E.; et al. Structural and functional analysis of E6 oncoprotein: Insights in the molecular pathways of human papillomavirus-mediated pathogenesis. Mol. Cell 2006, 21, 665–678. [Google Scholar] [CrossRef]
- Zanier, K.; Charbonnier, S.; Sidi, A.O.; McEwen, A.G.; Ferrario, M.G.; Poussin-Courmontagne, P.; Cura, V.; Brimer, N.; Babah, K.O.; Ansari, T.; et al. Structural basis for hijacking of cellular LxxLL motifs by papillomavirus E6 oncoproteins. Science 2013, 339, 694–698. [Google Scholar] [CrossRef] [Green Version]
- Zanier, K.; ould M’hamed ould Sidi, A.; Boulade-Ladame, C.; Rybin, V.; Chappelle, A.; Atkinson, A.; Kieffer, B.; Trave, G. Solution structure analysis of the HPV16 E6 oncoprotein reveals a self-association mechanism required for E6-mediated degradation of p53. Structure 2012, 20, 604–617. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.; Myers, M.P.; Massimi, P.; Guarnaccia, C.; Banks, L. Analysis of Multiple HPV E6 PDZ Interactions Defines Type-Specific PDZ Fingerprints That Predict Oncogenic Potential. PLoS Pathog. 2016, 12, e1005766. [Google Scholar] [CrossRef] [PubMed]
- Basukala, O.; Sarabia-Vega, V.; Banks, L. Human papillomavirus oncoproteins and post-translational modifications: Generating multifunctional hubs for overriding cellular homeostasis. Biol. Chem. 2020, 401, 585–599. [Google Scholar] [CrossRef]
- Boon, S.S.; Tomaic, V.; Thomas, M.; Roberts, S.; Banks, L. Cancer-causing human papillomavirus E6 proteins display major differences in the phospho-regulation of their PDZ interactions. J. Virol. 2015, 89, 1579–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boon, S.S.; Banks, L. High-risk human papillomavirus E6 oncoproteins interact with 14-3-3zeta in a PDZ binding motif-dependent manner. J. Virol. 2013, 87, 1586–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhne, C.; Gardiol, D.; Guarnaccia, C.; Amenitsch, H.; Banks, L. Differential regulation of human papillomavirus E6 by protein kinase A: Conditional degradation of human discs large protein by oncogenic E6. Oncogene 2000, 19, 5884–5891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thatte, J.; Massimi, P.; Thomas, M.; Boon, S.S.; Banks, L. The HPV E6 PDZ Binding Motif links DNA Damage Response Signaling to E6 Inhibition of p53 Transcriptional Activity. J. Virol. 2018, 92, e00465-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarabia-Vega, V.; Banks, L. Acquisition of a phospho-acceptor site enhances HPV E6 PDZ-binding motif functional promiscuity. J. Gen. Virol. 2020, 101, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Phelps, W.C.; Munger, K.; Yee, C.L.; Barnes, J.A.; Howley, P.M. Structure-function analysis of the human papillomavirus type 16 E7 oncoprotein. J. Virol. 1992, 66, 2418–2427. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Lee, E.E.; Kim, J.; Yang, R.; Chamseddin, B.; Ni, C.; Gusho, E.; Xie, Y.; Chiang, C.M.; Buszczak, M.; et al. Transforming activity of an oncoprotein-encoding circular RNA from human papillomavirus. Nat. Commun. 2019, 10, 2300. [Google Scholar] [CrossRef] [Green Version]
- Phelps, W.C.; Yee, C.L.; Munger, K.; Howley, P.M. The human papillomavirus type 16 E7 gene encodes transactivation and transformation functions similar to those of adenovirus E1A. Cell 1988, 53, 539–547. [Google Scholar] [CrossRef]
- Figge, J.; Smith, T.F. Cell-division sequence motif. Nature 1988, 334, 109. [Google Scholar] [CrossRef] [PubMed]
- Matlashewski, G.; Schneider, J.; Banks, L.; Jones, N.; Murray, A.; Crawford, L. Human papillomavirus type 16 DNA cooperates with activated ras in transforming primary cells. EMBO J. 1987, 6, 1741–1746. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.; Werness, B.A.; Dyson, N.; Phelps, W.C.; Harlow, E.; Howley, P.M. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989, 8, 4099–4105. [Google Scholar] [CrossRef] [PubMed]
- Dyson, N.; Howley, P.M.; Munger, K.; Harlow, E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 1989, 243, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Todorovic, B.; Hung, K.; Massimi, P.; Avvakumov, N.; Dick, F.A.; Shaw, G.S.; Banks, L.; Mymryk, J.S. Conserved region 3 of human papillomavirus 16 E7 contributes to deregulation of the retinoblastoma tumor suppressor. J. Virol. 2012, 86, 13313–13323. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.S.; Patrick, D.R.; Edwards, G.; Goodhart, P.J.; Huber, H.E.; Miles, L.; Garsky, V.M.; Oliff, A.; Heimbrook, D.C. Protein domains governing interactions between E2F, the retinoblastoma gene product, and human papillomavirus type 16 E7 protein. Mol. Cell. Biol. 1993, 13, 953–960. [Google Scholar] [CrossRef]
- Wu, E.W.; Clemens, K.E.; Heck, D.V.; Munger, K. The human papillomavirus E7 oncoprotein and the cellular transcription factor E2F bind to separate sites on the retinoblastoma tumor suppressor protein. J. Virol. 1993, 67, 2402–2407. [Google Scholar] [CrossRef] [Green Version]
- Firzlaff, J.M.; Galloway, D.A.; Eisenman, R.N.; Luscher, B. The E7 protein of human papillomavirus type 16 is phosphorylated by casein kinase II. New Biol. 1989, 1, 44–53. [Google Scholar]
- Barbosa, M.S.; Edmonds, C.; Fisher, C.; Schiller, J.T.; Lowy, D.R.; Vousden, K.H. The region of the HPV E7 oncoprotein homologous to adenovirus E1a and Sv40 large T antigen contains separate domains for Rb binding and casein kinase II phosphorylation. EMBO J. 1990, 9, 153–160. [Google Scholar] [CrossRef]
- McIntyre, M.C.; Frattini, M.G.; Grossman, S.R.; Laimins, L.A. Human papillomavirus type 18 E7 protein requires intact Cys-X-X-Cys motifs for zinc binding, dimerization, and transformation but not for Rb binding. J. Virol. 1993, 67, 3142–3150. [Google Scholar] [CrossRef] [Green Version]
- Calcada, E.O.; Felli, I.C.; Hosek, T.; Pierattelli, R. The heterogeneous structural behavior of E7 from HPV16 revealed by NMR spectroscopy. ChemBioChem 2013, 14, 1876–1882. [Google Scholar] [CrossRef]
- Ohlenschlager, O.; Seiboth, T.; Zengerling, H.; Briese, L.; Marchanka, A.; Ramachandran, R.; Baum, M.; Korbas, M.; Meyer-Klaucke, W.; Durst, M.; et al. Solution structure of the partially folded high-risk human papilloma virus 45 oncoprotein E7. Oncogene 2006, 25, 5953–5959. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Alai, M.M.; Alonso, L.G.; de Prat-Gay, G. The N-terminal module of HPV16 E7 is an intrinsically disordered domain that confers conformational and recognition plasticity to the oncoprotein. Biochemistry 2007, 46, 10405–10412. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Clements, A.; Zhao, K.; Marmorstein, R. Structure of the human Papillomavirus E7 oncoprotein and its mechanism for inactivation of the retinoblastoma tumor suppressor. J. Biol. Chem. 2006, 281, 578–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todorovic, B.; Massimi, P.; Hung, K.; Shaw, G.S.; Banks, L.; Mymryk, J.S. Systematic analysis of the amino acid residues of human papillomavirus type 16 E7 conserved region 3 involved in dimerization and transformation. J. Virol. 2011, 85, 10048–10057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemens, K.E.; Brent, R.; Gyuris, J.; Munger, K. Dimerization of the human papillomavirus E7 oncoprotein in vivo. Virology 1995, 214, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clements, A.; Johnston, K.; Mazzarelli, J.M.; Ricciardi, R.P.; Marmorstein, R. Oligomerization properties of the viral oncoproteins adenovirus E1A and human papillomavirus E7 and their complexes with the retinoblastoma protein. Biochemistry 2000, 39, 16033–16045. [Google Scholar] [CrossRef]
- Alonso, L.G.; Smal, C.; Garcia-Alai, M.M.; Chemes, L.; Salame, M.; de Prat-Gay, G. Chaperone holdase activity of human papillomavirus E7 oncoprotein. Biochemistry 2006, 45, 657–667. [Google Scholar] [CrossRef]
- Alonso, L.G.; Garcia-Alai, M.M.; Smal, C.; Centeno, J.M.; Iacono, R.; Castano, E.; Gualfetti, P.; de Prat-Gay, G. The HPV16 E7 viral oncoprotein self-assembles into defined spherical oligomers. Biochemistry 2004, 43, 3310–3317. [Google Scholar] [CrossRef]
- Smotkin, D.; Wettstein, F.O. The major human papillomavirus protein in cervical cancers is a cytoplasmic phosphoprotein. J. Virol. 1987, 61, 1686–1689. [Google Scholar] [CrossRef] [Green Version]
- Guccione, E.; Massimi, P.; Bernat, A.; Banks, L. Comparative analysis of the intracellular location of the high- and low-risk human papillomavirus oncoproteins. Virology 2002, 293, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, I.; Nickerson, J.; Penman, S.; Stanley, M. Human papillomavirus 16 E7 protein is associated with the nuclear matrix. Proc. Natl. Acad. Sci. USA 1991, 88, 11217–11221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, H.; Watanabe, S.; Furuno, A.; Yoshiike, K. Human papillomavirus type 16 E7 protein expressed in Escherichia coli and monkey COS-1 cells: Immunofluorescence detection of the nuclear E7 protein. Virology 1989, 170, 311–315. [Google Scholar] [CrossRef]
- Smith-McCune, K.; Kalman, D.; Robbins, C.; Shivakumar, S.; Yuschenkoff, L.; Bishop, J.M. Intranuclear localization of human papillomavirus 16 E7 during transformation and preferential binding of E7 to the Rb family member p130. Proc. Natl. Acad. Sci. USA 1999, 96, 6999–7004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdovinos-Torres, H.; Orozco-Morales, M.; Pedroza-Saavedra, A.; Padilla-Noriega, L.; Esquivel-Guadarrama, F.; Gutierrez-Xicotencatl, L. Different Isoforms of HPV-16 E7 Protein are Present in Cytoplasm and Nucleus. Open Virol. J. 2008, 2, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Yasumoto, S.; Burkhardt, A.L.; Doniger, J.; DiPaolo, J.A. Human papillomavirus type 16 DNA-induced malignant transformation of NIH 3T3 cells. J. Virol. 1986, 57, 572–577. [Google Scholar] [CrossRef] [Green Version]
- Bedell, M.A.; Jones, K.H.; Grossman, S.R.; Laimins, L.A. Identification of human papillomavirus type 18 transforming genes in immortalized and primary cells. J. Virol. 1989, 63, 1247–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanda, T.; Furuno, A.; Yoshiike, K. Human papillomavirus type 16 open reading frame E7 encodes a transforming gene for rat 3Y1 cells. J. Virol. 1988, 62, 610–613. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, A.; Noda, T.; Yajima, H.; Hatanaka, M.; Ito, Y. Identification of a transforming gene of human papillomavirus type 16. J. Virol. 1989, 63, 1465–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vousden, K.H.; Doniger, J.; DiPaolo, J.A.; Lowy, D.R. The E7 open reading frame of human papillomavirus type 16 encodes a transforming gene. Oncogene Res. 1988, 3, 167–175. [Google Scholar] [PubMed]
