
Optimisation of Neuraminidase Expression for Use in Drug Discovery by Using HEK293-6E Cells


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cloning
2.2. Protein Expression
2.3. Protein Purification
2.4. Protein Analysis
2.5. Neuraminidase Assay
2.6. Glycosylation and Western Blot Analysis
2.7. Crystallisation and Optimization
2.8. Data Collection and Processing
2.9. Molecular Replacement, Model Building and Refinement
2.10. Structural Analysis
3. Results
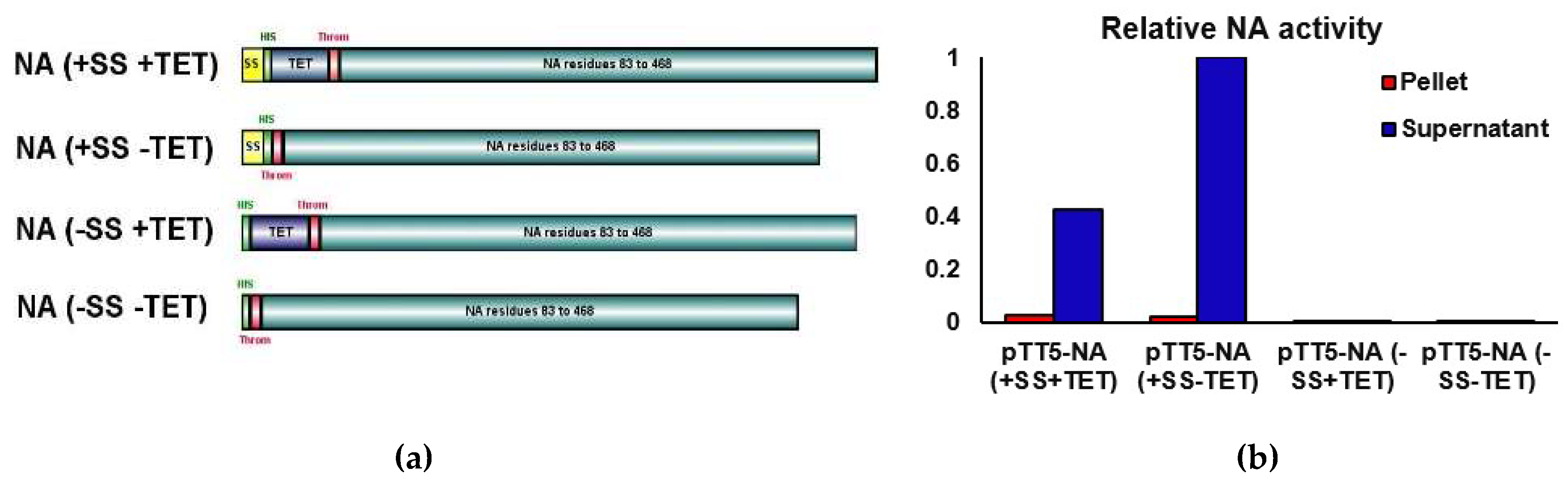
3.1. Choice of Genetic Construct for NA Expression
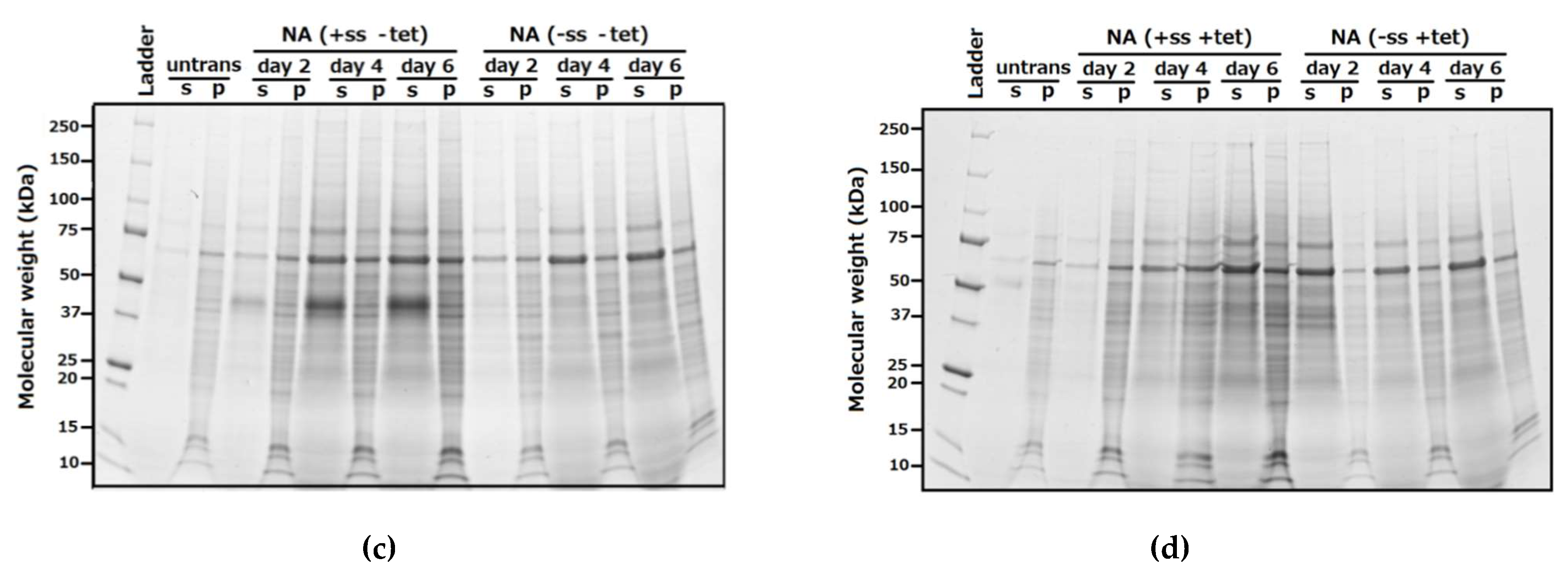
3.1.1. Analysis of Expression by SDS-PAGE
3.1.2. NA Activity Assay
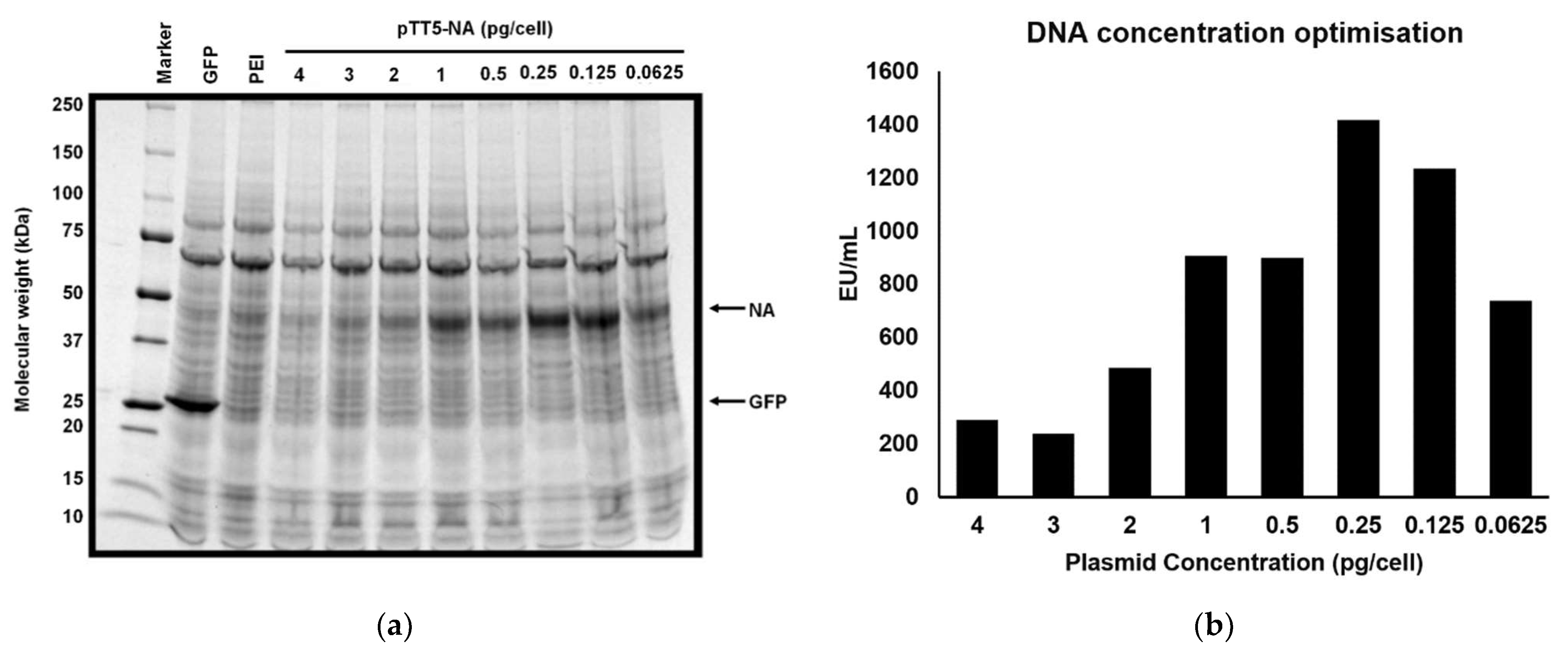
3.2. Optimisation of DNA Quantity and Up-Scaling for Larger Transfections
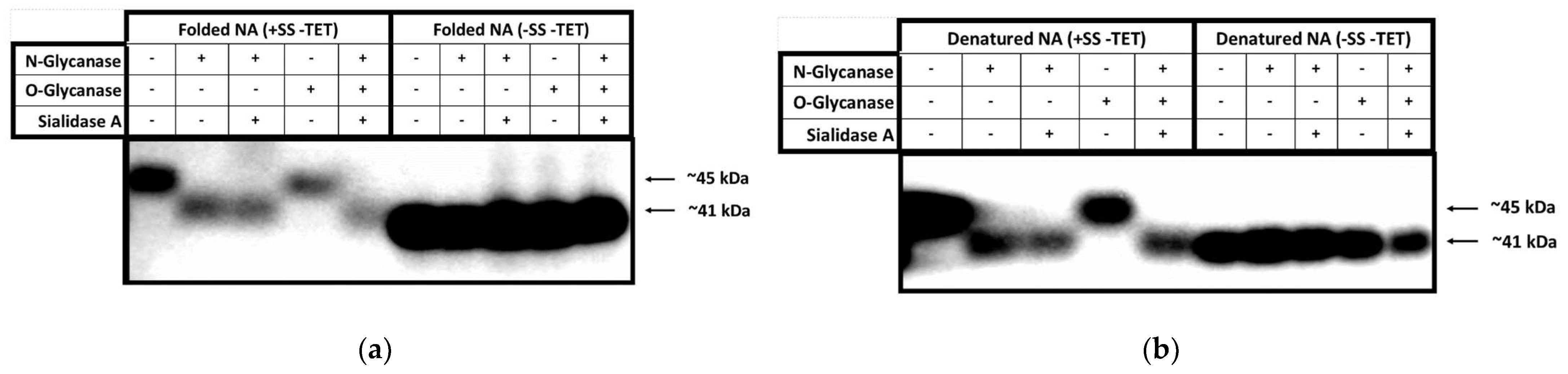
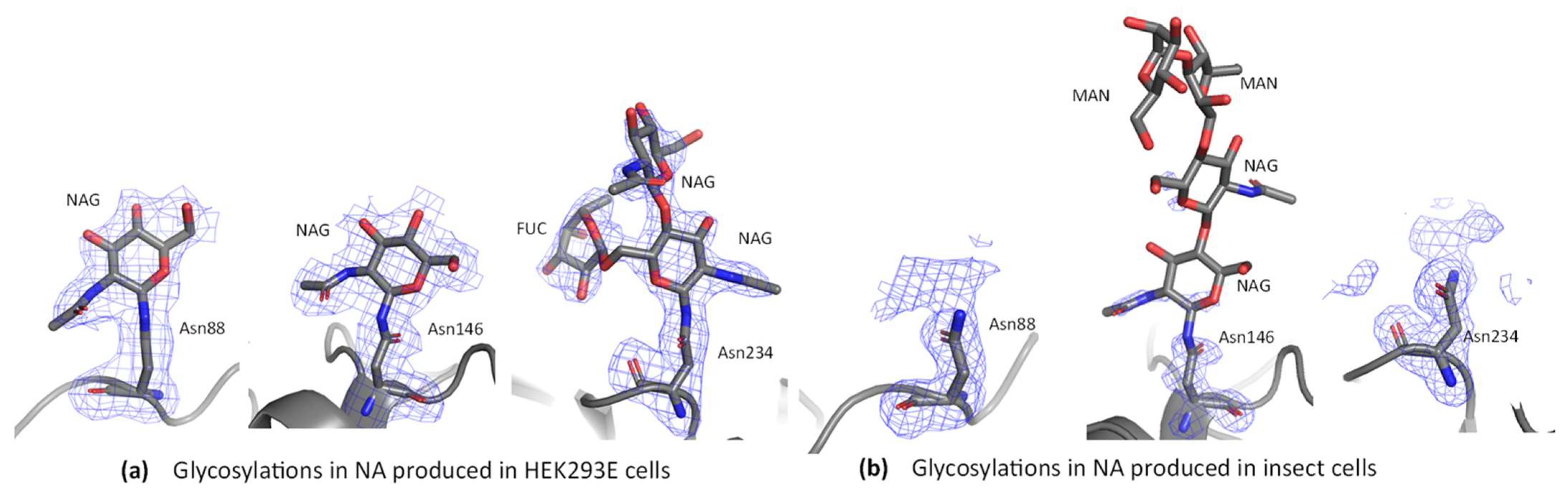
3.3. Glycosylation Analysis
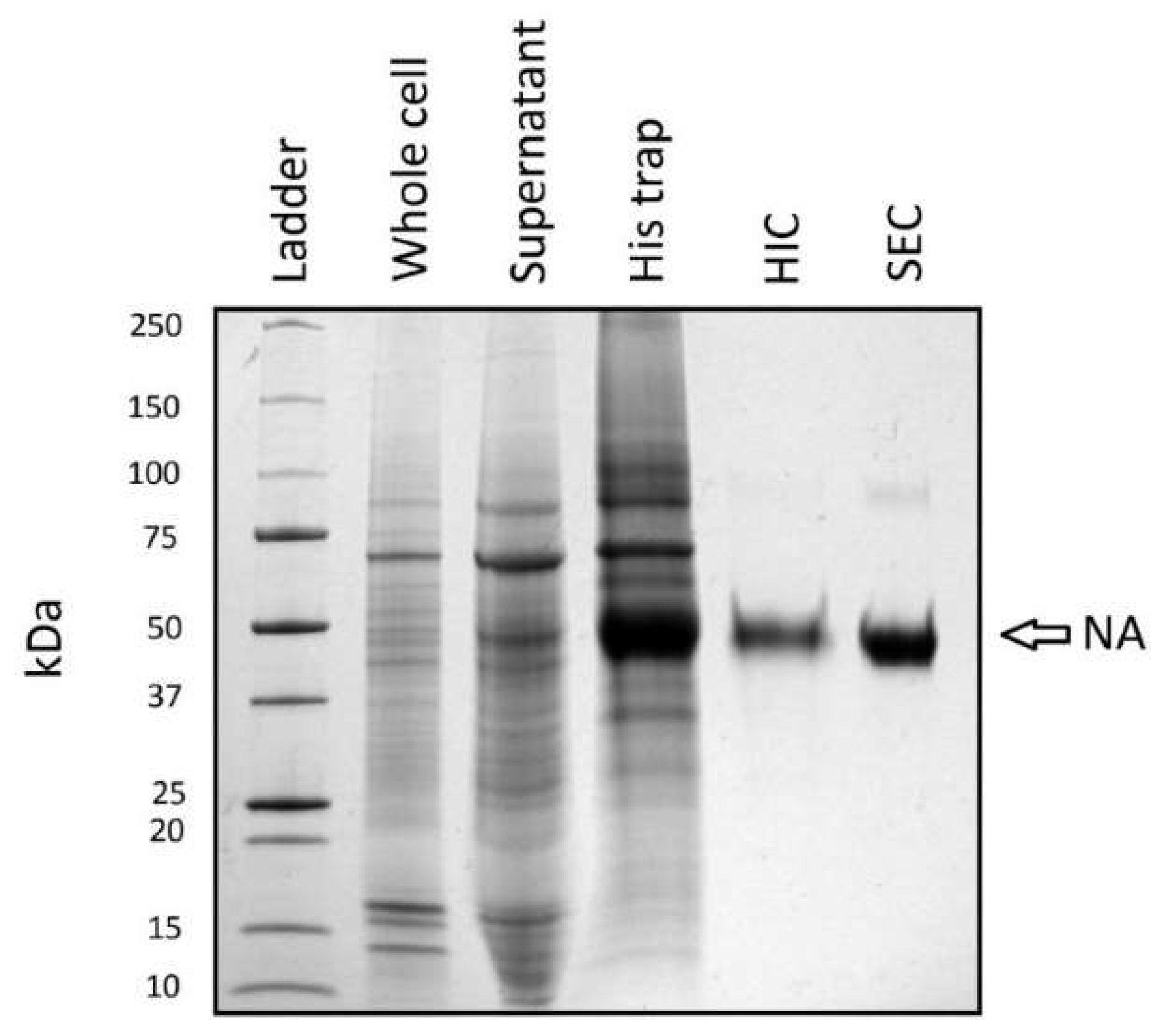
3.4. Purification
3.5. X-Ray Crystallography
3.5.1. Crystallisation
3.5.2. X-Ray Diffraction
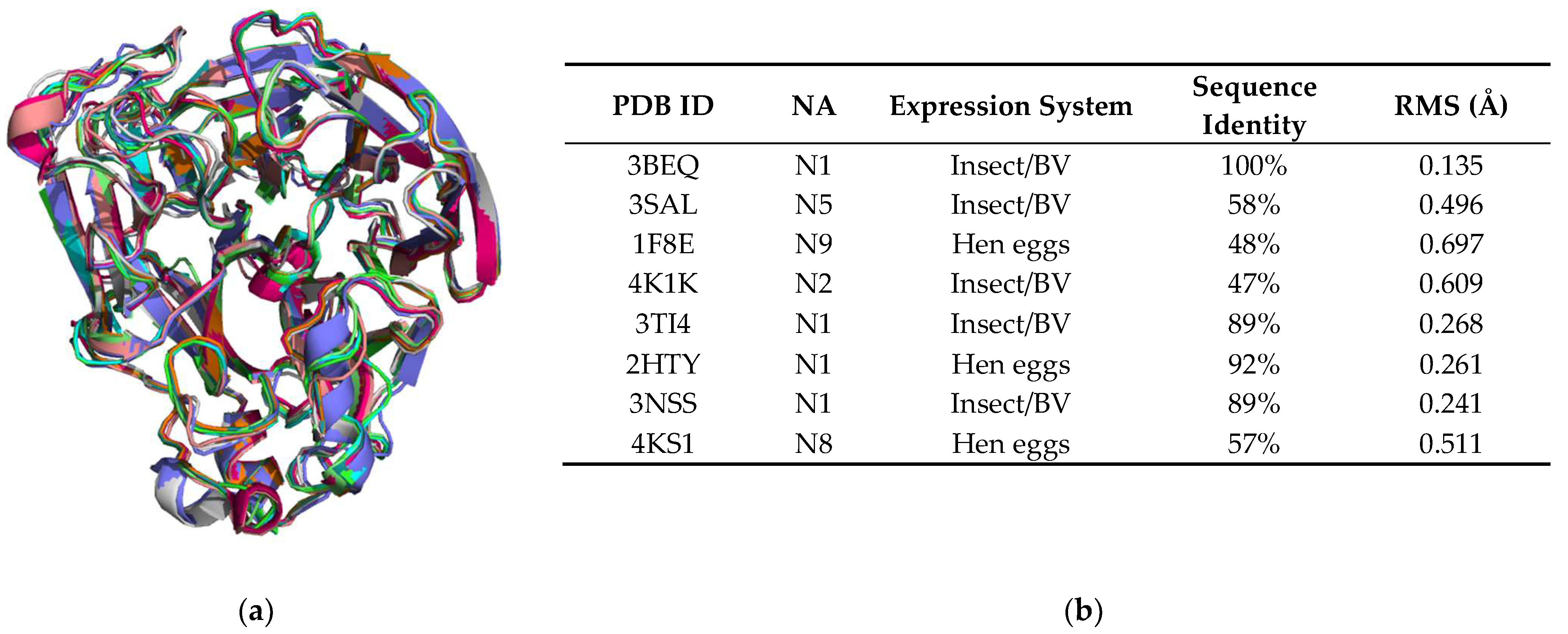
3.6. Structural Comparison with Other NA Structures
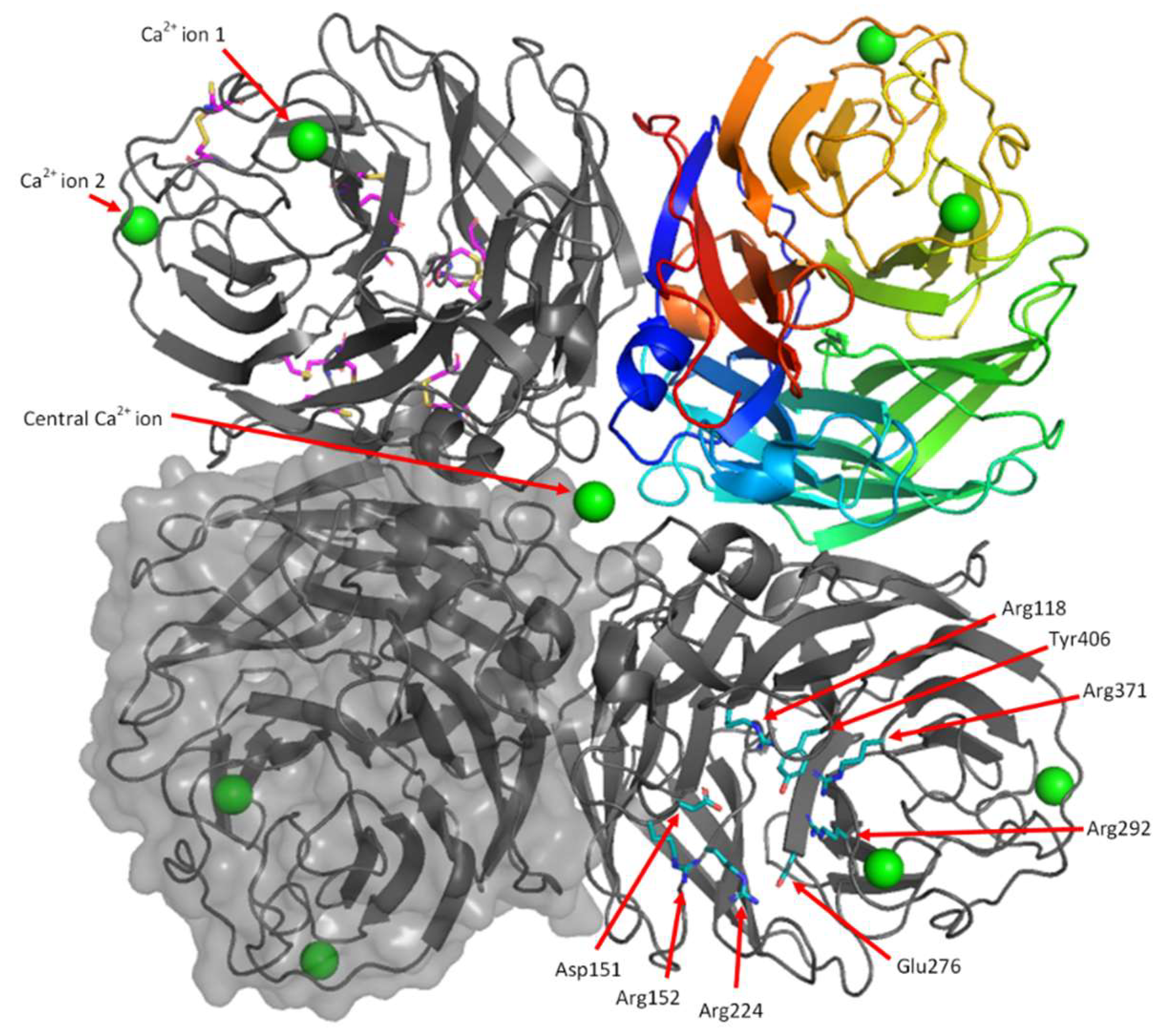
3.6.1. Overall Structure
3.6.2. Calcium Binding Sites
3.6.3. Active Site and 150-Cavity Structure
3.6.4. Electron Density Evidence for Glycosylation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pleschka, S. Overview of influenza viruses. Curr. Top. Microbiol. Immunol. 2013, 370, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Crusat, M.; de Jong, M.D. Neuraminidase inhibitors and their role in avian and pandemic influenza. Antivir. Ther. 2007, 12, 593–602. [Google Scholar] [PubMed]
- Tejada, S.; Jansson, M.; Solé-Lleonart, C.; Rello, J. Neuraminidase inhibitors are effective and safe in reducing influenza complications: Meta-analysis of randomized controlled trials. Eur. J. Intern. Med. 2021, 86, 54–65. [Google Scholar] [CrossRef]