- Watanabe, S.; Yoshiike, K. Transformation of rat 3Y1 cells by human papillomavirus type-18 DNA. Int. J. Cancer 1988, 41, 896–900. [Google Scholar] [CrossRef] [PubMed]
- Yutsudo, M.; Okamoto, Y.; Hakura, A. Functional dissociation of transforming genes of human papillomavirus type 16. Virology 1988, 166, 594–597. [Google Scholar] [CrossRef]
- Schlegel, R.; Phelps, W.C.; Zhang, Y.L.; Barbosa, M. Quantitative keratinocyte assay detects two biological activities of human papillomavirus DNA and identifies viral types associated with cervical carcinoma. EMBO J. 1988, 7, 3181–3187. [Google Scholar] [CrossRef]
- Woodworth, C.D.; Bowden, P.E.; Doniger, J.; Pirisi, L.; Barnes, W.; Lancaster, W.D.; DiPaolo, J.A. Characterization of normal human exocervical epithelial cells immortalized in vitro by papillomavirus types 16 and 18 DNA. Cancer Res. 1988, 48, 4620–4628. [Google Scholar] [PubMed]
- Woodworth, C.D.; Doniger, J.; DiPaolo, J.A. Immortalization of human foreskin keratinocytes by various human papillomavirus DNAs corresponds to their association with cervical carcinoma. J. Virol. 1989, 63, 159–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waggoner, S.E.; Woodworth, C.D.; Stoler, M.H.; Barnes, W.A.; Delgado, G.; DiPaolo, J.A. Human cervical cells immortalized in vitro with oncogenic human papillomavirus DNA differentiate dysplastically in vivo. Gynecol. Oncol. 1990, 38, 407–412. [Google Scholar] [CrossRef]
- McCance, D.J.; Kopan, R.; Fuchs, E.; Laimins, L.A. Human papillomavirus type 16 alters human epithelial cell differentiation in vitro. Proc. Natl. Acad. Sci. USA 1988, 85, 7169–7173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munger, K.; Phelps, W.C.; Bubb, V.; Howley, P.M.; Schlegel, R. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J. Virol. 1989, 63, 4417–4421. [Google Scholar] [CrossRef] [Green Version]
- Hudson, J.B.; Bedell, M.A.; McCance, D.J.; Laiminis, L.A. Immortalization and altered differentiation of human keratinocytes in vitro by the E6 and E7 open reading frames of human papillomavirus type 18. J. Virol. 1990, 64, 519–526. [Google Scholar] [CrossRef] [Green Version]
- Hawley-Nelson, P.; Vousden, K.H.; Hubbert, N.L.; Lowy, D.R.; Schiller, J.T. HPV16 E6 and E7 proteins cooperate to immortalize human foreskin keratinocytes. EMBO J. 1989, 8, 3905–3910. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.S.; Vass, W.C.; Lowy, D.R.; Schiller, J.T. In vitro biological activities of the E6 and E7 genes vary among human papillomaviruses of different oncogenic potential. J. Virol. 1991, 65, 292–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halbert, C.L.; Demers, G.W.; Galloway, D.A. The E6 and E7 genes of human papillomavirus type 6 have weak immortalizing activity in human epithelial cells. J. Virol. 1992, 66, 2125–2134. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Ghai, J.; Ostrow, R.S.; McGlennen, R.C.; Faras, A.J. The E6 gene of human papillomavirus type 16 is sufficient for transformation of baby rat kidney cells in cotransfection with activated Ha-ras. Virology 1994, 201, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Band, V.; De Caprio, J.A.; Delmolino, L.; Kulesa, V.; Sager, R. Loss of p53 protein in human papillomavirus type 16 E6-immortalized human mammary epithelial cells. J. Virol. 1991, 65, 6671–6676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halbert, C.L.; Demers, G.W.; Galloway, D.A. The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. J. Virol. 1991, 65, 473–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Liem, A.; Miller, J.A.; Lambert, P.F. Human papillomavirus types 16 E6 and E7 contribute differently to carcinogenesis. Virology 2000, 267, 141–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiMaio, D.; Guralski, D.; Schiller, J.T. Translation of open reading frame E5 of bovine papillomavirus is required for its transforming activity. Proc. Natl. Acad. Sci. USA 1986, 83, 1797–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.L.; Mounts, P. Transforming activity of E5a protein of human papillomavirus type 6 in NIH 3T3 and C127 cells. J. Virol. 1990, 64, 3226–3233. [Google Scholar] [CrossRef] [Green Version]
- Pim, D.; Collins, M.; Banks, L. Human papillomavirus type 16 E5 gene stimulates the transforming activity of the epidermal growth factor receptor. Oncogene 1992, 7, 27–32. [Google Scholar] [PubMed]
- Leechanachai, P.; Banks, L.; Moreau, F.; Matlashewski, G. The E5 gene from human papillomavirus type 16 is an oncogene which enhances growth factor-mediated signal transduction to the nucleus. Oncogene 1992, 7, 19–25. [Google Scholar]
- Leptak, C.; Ramon y Cajal, S.; Kulke, R.; Horwitz, B.H.; Riese, D.J., 2nd; Dotto, G.P.; DiMaio, D. Tumorigenic transformation of murine keratinocytes by the E5 genes of bovine papillomavirus type 1 and human papillomavirus type 16. J. Virol. 1991, 65, 7078–7083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valle, G.F.; Banks, L. The human papillomavirus (HPV)-6 and HPV-16 E5 proteins co-operate with HPV-16 E7 in the transformation of primary rodent cells. J. Gen. Virol. 1995, 76 Pt 5, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Bouvard, V.; Matlashewski, G.; Gu, Z.M.; Storey, A.; Banks, L. The human papillomavirus type 16 E5 gene cooperates with the E7 gene to stimulate proliferation of primary cells and increases viral gene expression. Virology 1994, 203, 73–80. [Google Scholar] [CrossRef]
- Stoppler, M.C.; Straight, S.W.; Tsao, G.; Schlegel, R.; McCance, D.J. The E5 gene of HPV-16 enhances keratinocyte immortalization by full-length DNA. Virology 1996, 223, 251–254. [Google Scholar] [CrossRef] [Green Version]
- Barbaresi, S.; Cortese, M.S.; Quinn, J.; Ashrafi, G.H.; Graham, S.V.; Campo, M.S. Effects of human papillomavirus type 16 E5 deletion mutants on epithelial morphology: Functional characterization of each transmembrane domain. J. Gen. Virol. 2010, 91, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Kivi, N.; Greco, D.; Auvinen, P.; Auvinen, E. Genes involved in cell adhesion, cell motility and mitogenic signaling are altered due to HPV 16 E5 protein expression. Oncogene 2008, 27, 2532–2541. [Google Scholar] [CrossRef] [Green Version]
- Genther Williams, S.M.; Disbrow, G.L.; Schlegel, R.; Lee, D.; Threadgill, D.W.; Lambert, P.F. Requirement of epidermal growth factor receptor for hyperplasia induced by E5, a high-risk human papillomavirus oncogene. Cancer Res. 2005, 65, 6534–6542. [Google Scholar] [CrossRef] [Green Version]
- Maufort, J.P.; Shai, A.; Pitot, H.C.; Lambert, P.F. A role for HPV16 E5 in cervical carcinogenesis. Cancer Res. 2010, 70, 2924–2931. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.L.; Tsao, Y.P.; Liu, D.W.; Huang, S.J.; Lee, W.H.; Chen, S.L. The expression of HPV-16 E5 protein in squamous neoplastic changes in the uterine cervix. J. Biomed. Sci. 2001, 8, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Kell, B.; Jewers, R.J.; Cason, J.; Pakarian, F.; Kaye, J.N.; Best, J.M. Detection of E5 oncoprotein in human papillomavirus type 16-positive cervical scrapes using antibodies raised to synthetic peptides. J. Gen. Virol. 1994, 75 Pt 9, 2451–2456. [Google Scholar] [CrossRef] [PubMed]
- Sahab, Z.; Sudarshan, S.R.; Liu, X.; Zhang, Y.; Kirilyuk, A.; Kamonjoh, C.M.; Simic, V.; Dai, Y.; Byers, S.W.; Doorbar, J.; et al. Quantitative measurement of human papillomavirus type 16 e5 oncoprotein levels in epithelial cell lines by mass spectrometry. J. Virol. 2012, 86, 9465–9473. [Google Scholar] [CrossRef] [Green Version]
- Um, S.H.; Mundi, N.; Yoo, J.; Palma, D.A.; Fung, K.; MacNeil, D.; Wehrli, B.; Mymryk, J.S.; Barrett, J.W.; Nichols, A.C. Variable expression of the forgotten oncogene E5 in HPV-positive oropharyngeal cancer. J. Clin. Virol. 2014, 61, 94–100. [Google Scholar] [CrossRef]
- Taberna, M.; Torres, M.; Alejo, M.; Mena, M.; Tous, S.; Marquez, S.; Pavon, M.A.; Leon, X.; Garcia, J.; Guix, M.; et al. The Use of HPV16-E5, EGFR, and pEGFR as Prognostic Biomarkers for Oropharyngeal Cancer Patients. Front. Oncol. 2018, 8, 589. [Google Scholar] [CrossRef] [PubMed]
- Nomine, Y.; Charbonnier, S.; Ristriani, T.; Stier, G.; Masson, M.; Cavusoglu, N.; Van Dorsselaer, A.; Weiss, E.; Kieffer, B.; Trave, G. Domain substructure of HPV E6 oncoprotein: Biophysical characterization of the E6 C-terminal DNA-binding domain. Biochemistry 2003, 42, 4909–4917. [Google Scholar] [CrossRef]
- Defeo-Jones, D.; Huang, P.S.; Jones, R.E.; Haskell, K.M.; Vuocolo, G.A.; Hanobik, M.G.; Huber, H.E.; Oliff, A. Cloning of cDNAs for cellular proteins that bind to the retinoblastoma gene product. Nature 1991, 352, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Roman, A.; Munger, K. The papillomavirus E7 proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vande Pol, S.B.; Klingelhutz, A.J. Papillomavirus E6 oncoproteins. Virology 2013, 445, 115–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiMaio, D.; Petti, L.M. The E5 proteins. Virology 2013, 445, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, E.; Olson, E. Cell differentiation. Curr. Opin. Cell Biol. 1996, 8, 823–825. [Google Scholar] [CrossRef]
- Goodwin, E.C.; DiMaio, D. Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc. Natl. Acad. Sci. USA 2000, 97, 12513–12518. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, E.C.; Yang, E.; Lee, C.J.; Lee, H.W.; DiMaio, D.; Hwang, E.S. Rapid induction of senescence in human cervical carcinoma cells. Proc. Natl. Acad. Sci. USA 2000, 97, 10978–10983. [Google Scholar] [CrossRef] [Green Version]
- Dyson, N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998, 12, 2245–2262. [Google Scholar] [CrossRef] [Green Version]
- Frolov, M.V.; Dyson, N.J. Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J. Cell Sci. 2004, 117, 2173–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyson, N.; Guida, P.; Munger, K.; Harlow, E. Homologous sequences in adenovirus E1A and human papillomavirus E7 proteins mediate interaction with the same set of cellular proteins. J. Virol. 1992, 66, 6893–6902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, S.N.; Wazer, D.E.; Band, V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996, 56, 4620–4624. [Google Scholar] [PubMed]
- Jones, D.L.; Thompson, D.A.; Munger, K. Destabilization of the RB tumor suppressor protein and stabilization of p53 contribute to HPV type 16 E7-induced apoptosis. Virology 1997, 239, 97–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, K.; Zhou, X.; Hayakawa, H.; Cho, J.Y.; Libermann, T.A.; Jin, J.; Harper, J.W.; Munger, K. Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J. Virol. 2007, 81, 9737–9747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gage, J.R.; Meyers, C.; Wettstein, F.O. The E7 proteins of the nononcogenic human papillomavirus type 6b (HPV-6b) and of the oncogenic HPV-16 differ in retinoblastoma protein binding and other properties. J. Virol. 1990, 64, 723–730. [Google Scholar] [CrossRef] [Green Version]
- Demers, G.W.; Foster, S.A.; Halbert, C.L.; Galloway, D.A. Growth arrest by induction of p53 in DNA damaged keratinocytes is bypassed by human papillomavirus 16 E7. Proc. Natl. Acad. Sci. USA 1994, 91, 4382–4386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, S.L.; Stremlau, M.; He, X.; Basile, J.R.; Munger, K. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. J. Virol. 2001, 75, 7583–7591. [Google Scholar] [CrossRef] [Green Version]
- Helt, A.M.; Galloway, D.A. Destabilization of the retinoblastoma tumor suppressor by human papillomavirus type 16 E7 is not sufficient to overcome cell cycle arrest in human keratinocytes. J. Virol. 2001, 75, 6737–6747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.L.; Munger, K. Analysis of the p53-mediated G1 growth arrest pathway in cells expressing the human papillomavirus type 16 E7 oncoprotein. J. Virol. 1997, 71, 2905–2912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Chen, W.; Roman, A. The E7 proteins of low- and high-risk human papillomaviruses share the ability to target the pRB family member p130 for degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 437–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrow-Laing, L.; Chen, W.; Roman, A. Low- and high-risk human papillomavirus E7 proteins regulate p130 differently. Virology 2010, 400, 233–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roman, A. The human papillomavirus E7 protein shines a spotlight on the pRB family member, p130. Cell Cycle 2006, 5, 567–568. [Google Scholar] [CrossRef] [PubMed]
- Zerfass, K.; Schulze, A.; Spitkovsky, D.; Friedman, V.; Henglein, B.; Jansen-Durr, P. Sequential activation of cyclin E and cyclin A gene expression by human papillomavirus type 16 E7 through sequences necessary for transformation. J. Virol. 1995, 69, 6389–6399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, C.L.; Munger, K. Direct association of the HPV16 E7 oncoprotein with cyclin A/CDK2 and cyclin E/CDK2 complexes. Virology 2008, 380, 21–25. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Staples, D.; Smith, C.; Fisher, C. Direct activation of cyclin-dependent kinase 2 by human papillomavirus E7. J. Virol. 2003, 77, 10566–10574. [Google Scholar] [CrossRef] [Green Version]
- Tommasino, M.; Adamczewski, J.P.; Carlotti, F.; Barth, C.F.; Manetti, R.; Contorni, M.; Cavalieri, F.; Hunt, T.; Crawford, L. HPV16 E7 protein associates with the protein kinase p33CDK2 and cyclin A. Oncogene 1993, 8, 195–202. [Google Scholar]
- Katich, S.C.; Zerfass-Thome, K.; Hoffmann, I. Regulation of the Cdc25A gene by the human papillomavirus Type 16 E7 oncogene. Oncogene 2001, 20, 543–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Zhou, Y.; Chen, J.J. Role of Cdc6 in re-replication in cells expressing human papillomavirus E7 oncogene. Carcinogenesis 2016, 37, 799–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kan, Q.; Jinno, S.; Yamamoto, H.; Kobayashi, K.; Okayama, H. ATP-dependent activation of p21WAF1/CIP1-associated Cdk2 by Cdc6. Proc. Natl. Acad. Sci. USA 2008, 105, 4757–4762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uranbileg, B.; Yamamoto, H.; Park, J.H.; Mohanty, A.R.; Arakawa-Takeuchi, S.; Jinno, S.; Okayama, H. Cdc6 protein activates p27KIP1-bound Cdk2 protein only after the bound p27 protein undergoes C-terminal phosphorylation. J. Biol. Chem. 2012, 287, 6275–6283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Zhang, Q.; Qiao, L.; Fan, X.; Zhang, W.; Zhao, W.; Chen, J.J. Cdc6 contributes to abrogating the G1 checkpoint under hypoxic conditions in HPV E7 expressing cells. Sci. Rep. 2017, 7, 2927. [Google Scholar] [CrossRef] [PubMed]
- Funk, J.O.; Waga, S.; Harry, J.B.; Espling, E.; Stillman, B.; Galloway, D.A. Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genes Dev. 1997, 11, 2090–2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.L.; Alani, R.M.; Munger, K. The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21Cip1-mediated inhibition of cdk2. Genes Dev. 1997, 11, 2101–2111. [Google Scholar] [CrossRef] [Green Version]
- Zerfass-Thome, K.; Zwerschke, W.; Mannhardt, B.; Tindle, R.; Botz, J.W.; Jansen-Durr, P. Inactivation of the cdk inhibitor p27KIP1 by the human papillomavirus type 16 E7 oncoprotein. Oncogene 1996, 13, 2323–2330. [Google Scholar]
- Firpo, E.J.; Koff, A.; Solomon, M.J.; Roberts, J.M. Inactivation of a Cdk2 inhibitor during interleukin 2-induced proliferation of human T lymphocytes. Mol. Cell. Biol. 1994, 14, 4889–4901. [Google Scholar] [CrossRef] [Green Version]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Assoian, R.K. Anchorage-dependent cell cycle progression. J. Cell Biol. 1997, 136, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Fang, F.; Orend, G.; Watanabe, N.; Hunter, T.; Ruoslahti, E. Dependence of cyclin E-CDK2 kinase activity on cell anchorage. Science 1996, 271, 499–502. [Google Scholar] [CrossRef]
- Jian, Y.; Schmidt-Grimminger, D.C.; Chien, W.M.; Wu, X.; Broker, T.R.; Chow, L.T. Post-transcriptional induction of p21cip1 protein by human papillomavirus E7 inhibits unscheduled DNA synthesis reactivated in differentiated keratinocytes. Oncogene 1998, 17, 2027–2038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.L.; Thompson, D.A.; Suh-Burgmann, E.; Grace, M.; Munger, K. Expression of the HPV E7 oncoprotein mimics but does not evoke a p53-dependent cellular DNA damage response pathway. Virology 1999, 258, 406–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noya, F.; Chien, W.M.; Broker, T.R.; Chow, L.T. p21cip1 Degradation in differentiated keratinocytes is abrogated by costabilization with cyclin E induced by human papillomavirus E7. J. Virol. 2001, 75, 6121–6134. [Google Scholar] [CrossRef] [Green Version]
- Ruesch, M.N.; Laimins, L.A. Initiation of DNA synthesis by human papillomavirus E7 oncoproteins is resistant to p21-mediated inhibition of cyclin E-cdk2 activity. J. Virol. 1997, 71, 5570–5578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helt, A.M.; Funk, J.O.; Galloway, D.A. Inactivation of both the retinoblastoma tumor suppressor and p21 by the human papillomavirus type 16 E7 oncoprotein is necessary to inhibit cell cycle arrest in human epithelial cells. J. Virol. 2002, 76, 10559–10568. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.G.; Lee, D.; Kim, J.; Seo, T.; Choe, J. Human papillomavirus type 16 E7 binds to E2F1 and activates E2F1-driven transcription in a retinoblastoma protein-independent manner. J. Biol. Chem. 2002, 277, 2923–2930. [Google Scholar] [CrossRef] [Green Version]
- Lyons, T.E.; Salih, M.; Tuana, B.S. Activating E2Fs mediate transcriptional regulation of human E2F6 repressor. Am. J. Physiol. Cell Physiol. 2006, 290, C189–C199. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin-Drubin, M.E.; Huh, K.W.; Munger, K. Human papillomavirus type 16 E7 oncoprotein associates with E2F6. J. Virol. 2008, 82, 8695–8705. [Google Scholar] [CrossRef] [Green Version]
- Evan, G.I.; Vousden, K.H. Proliferation, cell cycle and apoptosis in cancer. Nature 2001, 411, 342–348. [Google Scholar] [CrossRef]
- Werness, B.A.; Levine, A.J.; Howley, P.M. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 1990, 248, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Murray-Zmijewski, F.; Slee, E.A.; Lu, X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat. Rev. Mol. Cell Biol. 2008, 9, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Mietz, J.A.; Unger, T.; Huibregtse, J.M.; Howley, P.M. The transcriptional transactivation function of wild-type p53 is inhibited by SV40 large T-antigen and by HPV-16 E6 oncoprotein. EMBO J. 1992, 11, 5013–5020. [Google Scholar] [CrossRef]
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. Localization of the E6-AP regions that direct human papillomavirus E6 binding, association with p53, and ubiquitination of associated proteins. Mol. Cell. Biol. 1993, 13, 4918–4927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.J.; Hong, Y.; Rustamzadeh, E.; Baleja, J.D.; Androphy, E.J. Identification of an alpha helical motif sufficient for association with papillomavirus E6. J. Biol. Chem. 1998, 273, 13537–13544. [Google Scholar] [CrossRef] [Green Version]
- Kuballa, P.; Matentzoglu, K.; Scheffner, M. The role of the ubiquitin ligase E6-AP in human papillomavirus E6-mediated degradation of PDZ domain-containing proteins. J. Biol. Chem. 2007, 282, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Brimer, N.; Lyons, C.; Vande Pol, S.B. Association of E6AP (UBE3A) with human papillomavirus type 11 E6 protein. Virology 2007, 358, 303–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 1991, 10, 4129–4135. [Google Scholar] [CrossRef] [PubMed]