- Hayden, F.G.; Atmar, R.L.; Schilling, M.; Johnson, C.; Poretz, D.; Paar, D.; Huson, L.; Ward, P.; Mills, R.G. Use of the selective oral neuraminidase inhibitor oseltamivir to prevent influenza. N. Engl. J. Med. 1999, 341, 1336–1343. [Google Scholar] [CrossRef]
- Takashita, E.; Daniels, R.S.; Fujisaki, S.; Gregory, V.; Gubareva, L.V.; Huang, W.; Hurt, A.C.; Lackenby, A.; Nguyen, H.T.; Pereyaslov, D.; et al. Global update on the susceptibilities of human influenza viruses to neuraminidase inhibitors and the cap-dependent endonuclease inhibitor baloxavir, 2017–2018. Antivir. Res. 2020, 175, 104718. [Google Scholar] [CrossRef]
- Santesso, N.; Hsu, J.; Mustafa, R.; Brozek, J.; Chen, Y.L.; Hopkins, J.P.; Cheung, A.; Hovhannisyan, G.; Ivanova, L.; Flottorp, S.A.; et al. Antivirals for influenza: A summary of a systematic review and meta-analysis of observational studies. Influenza Other Respir. Viruses 2013, 7 (Suppl. 2), 76–81. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Hurt, A.C. Neuraminidase inhibitor resistance in influenza: A clinical perspective. Curr. Opin. Infect. Dis. 2018, 31, 520–526. [Google Scholar] [CrossRef]
- McKimm-Breschkin, J.L.; Selleck, P.W.; Usman, T.B.; Johnson, M.A. Reduced sensitivity of influenza A (H5N1) to oseltamivir. Emerg. Infect. Dis. 2007, 13, 1354–1357. [Google Scholar] [CrossRef]
- McKimm-Breschkin, J.L. Influenza neuraminidase inhibitors: Antiviral action and mechanisms of resistance. Influenza Other Respir. Viruses 2013, 7 (Suppl. 1), 25–36. [Google Scholar] [CrossRef] [Green Version]
- Colman, P.M.; Varghese, J.N.; Laver, W.G. Structure of the catalytic and antigenic sites in influenza virus neuraminidase. Nature 1983, 303, 41–44. [Google Scholar] [CrossRef]
- Baker, A.T.; Varghese, J.N.; Laver, W.G.; Air, G.M.; Colman, P.M. Three-dimensional structure of neuraminidase of subtype N9 from an avian influenza virus. Proteins 1987, 2, 111–117. [Google Scholar] [CrossRef]