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. Cloning and expression of the cDNA for E6-AP, a protein that mediates the interaction of the human papillomavirus E6 oncoprotein with p53. Mol. Cell. Biol. 1993, 13, 775–784. [Google Scholar] [CrossRef] [Green Version]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Hengstermann, A.; D’Silva, M.A.; Kuballa, P.; Butz, K.; Hoppe-Seyler, F.; Scheffner, M. Growth suppression induced by downregulation of E6-AP expression in human papillomavirus-positive cancer cell lines depends on p53. J. Virol. 2005, 79, 9296–9300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomaic, V.; Pim, D.; Banks, L. The stability of the human papillomavirus E6 oncoprotein is E6AP dependent. Virology 2009, 393, 7–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Chen, J.J.; Gao, Q.; Dalal, S.; Hong, Y.; Mansur, C.P.; Band, V.; Androphy, E.J. Multiple functions of human papillomavirus type 16 E6 contribute to the immortalization of mammary epithelial cells. J. Virol. 1999, 73, 7297–7307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, M.; Song, S.; Liem, A.; Androphy, E.; Liu, Y.; Lambert, P.F. A mutant of human papillomavirus type 16 E6 deficient in binding alpha-helix partners displays reduced oncogenic potential in vivo. J. Virol. 2002, 76, 13039–13048. [Google Scholar] [CrossRef] [Green Version]
- Vats, A.; Thatte, J.; Banks, L. Identification of E6AP-independent degradation targets of HPV E6. J. Gen. Virol. 2019, 100, 1674–1679. [Google Scholar] [CrossRef]
- Lechner, M.S.; Laimins, L.A. Inhibition of p53 DNA binding by human papillomavirus E6 proteins. J. Virol. 1994, 68, 4262–4273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.; Massimi, P.; Jenkins, J.; Banks, L. HPV-18 E6 mediated inhibition of p53 DNA binding activity is independent of E6 induced degradation. Oncogene 1995, 10, 261–268. [Google Scholar]
- Mantovani, F.; Banks, L. Inhibition of E6 induced degradation of p53 is not sufficient for stabilization of p53 protein in cervical tumour derived cell lines. Oncogene 1999, 18, 3309–3315. [Google Scholar] [CrossRef] [Green Version]
- Patel, D.; Huang, S.M.; Baglia, L.A.; McCance, D.J. The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. EMBO J. 1999, 18, 5061–5072. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, H.; Degenkolbe, R.; Bernard, H.U.; O’Connor, M.J. The human papillomavirus type 16 E6 oncoprotein can down-regulate p53 activity by targeting the transcriptional coactivator CBP/p300. J. Virol. 1999, 73, 6209–6219. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.C.; Chiang, C.M. E6 oncoprotein represses p53-dependent gene activation via inhibition of protein acetylation independently of inducing p53 degradation. Mol. Cell 2005, 17, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Chand, V.; John, R.; Jaiswal, N.; Johar, S.S.; Nag, A. High-risk HPV16E6 stimulates hADA3 degradation by enhancing its SUMOylation. Carcinogenesis 2014, 35, 1830–1839. [Google Scholar] [CrossRef] [PubMed]
- Sekaric, P.; Shamanin, V.A.; Luo, J.; Androphy, E.J. hAda3 regulates p14ARF-induced p53 acetylation and senescence. Oncogene 2007, 26, 6261–6268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Zhao, Y.; Meng, G.; Zeng, M.; Srinivasan, S.; Delmolino, L.M.; Gao, Q.; Dimri, G.; Weber, G.F.; Wazer, D.E.; et al. Human papillomavirus oncoprotein E6 inactivates the transcriptional coactivator human ADA3. Mol. Cell. Biol. 2002, 22, 5801–5812. [Google Scholar] [CrossRef] [Green Version]
- Jha, S.; Vande Pol, S.; Banerjee, N.S.; Dutta, A.B.; Chow, L.T.; Dutta, A. Destabilization of TIP60 by human papillomavirus E6 results in attenuation of TIP60-dependent transcriptional regulation and apoptotic pathway. Mol. Cell 2010, 38, 700–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.; Banks, L. Inhibition of Bak-induced apoptosis by HPV-18 E6. Oncogene 1998, 17, 2943–2954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.; Banks, L. Human papillomavirus (HPV) E6 interactions with Bak are conserved amongst E6 proteins from high and low risk HPV types. J. Gen. Virol. 1999, 80 Pt 6, 1513–1517. [Google Scholar] [CrossRef] [Green Version]
- Alfandari, J.; Shnitman Magal, S.; Jackman, A.; Schlegel, R.; Gonen, P.; Sherman, L. HPV16 E6 oncoprotein inhibits apoptosis induced during serum-calcium differentiation of foreskin human keratinocytes. Virology 1999, 257, 383–396. [Google Scholar] [CrossRef] [Green Version]
- Vogt, M.; Butz, K.; Dymalla, S.; Semzow, J.; Hoppe-Seyler, F. Inhibition of Bax activity is crucial for the antiapoptotic function of the human papillomavirus E6 oncoprotein. Oncogene 2006, 25, 4009–4015. [Google Scholar] [CrossRef] [Green Version]
- Filippova, M.; Parkhurst, L.; Duerksen-Hughes, P.J. The Human Papillomavirus 16 E6 Protein Binds to Fas-associated Death Domain and Protects Cells from Fas-triggered Apoptosis. J. Biol. Chem. 2004, 279, 25729–25744. [Google Scholar] [CrossRef] [Green Version]
- Filippova, M.; Johnson, M.M.; Bautista, M.; Filippov, V.; Fodor, N.; Tungteakkhun, S.S.; Williams, K.; Duerksen-Hughes, P.J. The Large and Small Isoforms of Human Papillomavirus Type 16 E6 Bind to and Differentially Affect Procaspase 8 Stability and Activity. J. Virol. 2007, 81, 4116–4129. [Google Scholar] [CrossRef] [Green Version]
- Nees, M.; Geoghegan, J.M.; Munson, P.; Prabhu, V.; Liu, Y.; Androphy, E.; Woodworth, C.D. Human papillomavirus type 16 E6 and E7 proteins inhibit differentiation-dependent expression of transforming growth factor-beta2 in cervical keratinocytes. Cancer Res. 2000, 60, 4289–4298. [Google Scholar] [PubMed]
- Oh, J.M.; Kim, S.H.; Cho, E.A.; Song, Y.S.; Kim, W.H.; Juhnn, Y.S. Human papillomavirus type 16 E5 protein inhibits hydrogen-peroxide-induced apoptosis by stimulating ubiquitin-proteasome-mediated degradation of Bax in human cervical cancer cells. Carcinogenesis 2010, 31, 402–410. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Spandau, D.F.; Roman, A. E5 protein of human papillomavirus type 16 protects human foreskin keratinocytes from UV B-irradiation-induced apoptosis. J. Virol. 2002, 76, 220–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabsch, K.; Mossadegh, N.; Kohl, A.; Komposch, G.; Schenkel, J.; Alonso, A.; Tomakidi, P. The HPV-16 E5 protein inhibits TRAIL- and FasL-mediated apoptosis in human keratinocyte raft cultures. Intervirology 2004, 47, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.A.; Zacny, V.; Belinsky, G.S.; Classon, M.; Jones, D.L.; Schlegel, R.; Munger, K. The HPV E7 oncoprotein inhibits tumor necrosis factor alpha-mediated apoptosis in normal human fibroblasts. Oncogene 2001, 20, 3629–3640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, S.M.; Screaton, R.A. Anoikis mechanisms. Curr. Opin. Cell Biol. 2001, 13, 555–562. [Google Scholar] [CrossRef]
- Huh, K.W.; DeMasi, J.; Ogawa, H.; Nakatani, Y.; Howley, P.M.; Munger, K. Association of the human papillomavirus type 16 E7 oncoprotein with the 600-kDa retinoblastoma protein-associated factor, p600. Proc. Natl. Acad. Sci. USA 2005, 102, 11492–11497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeMasi, J.; Huh, K.W.; Nakatani, Y.; Munger, K.; Howley, P.M. Bovine papillomavirus E7 transformation function correlates with cellular p600 protein binding. Proc. Natl. Acad. Sci. USA 2005, 102, 11486–11491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulliver, G.A.; Herber, R.L.; Liem, A.; Lambert, P.F. Both conserved region 1 (CR1) and CR2 of the human papillomavirus type 16 E7 oncogene are required for induction of epidermal hyperplasia and tumor formation in transgenic mice. J. Virol. 1997, 71, 5905–5914. [Google Scholar] [CrossRef] [Green Version]
- DeMasi, J.; Chao, M.C.; Kumar, A.S.; Howley, P.M. Bovine papillomavirus E7 oncoprotein inhibits anoikis. J. Virol. 2007, 81, 9419–9425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szalmas, A.; Tomaic, V.; Basukala, O.; Massimi, P.; Mittal, S.; Konya, J.; Banks, L. The PTPN14 Tumor Suppressor Is a Degradation Target of Human Papillomavirus E7. J. Virol. 2017, 91, e00057-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E.A.; Munger, K.; Howley, P.M. High-Risk Human Papillomavirus E7 Proteins Target PTPN14 for Degradation. mBio 2016, 7, e01530-16. [Google Scholar] [CrossRef] [Green Version]
- Hatterschide, J.; Bohidar, A.E.; Grace, M.; Nulton, T.J.; Kim, H.W.; Windle, B.; Morgan, I.M.; Munger, K.; White, E.A. PTPN14 degradation by high-risk human papillomavirus E7 limits keratinocyte differentiation and contributes to HPV-mediated oncogenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 7033–7042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatterschide, J.; Brantly, A.C.; Grace, M.; Munger, K.; White, E.A. A Conserved Amino Acid in the C Terminus of Human Papillomavirus E7 Mediates Binding to PTPN14 and Repression of Epithelial Differentiation. J. Virol. 2020, 94, e01024-20. [Google Scholar] [CrossRef]
- Straight, S.W.; Hinkle, P.M.; Jewers, R.J.; McCance, D.J. The E5 oncoprotein of human papillomavirus type 16 transforms fibroblasts and effects the downregulation of the epidermal growth factor receptor in keratinocytes. J. Virol. 1993, 67, 4521–4532. [Google Scholar] [CrossRef] [Green Version]
- Tomakidi, P.; Cheng, H.; Kohl, A.; Komposch, G.; Alonso, A. Modulation of the epidermal growth factor receptor by the human papillomavirus type 16 E5 protein in raft cultures of human keratinocytes. Eur. J. Cell Biol. 2000, 79, 407–412. [Google Scholar] [CrossRef]
- Crusius, K.; Auvinen, E.; Steuer, B.; Gaissert, H.; Alonso, A. The human papillomavirus type 16 E5-protein modulates ligand-dependent activation of the EGF receptor family in the human epithelial cell line HaCaT. Exp. Cell Res. 1998, 241, 76–83. [Google Scholar] [CrossRef]
- Straight, S.W.; Herman, B.; McCance, D.J. The E5 oncoprotein of human papillomavirus type 16 inhibits the acidification of endosomes in human keratinocytes. J. Virol. 1995, 69, 3185–3192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, M.I.; Finbow, M.E.; Alonso, A. Binding of human papillomavirus 16 E5 to the 16 kDa subunit c (proteolipid) of the vacuolar H+-ATPase can be dissociated from the E5-mediated epidermal growth factor receptor overactivation. Oncogene 2000, 19, 3727–3732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suprynowicz, F.A.; Krawczyk, E.; Hebert, J.D.; Sudarshan, S.R.; Simic, V.; Kamonjoh, C.M.; Schlegel, R. The human papillomavirus type 16 E5 oncoprotein inhibits epidermal growth factor trafficking independently of endosome acidification. J. Virol. 2010, 84, 10619–10629. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Srirangam, A.; Potter, D.A.; Roman, A. HPV16 E5 protein disrupts the c-Cbl-EGFR interaction and EGFR ubiquitination in human foreskin keratinocytes. Oncogene 2005, 24, 2585–2588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venuti, A.; Salani, D.; Poggiali, F.; Manni, V.; Bagnato, A. The E5 oncoprotein of human papillomavirus type 16 enhances endothelin-1-induced keratinocyte growth. Virology 1998, 248, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Simeone, P.; Teson, M.; Latini, A.; Carducci, M.; Venuti, A. Endothelin-1 could be one of the targets of psoriasis therapy. Br. J. Dermatol. 2004, 151, 1273–1275. [Google Scholar] [CrossRef] [PubMed]
- Bagnato, A.; Loizidou, M.; Pflug, B.R.; Curwen, J.; Growcott, J. Role of the endothelin axis and its antagonists in the treatment of cancer. Br. J. Pharm. 2011, 163, 220–233. [Google Scholar] [CrossRef] [Green Version]
- Scott, M.L.; Coleman, D.T.; Kelly, K.C.; Carroll, J.L.; Woodby, B.; Songock, W.K.; Cardelli, J.A.; Bodily, J.M. Human papillomavirus type 16 E5-mediated upregulation of Met in human keratinocytes. Virology 2018, 519, 1–11. [Google Scholar] [CrossRef]
- Ranieri, D.; Belleudi, F.; Magenta, A.; Torrisi, M.R. HPV16 E5 expression induces switching from FGFR2b to FGFR2c and epithelial-mesenchymal transition. Int. J. Cancer 2015, 137, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belleudi, F.; Leone, L.; Purpura, V.; Cannella, F.; Scrofani, C.; Torrisi, M.R. HPV16 E5 affects the KGFR/FGFR2b-mediated epithelial growth through alteration of the receptor expression, signaling and endocytic traffic. Oncogene 2011, 30, 4963–4976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crusius, K.; Rodriguez, I.; Alonso, A. The human papillomavirus type 16 E5 protein modulates ERK1/2 and p38 MAP kinase activation by an EGFR-independent process in stressed human keratinocytes. Virus Genes 2000, 20, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Crusius, K.; Auvinen, E.; Alonso, A. Enhancement of EGF- and PMA-mediated MAP kinase activation in cells expressing the human papillomavirus type 16 E5 protein. Oncogene 1997, 15, 1437–1444. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Matlashewski, G. Effect of human papillomavirus type 16 oncogenes on MAP kinase activity. J. Virol. 1995, 69, 8051–8056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.L.; Huang, C.H.; Tsai, T.C.; Lu, K.Y.; Tsao, Y.P. The regulation mechanism of c-jun and junB by human papillomavirus type 16 E5 oncoprotein. Arch. Virol. 1996, 141, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.L.; Lin, Y.K.; Li, L.Y.; Tsao, Y.P.; Lo, H.Y.; Wang, W.B.; Tsai, T.C. E5 proteins of human papillomavirus types 11 and 16 transactivate the c-fos promoter through the NF1 binding element. J. Virol. 1996, 70, 8558–8563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsao, Y.P.; Li, L.Y.; Tsai, T.C.; Chen, S.L. Human papillomavirus type 11 and 16 E5 represses p21(WafI/SdiI/CipI) gene expression in fibroblasts and keratinocytes. J. Virol. 1996, 70, 7535–7539. [Google Scholar] [CrossRef] [Green Version]
- Pedroza-Saavedra, A.; Lam, E.W.; Esquivel-Guadarrama, F.; Gutierrez-Xicotencatl, L. The human papillomavirus type 16 E5 oncoprotein synergizes with EGF-receptor signaling to enhance cell cycle progression and the down-regulation of p27(Kip1). Virology 2010, 400, 44–52. [Google Scholar] [CrossRef] [Green Version]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT signaling pathway and cancer: An updated review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef]
- Contreras-Paredes, A.; De la Cruz-Hernandez, E.; Martinez-Ramirez, I.; Duenas-Gonzalez, A.; Lizano, M. E6 variants of human papillomavirus 18 differentially modulate the protein kinase B/phosphatidylinositol 3-kinase (akt/PI3K) signaling pathway. Virology 2009, 383, 78–85. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Hu, X.; Li, Y.; Zheng, L.; Zhou, Y.; Jiang, H.; Ning, T.; Basang, Z.; Zhang, C.; Ke, Y. Human papillomavirus 16 E6 oncoprotein interferences with insulin signaling pathway by binding to tuberin. J. Biol. Chem. 2004, 279, 35664–35670. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Ding, H.; Lu, Z.; Li, Y.; Pan, Y.; Ning, T.; Ke, Y. E3 ubiquitin ligase E6AP-mediated TSC2 turnover in the presence and absence of HPV16 E6. Genes Cells 2008, 13, 285–294. [Google Scholar] [CrossRef]
- Spangle, J.M.; Munger, K. The human papillomavirus type 16 E6 oncoprotein activates mTORC1 signaling and increases protein synthesis. J. Virol. 2010, 84, 9398–9407. [Google Scholar] [CrossRef] [Green Version]
- Spangle, J.M.; Munger, K. The HPV16 E6 oncoprotein causes prolonged receptor protein tyrosine kinase signaling and enhances internalization of phosphorylated receptor species. PLoS Pathog. 2013, 9, e1003237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichtig, H.; Gilboa, D.A.; Jackman, A.; Gonen, P.; Levav-Cohen, Y.; Haupt, Y.; Sherman, L. HPV16 E6 augments Wnt signaling in an E6AP-dependent manner. Virology 2010, 396, 47–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rampias, T.; Boutati, E.; Pectasides, E.; Sasaki, C.; Kountourakis, P.; Weinberger, P.; Psyrri, A. Activation of Wnt signaling pathway by human papillomavirus E6 and E7 oncogenes in HPV16-positive oropharyngeal squamous carcinoma cells. Mol. Cancer Res. 2010, 8, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Bonilla-Delgado, J.; Bulut, G.; Liu, X.; Cortes-Malagon, E.M.; Schlegel, R.; Flores-Maldonado, C.; Contreras, R.G.; Chung, S.H.; Lambert, P.F.; Uren, A.; et al. The E6 oncoprotein from HPV16 enhances the canonical Wnt/beta-catenin pathway in skin epidermis in vivo. Mol. Cancer Res. 2012, 10, 250–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vliet-Gregg, P.A.; Hamilton, J.R.; Katzenellenbogen, R.A. NFX1-123 and human papillomavirus 16E6 increase Notch expression in keratinocytes. J. Virol. 2013, 87, 13741–13750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weijzen, S.; Zlobin, A.; Braid, M.; Miele, L.; Kast, W.M. HPV16 E6 and E7 oncoproteins regulate Notch-1 expression and cooperate to induce transformation. J. Cell. Physiol. 2003, 194, 356–362. [Google Scholar] [CrossRef]
- Kranjec, C.; Holleywood, C.; Libert, D.; Griffin, H.; Mahmood, R.; Isaacson, E.; Doorbar, J. Modulation of basal cell fate during productive and transforming HPV-16 infection is mediated by progressive E6-driven depletion of Notch. J. Pathol. 2017, 242, 448–462. [Google Scholar] [CrossRef]
- White, E.A.; Howley, P.M. Proteomic approaches to the study of papillomavirus-host interactions. Virology 2013, 435, 57–69. [Google Scholar] [CrossRef] [Green Version]
- Brimer, N.; Drews, C.M.; Vande Pol, S.B. Association of papillomavirus E6 proteins with either MAML1 or E6AP clusters E6 proteins by structure, function, and evolutionary relatedness. PLoS Pathog. 2017, 13, e1006781. [Google Scholar] [CrossRef]
- Brimer, N.; Lyons, C.; Wallberg, A.E.; Vande Pol, S.B. Cutaneous papillomavirus E6 oncoproteins associate with MAML1 to repress transactivation and NOTCH signaling. Oncogene 2012, 31, 4639–4646. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.J.; White, E.A.; Sowa, M.E.; Harper, J.W.; Aster, J.C.; Howley, P.M. Cutaneous beta-human papillomavirus E6 proteins bind Mastermind-like coactivators and repress Notch signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E1473–E1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayflick, L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Klingelhutz, A.J.; Foster, S.A.; McDougall, J.K. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature 1996, 380, 79–82. [Google Scholar] [CrossRef]
- Klingelhutz, A.J.; Barber, S.A.; Smith, P.P.; Dyer, K.; McDougall, J.K. Restoration of telomeres in human papillomavirus-immortalized human anogenital epithelial cells. Mol. Cell. Biol. 1994, 14, 961–969. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.J.; Grandori, C.; Amacker, M.; Simon-Vermot, N.; Polack, A.; Lingner, J.; Dalla-Favera, R. Direct activation of TERT transcription by c-MYC. Nat. Genet. 1999, 21, 220–224. [Google Scholar] [CrossRef]
- Oh, S.T.; Kyo, S.; Laimins, L.A. Telomerase activation by human papillomavirus type 16 E6 protein: Induction of human telomerase reverse transcriptase expression through Myc and GC-rich Sp1 binding sites. J. Virol. 2001, 75, 5559–5566. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Yuan, H.; Fu, B.; Disbrow, G.L.; Apolinario, T.; Tomaic, V.; Kelley, M.L.; Baker, C.C.; Huibregtse, J.; Schlegel, R. The E6AP ubiquitin ligase is required for transactivation of the hTERT promoter by the human papillomavirus E6 oncoprotein. J. Biol. Chem. 2005, 280, 10807–10816. [Google Scholar] [CrossRef] [Green Version]
- McMurray, H.R.; McCance, D.J. Human papillomavirus type 16 E6 activates TERT gene transcription through induction of c-Myc and release of USF-mediated repression. J. Virol. 2003, 77, 9852–9861. [Google Scholar] [CrossRef] [Green Version]
- Takakura, M.; Kyo, S.; Kanaya, T.; Hirano, H.; Takeda, J.; Yutsudo, M.; Inoue, M. Cloning of human telomerase catalytic subunit (hTERT) gene promoter and identification of proximal core promoter sequences essential for transcriptional activation in immortalized and cancer cells. Cancer Res. 1999, 59, 551–557. [Google Scholar]
- James, M.A.; Lee, J.H.; Klingelhutz, A.J. HPV16-E6 associated hTERT promoter acetylation is E6AP dependent, increased in later passage cells and enhanced by loss of p300. Int. J. Cancer 2006, 119, 1878–1885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Luo, W.; Elzi, D.J.; Grandori, C.; Galloway, D.A. NFX1 interacts with mSin3A/histone deacetylase to repress hTERT transcription in keratinocytes. Mol. Cell. Biol. 2008, 28, 4819–4828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gewin, L.; Myers, H.; Kiyono, T.; Galloway, D.A. Identification of a novel telomerase repressor that interacts with the human papillomavirus type-16 E6/E6-AP complex. Genes Dev. 2004, 18, 2269–2282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katzenellenbogen, R.A.; Egelkrout, E.M.; Vliet-Gregg, P.; Gewin, L.C.; Gafken, P.R.; Galloway, D.A. NFX1-123 and poly(A) binding proteins synergistically augment activation of telomerase in human papillomavirus type 16 E6-expressing cells. J. Virol. 2007, 81, 3786–3796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Costa, Z.J.; Jolly, C.; Androphy, E.J.; Mercer, A.; Matthews, C.M.; Hibma, M.H. Transcriptional repression of E-cadherin by human papillomavirus type 16 E6. PLoS ONE 2012, 7, e48954. [Google Scholar] [CrossRef] [PubMed]