- Von Itzstein, M.; Wu, W.-Y.; Kok, G.B.; Pegg, M.S.; Dyason, J.C.; Jin, B.; Phan, T.V.; Smythe, M.L.; White, H.F.; Oliver, S.W.; et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 1993, 363, 418–423. [Google Scholar] [CrossRef]
- Colman, P.M. Influenza virus neuraminidase: Structure, antibodies, and inhibitors. Protein Sci. A Publ. Protein Soc. 1994, 3, 1687–1696. [Google Scholar] [CrossRef] [Green Version]
- McKimm-Breschkin, J.L.; Sahasrabudhe, A.; Blick, T.J.; McDonald, M.; Colman, P.M.; Hart, G.J.; Bethell, R.C.; Varghese, J.N. Mutations in a conserved residue in the influenza virus neuraminidase active site decreases sensitivity to Neu5Ac2en-derived inhibitors. J. Virol. 1998, 72, 2456–2462. [Google Scholar] [CrossRef] [Green Version]
- Colman, P.M. Neuraminidase inhibitors as antivirals. Vaccine 2002, 20 (Suppl. 2), S55–S58. [Google Scholar] [CrossRef]
- Smith, B.J.; McKimm-Breshkin, J.L.; McDonald, M.; Fernley, R.T.; Varghese, J.N.; Colman, P.M. Structural studies of the resistance of influenza virus neuramindase to inhibitors. J. Med. Chem. 2002, 45, 2207–2212. [Google Scholar] [CrossRef] [PubMed]
- Von Itzstein, M.; Thomson, R. Anti-influenza drugs: The development of sialidase inhibitors. Handb. Exp. Pharmacol. 2009, 111–154. [Google Scholar] [CrossRef]
- McKimm-Breschkin, J.L.; Caldwell, J.B.; Guthrie, R.E.; Kortt, A.A. A new method for the purification of the influenza A virus neuraminidase. J. Virol. Methods 1991, 32, 121–124. [Google Scholar] [CrossRef]
- Schmidt, P.M.; Attwood, R.M.; Mohr, P.G.; Barrett, S.A.; McKimm-Breschkin, J.L. A generic system for the expression and purification of soluble and stable influenza neuraminidase. PLoS ONE 2011, 6, e16284. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Reid, A.H.; Fanning, T.G. The 1918 influenza virus: A killer comes into view. Virology 2000, 274, 241–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.L.; Chang, S.H.; Gong, X.; Wu, J.; Liu, B. Expression, purification and characterization of low-glycosylation influenza neuraminidase in alpha-1,6-mannosyltransferase defective Pichia pastoris. Mol. Biol. Rep. 2012, 39, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Lee, C.J.; Park, J.S. Strategies for Optimizing the Production of Proteins and Peptides with Multiple Disulfide Bonds. Antibiotics 2020, 9, 541. [Google Scholar] [CrossRef] [PubMed]
- Martinet, W.; Saelens, X.; Deroo, T.; Neirynck, S.; Contreras, R.; Min Jou, W.; Fiers, W. Protection of mice against a lethal influenza challenge by immunization with yeast-derived recombinant influenza neuraminidase. Eur. J. Biochem. 1997, 247, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Shigemori, T.; Nagayama, M.; Yamada, J.; Miura, N.; Yongkiettrakul, S.; Kuroda, K.; Katsuragi, T.; Ueda, M. Construction of a convenient system for easily screening inhibitors of mutated influenza virus neuraminidases. FEBS Open Bio 2013, 3, 484–489. [Google Scholar] [CrossRef] [Green Version]
- Pua, T.L.; Loh, H.S.; Massawe, F.; Tan, C.S.; Omar, A.R. Expression of Insoluble Influenza Neuraminidase Type 1 (NA1) Protein in Tobacco. J. Trop. Life Sci. 2012, 2, 62–71. [Google Scholar]
- Xu, X.; Zhu, X.; Dwek, R.A.; Stevens, J.; Wilson, I.A. Structural characterization of the 1918 influenza virus H1N1 neuraminidase. J. Virol. 2008, 82, 10493–10501. [Google Scholar] [CrossRef] [Green Version]
- Streltsov, V.A.; Schmidt, P.M.; McKimm-Breschkin, J.L. Structure of an Influenza A virus N9 neuraminidase with a tetrabrachion-domain stalk. Acta Cryst. F Struct. Biol. Commun. 2019, 75, 89–97. [Google Scholar] [CrossRef]
- Wan, H.; Gao, J.; Yang, H.; Yang, S.; Harvey, R.; Chen, Y.-Q.; Zheng, N.-Y.; Chang, J.; Carney, P.J.; Li, X.; et al. The neuraminidase of A(H3N2) influenza viruses circulating since 2016 is antigenically distinct from the A/Hong Kong/4801/2014 vaccine strain. Nat. Microbiol. 2019, 4, 2216–2225. [Google Scholar] [CrossRef]