- Hellner, K.; Mar, J.; Fang, F.; Quackenbush, J.; Münger, K. HPV16 E7 oncogene expression in normal human epithelial cells causes molecular changes indicative of an epithelial to mesenchymal transition. Virology 2009, 391, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Klein, T.; Bischoff, R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids 2011, 41, 271–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, K.; Pickard, A.; McDade, S.; McCance, D.J. p63 drives invasion in keratinocytes expressing HPV16 E6/E7 genes through regulation of Src-FAK signalling. Oncotarget 2017, 8, 16202–16219. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhang, X.; Huang, L.; Li, J.; Qu, S.; Pan, F. Matrix metalloproteinase-14 expression and its prognostic value in cervical carcinoma. Cell Biochem. Biophys. 2014, 70, 729–734. [Google Scholar] [CrossRef]
- Zhai, Y.; Hotary, K.B.; Nan, B.; Bosch, F.X.; Munoz, N.; Weiss, S.J.; Cho, K.R. Expression of membrane type 1 matrix metalloproteinase is associated with cervical carcinoma progression and invasion. Cancer Res. 2005, 65, 6543–6550. [Google Scholar] [CrossRef] [Green Version]
- Kaewprag, J.; Umnajvijit, W.; Ngamkham, J.; Ponglikitmongkol, M. HPV16 oncoproteins promote cervical cancer invasiveness by upregulating specific matrix metalloproteinases. PLoS ONE 2013, 8, e71611. [Google Scholar] [CrossRef]
- Zhu, D.; Ye, M.; Zhang, W. E6/E7 oncoproteins of high risk HPV-16 upregulate MT1-MMP, MMP-2 and MMP-9 and promote the migration of cervical cancer cells. Int. J. Clin. Exp. Pathol. 2015, 8, 4981–4989. [Google Scholar]
- Menges, C.W.; Baglia, L.A.; Lapoint, R.; McCance, D.J. Human papillomavirus type 16 E7 up-regulates AKT activity through the retinoblastoma protein. Cancer Res. 2006, 66, 5555–5559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pim, D.; Massimi, P.; Dilworth, S.M.; Banks, L. Activation of the protein kinase B pathway by the HPV-16 E7 oncoprotein occurs through a mechanism involving interaction with PP2A. Oncogene 2005, 24, 7830–7838. [Google Scholar] [CrossRef] [Green Version]
- Basukala, O.; Mittal, S.; Massimi, P.; Bestagno, M.; Banks, L. The HPV-18 E7 CKII phospho acceptor site is required for maintaining the transformed phenotype of cervical tumour-derived cells. PLoS Pathog. 2019, 15, e1007769. [Google Scholar] [CrossRef]
- Kiyono, T.; Hiraiwa, A.; Fujita, M.; Hayashi, Y.; Akiyama, T.; Ishibashi, M. Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the Drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. USA 1997, 94, 11612–11616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.S.; Weiss, R.S.; Javier, R.T. Binding of human virus oncoproteins to hDlg/SAP97, a mammalian homolog of the Drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. USA 1997, 94, 6670–6675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Songyang, Z.; Fanning, A.S.; Fu, C.; Xu, J.; Marfatia, S.M.; Chishti, A.H.; Crompton, A.; Chan, A.C.; Anderson, J.M.; Cantley, L.C. Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science 1997, 275, 73–77. [Google Scholar] [CrossRef]
- James, C.D.; Roberts, S. Viral Interactions with PDZ Domain-Containing Proteins-An Oncogenic Trait? Pathogens 2016, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, S.; Huibregtse, J.M. Human scribble (Vartul) is targeted for ubiquitin-mediated degradation by the high-risk papillomavirus E6 proteins and the E6AP ubiquitin-protein ligase. Mol. Cell. Biol. 2000, 20, 8244–8253. [Google Scholar] [CrossRef] [PubMed]
- Dobrosotskaya, I.; Guy, R.K.; James, G.L. MAGI-1, a Membrane-associated Guanylate Kinase with a Unique Arrangement of Protein-Protein Interaction Domains. J. Biol. Chem. 1997, 272, 31589–31597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ide, N.; Hata, Y.; Nishioka, H.; Hirao, K.; Yao, I.; Deguchi, M.; Mizoguchi, A.; Nishimori, H.; Tokino, T.; Nakamura, Y.; et al. Localization of membrane-associated guanylate kinase (MAGI)-1/BAI-associated protein (BAP) 1 at tight junctions of epithelial cells. Oncogene 1999, 18, 7810–7815. [Google Scholar] [CrossRef] [Green Version]
- Javier, R.T. Cell polarity proteins: Common targets for tumorigenic human viruses. Oncogene 2008, 27, 7031–7046. [Google Scholar] [CrossRef] [Green Version]
- Subbaiah, V.K.; Kranjec, C.; Thomas, M.; Banks, L. PDZ domains: The building blocks regulating tumorigenesis. Biochem. J. 2011, 439, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Glaunsinger, B.A.; Lee, S.S.; Thomas, M.; Banks, L.; Javier, R. Interactions of the PDZ-protein MAGI-1 with adenovirus E4-ORF1 and high-risk papillomavirus E6 oncoproteins. Oncogene 2000, 19, 5270–5280. [Google Scholar] [CrossRef] [Green Version]
- Gardiol, D.; Kuhne, C.; Glaunsinger, B.; Lee, S.S.; Javier, R.; Banks, L. Oncogenic human papillomavirus E6 proteins target the discs large tumour suppressor for proteasome-mediated degradation. Oncogene 1999, 18, 5487–5496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.; Narayan, N.; Pim, D.; Tomaic, V.; Massimi, P.; Nagasaka, K.; Kranjec, C.; Gammoh, N.; Banks, L. Human papillomaviruses, cervical cancer and cell polarity. Oncogene 2008, 27, 7018–7030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pim, D.; Bergant, M.; Boon, S.S.; Ganti, K.; Kranjec, C.; Massimi, P.; Subbaiah, V.K.; Thomas, M.; Tomaic, V.; Banks, L. Human papillomaviruses and the specificity of PDZ domain targeting. FEBS J. 2012, 279, 3530–3537. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Massimi, P.; Navarro, C.; Borg, J.P.; Banks, L. The hScrib/Dlg apico-basal control complex is differentially targeted by HPV-16 and HPV-18 E6 proteins. Oncogene 2005, 24, 6222–6230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimatsu, Y.; Nakahara, T.; Tanaka, K.; Inagawa, Y.; Narisawa-Saito, M.; Yugawa, T.; Ohno, S.I.; Fujita, M.; Nakagama, H.; Kiyono, T. Roles of the PDZ-binding motif of HPV 16 E6 protein in oncogenic transformation of human cervical keratinocytes. Cancer Sci. 2017, 108, 1303–1309. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Laimins, L.A. Role of the PDZ domain-binding motif of the oncoprotein E6 in the pathogenesis of human papillomavirus type 31. J. Virol. 2004, 78, 12366–12377. [Google Scholar] [CrossRef] [Green Version]
- Marsh, E.K.; Delury, C.P.; Davies, N.J.; Weston, C.J.; Miah, M.A.L.; Banks, L.; Parish, J.L.; Higgs, M.R.; Roberts, S. Mitotic control of human papillomavirus genome-containing cells is regulated by the function of the PDZ-binding motif of the E6 oncoprotein. Oncotarget 2017, 8, 19491–19506. [Google Scholar] [CrossRef]
- Nicolaides, L.; Davy, C.; Raj, K.; Kranjec, C.; Banks, L.; Doorbar, J. Stabilization of HPV16 E6 protein by PDZ proteins, and potential implications for genome maintenance. Virology 2011, 414, 137–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechler, T.; Fuchs, E. Asymmetric cell divisions promote stratification and differentiation of mammalian skin. Nature 2005, 437, 275–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulson, N.D.; Lechler, T. Robust control of mitotic spindle orientation in the developing epidermis. J. Cell Biol. 2010, 191, 915–922. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Munger, K. Human papillomavirus type 16 E7 oncoprotein inhibits the anaphase promoting complex/cyclosome activity by dysregulating EMI1 expression in mitosis. Virology 2013, 446, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Munger, K. Human papillomavirus type 16 E7 oncoprotein engages but does not abrogate the mitotic spindle assembly checkpoint. Virology 2012, 432, 120–126. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.; Du, Q.; Chen, X.; Zheng, Z.; Balsbaugh, J.L.; Maitra, S.; Shabanowitz, J.; Hunt, D.F.; Macara, I.G. Par3 controls epithelial spindle orientation by aPKC-mediated phosphorylation of apical Pins. Curr. Biol. 2010, 20, 1809–1818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, C.A.; Hirono, K.; Prehoda, K.E.; Doe, C.Q. Identification of an Aurora-A/PinsLINKER/Dlg spindle orientation pathway using induced cell polarity in S2 cells. Cell 2009, 138, 1150–1163. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Toussaint-Smith, E.; Donner, D.B.; Roman, A. Expression of human papillomavirus type 16 E6 and E7 oncoproteins in primary foreskin keratinocytes is sufficient to alter the expression of angiogenic factors. Oncogene 2004, 23, 2988–2995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravi, R.; Mookerjee, B.; Bhujwalla, Z.M.; Sutter, C.H.; Artemov, D.; Zeng, Q.; Dillehay, L.E.; Madan, A.; Semenza, G.L.; Bedi, A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000, 14, 34–44. [Google Scholar] [PubMed]
- Zou, Z.; Gao, C.; Nagaich, A.K.; Connell, T.; Saito, S.; Moul, J.W.; Seth, P.; Appella, E.; Srivastava, S. p53 regulates the expression of the tumor suppressor gene maspin. J. Biol. Chem. 2000, 275, 6051–6054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, D.; Tsiokas, L.; Sukhatme, V.P. Wild-type p53 and v-Src exert opposing influences on human vascular endothelial growth factor gene expression. Cancer Res. 1995, 55, 6161–6165. [Google Scholar] [PubMed]
- Chen, W.; Li, F.; Mead, L.; White, H.; Walker, J.; Ingram, D.A.; Roman, A. Human papillomavirus causes an angiogenic switch in keratinocytes which is sufficient to alter endothelial cell behavior. Virology 2007, 367, 168–174. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Ocejo, O.; Viloria-Petit, A.; Bequet-Romero, M.; Mukhopadhyay, D.; Rak, J.; Kerbel, R.S. Oncogenes and tumor angiogenesis: The HPV-16 E6 oncoprotein activates the vascular endothelial growth factor (VEGF) gene promoter in a p53 independent manner. Oncogene 2000, 19, 4611–4620. [Google Scholar] [CrossRef] [Green Version]
- Tischer, E.; Mitchell, R.; Hartman, T.; Silva, M.; Gospodarowicz, D.; Fiddes, J.C.; Abraham, J.A. The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J. Biol. Chem. 1991, 266, 11947–11954. [Google Scholar] [CrossRef]
- Antinore, M.J.; Birrer, M.J.; Patel, D.; Nader, L.; McCance, D.J. The human papillomavirus type 16 E7 gene product interacts with and trans-activates the AP1 family of transcription factors. EMBO J. 1996, 15, 1950–1960. [Google Scholar] [CrossRef]
- Wang, N.; Zhan, T.; Ke, T.; Huang, X.; Ke, D.; Wang, Q.; Li, H. Increased expression of RRM2 by human papillomavirus E7 oncoprotein promotes angiogenesis in cervical cancer. Br. J. Cancer 2014, 110, 1034–1044. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Cui, J. Human telomerase reverse transcriptase regulates vascular endothelial growth factor expression via human papillomavirus oncogene E7 in HPV-18-positive cervical cancer cells. Med Oncol. 2015, 32, 199. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- Zwerschke, W.; Mazurek, S.; Massimi, P.; Banks, L.; Eigenbrodt, E.; Jansen-Durr, P. Modulation of type M2 pyruvate kinase activity by the human papillomavirus type 16 E7 oncoprotein. Proc. Natl. Acad. Sci. USA 1999, 96, 1291–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganti, K.; Massimi, P.; Manzo-Merino, J.; Tomaic, V.; Pim, D.; Playford, M.P.; Lizano, M.; Roberts, S.; Kranjec, C.; Doorbar, J.; et al. Interaction of the Human Papillomavirus E6 Oncoprotein with Sorting Nexin 27 Modulates Endocytic Cargo Transport Pathways. PLoS Pathog. 2016, 12, e1005854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krupar, R.; Robold, K.; Gaag, D.; Spanier, G.; Kreutz, M.; Renner, K.; Hellerbrand, C.; Hofstaedter, F.; Bosserhoff, A.K. Immunologic and metabolic characteristics of HPV-negative and HPV-positive head and neck squamous cell carcinomas are strikingly different. Virchows Arch. Int. J. Pathol. 2014, 465, 299–312. [Google Scholar] [CrossRef] [PubMed]
- DiPaolo, J.A.; Woodworth, C.D.; Popescu, N.C.; Notario, V.; Doniger, J. Induction of human cervical squamous cell carcinoma by sequential transfection with human papillomavirus 16 DNA and viral Harvey ras. Oncogene 1989, 4, 395–399. [Google Scholar] [PubMed]
- Durst, M.; Gallahan, D.; Jay, G.; Rhim, J.S. Glucocorticoid-enhanced neoplastic transformation of human keratinocytes by human papillomavirus type 16 and an activated ras oncogene. Virology 1989, 173, 767–771. [Google Scholar] [CrossRef]
- Hurlin, P.J.; Kaur, P.; Smith, P.P.; Perez-Reyes, N.; Blanton, R.A.; McDougall, J.K. Progression of human papillomavirus type 18-immortalized human keratinocytes to a malignant phenotype. Proc. Natl. Acad. Sci. USA 1991, 88, 570–574. [Google Scholar] [CrossRef] [Green Version]
- Pei, X.F.; Meck, J.M.; Greenhalgh, D.; Schlegel, R. Cotransfection of HPV-18 and v-fos DNA induces tumorigenicity of primary human keratinocytes. Virology 1993, 196, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Duensing, S.; Lee, L.Y.; Duensing, A.; Basile, J.; Piboonniyom, S.; Gonzalez, S.; Crum, C.P.; Munger, K. The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc. Natl. Acad. Sci. USA 2000, 97, 10002–10007. [Google Scholar] [CrossRef] [Green Version]
- Duensing, S.; Munger, K. Human papillomavirus type 16 E7 oncoprotein can induce abnormal centrosome duplication through a mechanism independent of inactivation of retinoblastoma protein family members. J. Virol. 2003, 77, 12331–12335. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, C.L.; Eichwald, C.; Nibert, M.L.; Munger, K. Human papillomavirus type 16 E7 oncoprotein associates with the centrosomal component gamma-tubulin. J. Virol. 2007, 81, 13533–13543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korzeniewski, N.; Treat, B.; Duensing, S. The HPV-16 E7 oncoprotein induces centriole multiplication through deregulation of Polo-like kinase 4 expression. Mol. Cancer 2011, 10, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, C.L.; McLaughlin-Drubin, M.E.; Munger, K. Delocalization of the microtubule motor Dynein from mitotic spindles by the human papillomavirus E7 oncoprotein is not sufficient for induction of multipolar mitoses. Cancer Res. 2008, 68, 8715–8722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, C.L.; Munger, K. Human papillomavirus E7 protein deregulates mitosis via an association with nuclear mitotic apparatus protein 1. J. Virol. 2009, 83, 1700–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moody, C.A.; Laimins, L.A. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, N.S.; Wang, H.K.; Broker, T.R.; Chow, L.T. Human papillomavirus (HPV) E7 induces prolonged G2 following S phase reentry in differentiated human keratinocytes. J. Biol. Chem. 2011, 286, 15473–15482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spardy, N.; Covella, K.; Cha, E.; Hoskins, E.E.; Wells, S.I.; Duensing, A.; Duensing, S. Human papillomavirus 16 E7 oncoprotein attenuates DNA damage checkpoint control by increasing the proteolytic turnover of claspin. Cancer Res. 2009, 69, 7022–7029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, N.A.; Khanal, S.; Robinson, K.L.; Wendel, S.O.; Messer, J.J.; Galloway, D.A. High-Risk Alphapapillomavirus Oncogenes Impair the Homologous Recombination Pathway. J. Virol. 2017, 91, e01084-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitz, J.; Blanchet, S.A.; Gameiro, S.F.; Biquand, E.; Morgan, T.M.; Galloy, M.; Dessapt, J.; Lavoie, E.G.; Blondeau, A.; Smith, B.C.; et al. Human papillomavirus E7 oncoprotein targets RNF168 to hijack the host DNA damage response. Proc. Natl. Acad. Sci. USA 2019, 116, 19552–19562. [Google Scholar] [CrossRef] [Green Version]
- Kadaja, M.; Isok-Paas, H.; Laos, T.; Ustav, E.; Ustav, M. Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses. PLoS Pathog. 2009, 5, e1000397. [Google Scholar] [CrossRef]
- Burgers, W.A.; Blanchon, L.; Pradhan, S.; de Launoit, Y.; Kouzarides, T.; Fuks, F. Viral oncoproteins target the DNA methyltransferases. Oncogene 2007, 26, 1650–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avvakumov, N.; Torchia, J.; Mymryk, J.S. Interaction of the HPV E7 proteins with the pCAF acetyltransferase. Oncogene 2003, 22, 3833–3841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.M.; McCance, D.J. Down regulation of the interleukin-8 promoter by human papillomavirus type 16 E6 and E7 through effects on CREB binding protein/p300 and P/CAF. J. Virol. 2002, 76, 8710–8721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brehm, A.; Nielsen, S.J.; Miska, E.A.; McCance, D.J.; Reid, J.L.; Bannister, A.J.; Kouzarides, T. The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. EMBO J. 1999, 18, 2449–2458. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.X.; Westbrook, T.F.; McCance, D.J. Human papillomavirus type 16 E7 maintains elevated levels of the cdc25A tyrosine phosphatase during deregulation of cell cycle arrest. J. Virol. 2002, 76, 619–632. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Kim, E.J.; Kwon, H.J.; Hwang, E.S.; Namkoong, S.E.; Um, S.J. Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis. J. Biol. Chem. 2000, 275, 6764–6769. [Google Scholar] [CrossRef] [Green Version]
- Longworth, M.S.; Wilson, R.; Laimins, L.A. HPV31 E7 facilitates replication by activating E2F2 transcription through its interaction with HDACs. EMBO J. 2005, 24, 1821–1830. [Google Scholar] [CrossRef] [Green Version]
- Bodily, J.M.; Mehta, K.P.; Laimins, L.A. Human papillomavirus E7 enhances hypoxia-inducible factor 1-mediated transcription by inhibiting binding of histone deacetylases. Cancer Res. 2011, 71, 1187–1195. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin-Drubin, M.E.; Crum, C.P.; Munger, K. Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming. Proc. Natl. Acad. Sci. USA 2011, 108, 2130–2135. [Google Scholar] [CrossRef] [Green Version]
- Munger, K.; Gwin, T.K.; McLaughlin-Drubin, M.E. p16 in HPV-associated cancers. Oncotarget 2013, 4, 1864–1865. [Google Scholar] [CrossRef]
- McLaughlin-Drubin, M.E.; Park, D.; Munger, K. Tumor suppressor p16INK4A is necessary for survival of cervical carcinoma cell lines. Proc. Natl. Acad. Sci. USA 2013, 110, 16175–16180. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin-Drubin, M.E.; Munger, K. Biochemical and functional interactions of human papillomavirus proteins with polycomb group proteins. Viruses 2013, 5, 1231–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto, D.R.; Barton, C.; Munger, K.; McLaughlin-Drubin, M.E. KDM6A addiction of cervical carcinoma cell lines is triggered by E7 and mediated by p21CIP1 suppression of replication stress. PLoS Pathog. 2017, 13, e1006661. [Google Scholar] [CrossRef]
- Bracken, A.P.; Helin, K. Polycomb group proteins: Navigators of lineage pathways led astray in cancer. Nat. Rev. Cancer 2009, 9, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Holland, D.; Hoppe-Seyler, K.; Schuller, B.; Lohrey, C.; Maroldt, J.; Durst, M.; Hoppe-Seyler, F. Activation of the enhancer of zeste homologue 2 gene by the human papillomavirus E7 oncoprotein. Cancer Res. 2008, 68, 9964–9972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzmichev, A.; Margueron, R.; Vaquero, A.; Preissner, T.S.; Scher, M.; Kirmizis, A.; Ouyang, X.; Brockdorff, N.; Abate-Shen, C.; Farnham, P.; et al. Composition and histone substrates of polycomb repressive group complexes change during cellular differentiation. Proc. Natl. Acad. Sci. USA 2005, 102, 1859–1864. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Maruyama, R.; Yamamoto, E.; Kai, M. DNA methylation and microRNA dysregulation in cancer. Mol. Oncol. 2012, 6, 567–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melar-New, M.; Laimins, L.A. Human papillomaviruses modulate expression of microRNA 203 upon epithelial differentiation to control levels of p63 proteins. J. Virol. 2010, 84, 5212–5221. [Google Scholar] [CrossRef] [Green Version]
- Harden, M.E.; Prasad, N.; Griffiths, A.; Munger, K. Modulation of microRNA-mRNA Target Pairs by Human Papillomavirus 16 Oncoproteins. mBio 2017, 8, e02170-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yablonska, S.; Hoskins, E.E.; Wells, S.I.; Khan, S.A. Identification of miRNAs dysregulated in human foreskin keratinocytes (HFKs) expressing the human papillomavirus (HPV) Type 16 E6 and E7 oncoproteins. Microrna 2013, 2, 2–13. [Google Scholar] [CrossRef]