- Yang, H.; Carney, P.J.; Mishin, V.P.; Guo, Z.; Chang, J.C.; Wentworth, D.E.; Gubareva, L.V.; Stevens, J. Molecular Characterizations of Surface Proteins Hemagglutinin and Neuraminidase from Recent H5Nx Avian Influenza Viruses. J. Virol. 2016, 90, 5770–5784. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Turner, H.L.; Lang, S.; McBride, R.; Bangaru, S.; Gilchuk, I.M.; Yu, W.; Paulson, J.C.; Crowe, J.E., Jr.; Ward, A.B.; et al. Structural Basis of Protection against H7N9 Influenza Virus by Human Anti-N9 Neuraminidase Antibodies. Cell Host Microbe 2019, 26, 729–738.e724. [Google Scholar] [CrossRef]
- Madsen, A.; Dai, Y.N.; McMahon, M.; Schmitz, A.J.; Turner, J.S.; Tan, J.; Lei, T.; Alsoussi, W.B.; Strohmeier, S.; Amor, M.; et al. Human Antibodies Targeting Influenza B Virus Neuraminidase Active Site Are Broadly Protective. Immunity 2020, 53, 852–863.e857. [Google Scholar] [CrossRef]
- Nivitchanyong, T.; Yongkiettrakul, S.; Kramyu, J.; Pannengpetch, S.; Wanasen, N. Enhanced expression of secretable influenza virus neuraminidase in suspension mammalian cells by influenza virus nonstructural protein 1. J. Virol. Methods 2011, 178, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Ecker, J.W.; Kirchenbaum, G.A.; Pierce, S.R.; Skarlupka, A.L.; Abreu, R.B.; Cooper, R.E.; Taylor-Mulneix, D.; Ross, T.M.; Sautto, G.A. High-Yield Expression and Purification of Recombinant Influenza Virus Proteins from Stably-Transfected Mammalian Cell Lines. Vaccines 2020, 8, 462. [Google Scholar] [CrossRef]
- Van der Woude, R.; Turner, H.L.; Tomris, I.; Bouwman, K.M.; Ward, A.B.; de Vries, R.P. Drivers of recombinant soluble influenza A virus hemagglutinin and neuraminidase expression in mammalian cells. Protein Sci. A Publ. Protein Soc. 2020, 29, 1975–1982. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Smart, T.G. HEK293 cell line: A vehicle for the expression of recombinant proteins. J. Pharmacol. Toxicol. Methods 2005, 51, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Kongkamnerd, J.; Milani, A.; Cattoli, G.; Terregino, C.; Capua, I.; Beneduce, L.; Gallotta, A.; Pengo, P.; Fassina, G.; Monthakantirat, O.; et al. The quenching effect of flavonoids on 4-methylumbelliferone, a potential pitfall in fluorimetric neuraminidase inhibition assays. J. Biomol. Screen. 2011, 16, 755–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durocher, Y.; Perret, S.; Kamen, A. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 2002, 30, E9. [Google Scholar] [CrossRef] [PubMed]
- Couñago, R.M.; Fleming, S.B.; Mercer, A.A.; Krause, K.L. Crystallization and preliminary X-ray analysis of the chemokine-binding protein from orf virus (Poxviridae). Acta Cryst. Sect. F Struct. Biol. Cryst. Commun. 2010, 66, 819–823. [Google Scholar] [CrossRef]
- Quan, J.; Tian, J. Circular polymerase extension cloning for high-throughput cloning of complex and combinatorial DNA libraries. Nat. Protoc. 2011, 6, 242–251. [Google Scholar] [CrossRef]
- Kuhnel, K.; Jarchau, T.; Wolf, E.; Schlichting, I.; Walter, U.; Wittinghofer, A.; Strelkov, S.V. The VASP tetramerization domain is a right-handed coiled coil based on a 15-residue repeat. Proc. Natl. Acad. Sci. USA 2004, 101, 17027–17032. [Google Scholar] [CrossRef] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Leang, S.-K.; Hurt, A.C. Fluorescence-based Neuraminidase Inhibition Assay to Assess the Susceptibility of Influenza Viruses to The Neuraminidase Inhibitor Class of Antivirals. J. Vis. Exp. 2017, 55570. [Google Scholar] [CrossRef] [PubMed]
- Potier, M.; Mameli, L.; Belisle, M.; Dallaire, L.; Melancon, S.B. Fluorometric assay of neuraminidase with a sodium (4-methylumbelliferyl-alpha-d-N-acetylneuraminate) substrate. Anal. Biochem. 1979, 94, 287–296. [Google Scholar] [CrossRef]