- Ashrafi, G.H.; Haghshenas, M.R.; Marchetti, B.; O’Brien, P.M.; Campo, M.S. E5 protein of human papillomavirus type 16 selectively downregulates surface HLA class I. Int. J. Cancer 2005, 113, 276–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campo, M.S.; Graham, S.V.; Cortese, M.S.; Ashrafi, G.H.; Araibi, E.H.; Dornan, E.S.; Miners, K.; Nunes, C.; Man, S. HPV-16 E5 down-regulates expression of surface HLA class I and reduces recognition by CD8 T cells. Virology 2010, 407, 137–142. [Google Scholar] [CrossRef] [Green Version]
- Cortese, M.S.; Ashrafi, G.H.; Campo, M.S. All 4 di-leucine motifs in the first hydrophobic domain of the E5 oncoprotein of human papillomavirus type 16 are essential for surface MHC class I downregulation activity and E5 endomembrane localization. Int. J. Cancer 2010, 126, 1675–1682. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.H.; Haghshenas, M.; Marchetti, B.; Campo, M.S. E5 protein of human papillomavirus 16 downregulates HLA class I and interacts with the heavy chain via its first hydrophobic domain. Int. J. Cancer 2006, 119, 2105–2112. [Google Scholar] [CrossRef]
- Gruener, M.; Bravo, I.G.; Momburg, F.; Alonso, A.; Tomakidi, P. The E5 protein of the human papillomavirus type 16 down-regulates HLA-I surface expression in calnexin-expressing but not in calnexin-deficient cells. Virol. J. 2007, 4, 116. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Li, P.; Wang, E.; Brahmi, Z.; Dunn, K.W.; Blum, J.S.; Roman, A. The E5 protein of human papillomavirus type 16 perturbs MHC class II antigen maturation in human foreskin keratinocytes treated with interferon-gamma. Virology 2003, 310, 100–108. [Google Scholar] [CrossRef] [Green Version]
- Miura, S.; Kawana, K.; Schust, D.J.; Fujii, T.; Yokoyama, T.; Iwasawa, Y.; Nagamatsu, T.; Adachi, K.; Tomio, A.; Tomio, K.; et al. CD1d, a sentinel molecule bridging innate and adaptive immunity, is downregulated by the human papillomavirus (HPV) E5 protein: A possible mechanism for immune evasion by HPV. J. Virol. 2010, 84, 11614–11623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyauchi, S.; Sanders, P.D.; Guram, K.; Kim, S.S.; Paolini, F.; Venuti, A.; Cohen, E.E.W.; Gutkind, J.S.; Califano, J.A.; Sharabi, A.B. HPV16 E5 Mediates Resistance to PD-L1 Blockade and Can Be Targeted with Rimantadine in Head and Neck Cancer. Cancer Res. 2020, 80, 732–746. [Google Scholar] [CrossRef] [Green Version]
- Scott, M.L.; Woodby, B.L.; Ulicny, J.; Raikhy, G.; Orr, A.W.; Songock, W.K.; Bodily, J.M. Human Papillomavirus 16 E5 Inhibits Interferon Signaling and Supports Episomal Viral Maintenance. J. Virol. 2020, 94, e01582-19. [Google Scholar] [CrossRef]
- Ronco, L.V.; Karpova, A.Y.; Vidal, M.; Howley, P.M. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998, 12, 2061–2072. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Labrecque, S.; Gauzzi, M.C.; Cuddihy, A.R.; Wong, A.H.; Pellegrini, S.; Matlashewski, G.J.; Koromilas, A.E. The human papilloma virus (HPV)-18 E6 oncoprotein physically associates with Tyk2 and impairs Jak-STAT activation by interferon-alpha. Oncogene 1999, 18, 5727–5737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, C.; Pauli, E.K.; Biryukov, J.; Feister, K.F.; Meng, M.; White, E.A.; Munger, K.; Howley, P.M.; Meyers, C.; Gack, M.U. The Human Papillomavirus E6 Oncoprotein Targets USP15 and TRIM25 To Suppress RIG-I-Mediated Innate Immune Signaling. J. Virol. 2018, 92, e01737-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiser, J.; Hurst, J.; Voges, M.; Krauss, P.; Munch, P.; Iftner, T.; Stubenrauch, F. High-risk human papillomaviruses repress constitutive kappa interferon transcription via E6 to prevent pathogen recognition receptor and antiviral-gene expression. J. Virol. 2011, 85, 11372–11380. [Google Scholar] [CrossRef] [Green Version]
- Lau, L.; Gray, E.E.; Brunette, R.L.; Stetson, D.B. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 2015, 350, 568–571. [Google Scholar] [CrossRef] [Green Version]
- Lo Cigno, I.; Calati, F.; Borgogna, C.; Zevini, A.; Albertini, S.; Martuscelli, L.; De Andrea, M.; Hiscott, J.; Landolfo, S.; Gariglio, M. Human Papillomavirus E7 Oncoprotein Subverts Host Innate Immunity via SUV39H1-Mediated Epigenetic Silencing of Immune Sensor Genes. J. Virol. 2020, 94, e01812-19. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Cao, J.; Cai, W.L.; Lang, S.M.; Horton, J.R.; Jansen, D.J.; Liu, Z.Z.; Chen, J.F.; Zhang, M.; Mott, B.T.; et al. KDM5 histone demethylases repress immune response via suppression of STING. PLoS Biol. 2018, 16, e2006134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, U.A.; Zannetti, C.; Parroche, P.; Goutagny, N.; Malfroy, M.; Roblot, G.; Carreira, C.; Hussain, I.; Muller, M.; Taylor-Papadimitriou, J.; et al. The human papillomavirus type 16 E7 oncoprotein induces a transcriptional repressor complex on the Toll-like receptor 9 promoter. J. Exp. Med. 2013, 210, 1369–1387. [Google Scholar] [CrossRef]
- Ren, C.; Cheng, X.; Lu, B.; Yang, G. Activation of interleukin-6/signal transducer and activator of transcription 3 by human papillomavirus early proteins 6 induces fibroblast senescence to promote cervical tumourigenesis through autocrine and paracrine pathways in tumour microenvironment. Eur. J. Cancer 2013, 49, 3889–3899. [Google Scholar] [CrossRef]
- Richards, K.H.; Doble, R.; Wasson, C.W.; Haider, M.; Blair, G.E.; Wittmann, M.; Macdonald, A. Human papillomavirus E7 oncoprotein increases production of the anti-inflammatory interleukin-18 binding protein in keratinocytes. J. Virol. 2014, 88, 4173–4179. [Google Scholar] [CrossRef] [Green Version]
- Hammes, L.S.; Tekmal, R.R.; Naud, P.; Edelweiss, M.I.; Kirma, N.; Valente, P.T.; Syrjänen, K.J.; Cunha-Filho, J.S. Macrophages, inflammation and risk of cervical intraepithelial neoplasia (CIN) progression—Clinicopathological correlation. Gynecol. Oncol. 2007, 105, 157–165. [Google Scholar] [CrossRef]
- Kobayashi, A.; Weinberg, V.; Darragh, T.; Smith-McCune, K. Evolving immunosuppressive microenvironment during human cervical carcinogenesis. Mucosal Immunol. 2008, 1, 412–420. [Google Scholar] [CrossRef]
- Mazibrada, J.; Ritta, M.; Mondini, M.; De Andrea, M.; Azzimonti, B.; Borgogna, C.; Ciotti, M.; Orlando, A.; Surico, N.; Chiusa, L.; et al. Interaction between inflammation and angiogenesis during different stages of cervical carcinogenesis. Gynecol. Oncol. 2008, 108, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Pahler, J.C.; Tazzyman, S.; Erez, N.; Chen, Y.Y.; Murdoch, C.; Nozawa, H.; Lewis, C.E.; Hanahan, D. Plasticity in tumor-promoting inflammation: Impairment of macrophage recruitment evokes a compensatory neutrophil response. Neoplasia 2008, 10, 329–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giraudo, E.; Inoue, M.; Hanahan, D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J. Clin. Investig. 2004, 114, 623–633. [Google Scholar] [CrossRef] [PubMed]
- De Visser, K.E.; Korets, L.V.; Coussens, L.M. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell 2005, 7, 411–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiraku, Y.; Tabata, T.; Ma, N.; Murata, M.; Ding, X.; Kawanishi, S. Nitrative and oxidative DNA damage in cervical intraepithelial neoplasia associated with human papilloma virus infection. Cancer Sci. 2007, 98, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Leong, C.M.; Doorbar, J.; Nindl, I.; Yoon, H.S.; Hibma, M.H. Loss of epidermal Langerhans cells occurs in human papillomavirus alpha, gamma, and mu but not beta genus infections. J. Investig. Dermatol. 2010, 130, 472–480. [Google Scholar] [CrossRef] [Green Version]
- Matthews, K.; Leong, C.M.; Baxter, L.; Inglis, E.; Yun, K.; Backstrom, B.T.; Doorbar, J.; Hibma, M. Depletion of Langerhans cells in human papillomavirus type 16-infected skin is associated with E6-mediated down regulation of E-cadherin. J. Virol. 2003, 77, 8378–8385. [Google Scholar] [CrossRef] [Green Version]
- Fausch, S.C.; Da Silva, D.M.; Rudolf, M.P.; Kast, W.M. Human papillomavirus virus-like particles do not activate Langerhans cells: A possible immune escape mechanism used by human papillomaviruses. J. Immunol. 2002, 169, 3242–3249. [Google Scholar] [CrossRef] [Green Version]
- Fausch, S.C.; Fahey, L.M.; Da Silva, D.M.; Kast, W.M. Human papillomavirus can escape immune recognition through Langerhans cell phosphoinositide 3-kinase activation. J. Immunol. 2005, 174, 7172–7178. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.S.; Herrero, R.; Bosetti, C.; Munoz, N.; Bosch, F.X.; Eluf-Neto, J.; Castellsague, X.; Meijer, C.J.; Van den Brule, A.J.; Franceschi, S.; et al. Herpes simplex virus-2 as a human papillomavirus cofactor in the etiology of invasive cervical cancer. J. Natl. Cancer Inst. 2002, 94, 1604–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.S.; Munoz, N.; Herrero, R.; Eluf-Neto, J.; Ngelangel, C.; Franceschi, S.; Bosch, F.X.; Walboomers, J.M.; Peeling, R.W. Evidence for Chlamydia trachomatis as a human papillomavirus cofactor in the etiology of invasive cervical cancer in Brazil and the Philippines. J. Infect. Dis. 2002, 185, 324–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vats, A.; Trejo-Cerro, O.; Thomas, M.; Banks, L. Human papillomavirus E6 and E7: What remains? Tumour Virus Res. 2021, 11, 200213. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basukala, O.; Banks, L. The Not-So-Good, the Bad and the Ugly: HPV E5, E6 and E7 Oncoproteins in the Orchestration of Carcinogenesis. Viruses 2021, 13, 1892. https://doi.org/10.3390/v13101892
Basukala O, Banks L. The Not-So-Good, the Bad and the Ugly: HPV E5, E6 and E7 Oncoproteins in the Orchestration of Carcinogenesis. Viruses. 2021; 13(10):1892. https://doi.org/10.3390/v13101892
Chicago/Turabian StyleBasukala, Om, and Lawrence Banks. 2021. "The Not-So-Good, the Bad and the Ugly: HPV E5, E6 and E7 Oncoproteins in the Orchestration of Carcinogenesis" Viruses 13, no. 10: 1892. https://doi.org/10.3390/v13101892
APA StyleBasukala, O., & Banks, L. (2021). The Not-So-Good, the Bad and the Ugly: HPV E5, E6 and E7 Oncoproteins in the Orchestration of Carcinogenesis. Viruses, 13(10), 1892. https://doi.org/10.3390/v13101892