- Gupta, R.; Brunak, S. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac. Symp. Biocomput. 2002, 310–322. [Google Scholar]
- Nielsen, H. Predicting Secretory Proteins with SignalP. Methods Mol. Biol. 2017, 1611, 59–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McPhillips, T.M.; McPhillips, S.E.; Chiu, H.J.; Cohen, A.E.; Deacon, A.M.; Ellis, P.J.; Garman, E.; Gonzalez, A.; Sauter, N.K.; Phizackerley, R.P.; et al. Blu-Ice and the Distributed Control System: Software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Radiat. 2002, 9, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Aragão, D.; Aishima, J.; Cherukuvada, H.; Clarken, R.; Clift, M.; Cowieson, N.P.; Ericsson, D.J.; Gee, C.L.; Macedo, S.; Mudie, N.; et al. MX2: A high-flux undulator microfocus beamline serving both the chemical and macromolecular crystallography communities at the Australian Synchrotron. J. Synchrotron Radiat. 2018, 25, 885–891. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef]
- Vagin, A.; Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 22–25. [Google Scholar] [CrossRef]
- Matthews, B.W. Solvent content of protein crystals. J. Mol. Biol. 1968, 33, 491–497. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Cryst. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, R. Ligand fitting with CCP4. Acta Crystallogr. Sect. D 2017, 73, 158–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Lütteke, T.; von der Lieth, C.W. pdb-care (PDB carbohydrate residue check): A program to support annotation of complex carbohydrate structures in PDB files. BMC Bioinform. 2004, 5, 69. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Cooper, D.R.; Porebski, P.J.; Shabalin, I.G.; Handing, K.B.; Minor, W. CheckMyMetal: A macromolecular metal-binding validation tool. Acta Cryst. D Struct. Biol. 2017, 73, 223–233. [Google Scholar] [CrossRef] [Green Version]
- Gore, S.; Sanz García, E.; Hendrickx, P.M.S.; Gutmanas, A.; Westbrook, J.D.; Yang, H.; Feng, Z.; Baskaran, K.; Berrisford, J.M.; Hudson, B.P.; et al. Validation of Structures in the Protein Data Bank. Structure 2017, 25, 1916–1927. [Google Scholar] [CrossRef] [Green Version]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A. PDBsum new things. Nucleic Acids Res. 2009, 37, D355–D359. [Google Scholar] [CrossRef] [PubMed]
- Kleywegt, G.J.; Harris, M.R.; Zou, J.Y.; Taylor, T.C.; Wählby, A.; Jones, T.A. The Uppsala Electron-Density Server. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2240–2249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebschner, D.; Afonine, P.V.; Moriarty, N.W.; Poon, B.K.; Sobolev, O.V.; Terwilliger, T.C.; Adams, P.D. Polder maps: Improving OMIT maps by excluding bulk solvent. Acta Cryst. D Struct. Biol. 2017, 73, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Xie, Y.; Ma, J.; Luo, X.; Nie, P.; Zuo, Z.; Lahrmann, U.; Zhao, Q.; Zheng, Y.; Zhao, Y.; et al. IBS: An illustrator for the presentation and visualization of biological sequences. Bioinformatics 2015, 31, 3359–3361. [Google Scholar] [CrossRef] [Green Version]
- Fang, Q.; Shen, B. Optimization of polyethylenimine-mediated transient transfection using response surface methodology design. Electron. J. Biotechnol. 2010, 13. [Google Scholar] [CrossRef] [Green Version]
- Bollin, F.; Dechavanne, V.; Chevalet, L. Design of Experiment in CHO and HEK transient transfection condition optimization. Protein Expr. Purif. 2011, 78, 61–68. [Google Scholar] [CrossRef]
- De Los Milagros Bassani Molinas, M.; Beer, C.; Hesse, F.; Wirth, M.; Wagner, R. Optimizing the transient transfection process of HEK-293 suspension cells for protein production by nucleotide ratio monitoring. Cytotechnology 2013, 66, 493–514. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Wang, Q.; Zhao, F.; Chen, W.; Li, Z. Glycosylation Site Alteration in the Evolution of Influenza A (H1N1) Viruses. PLoS ONE 2011, 6, e22844. [Google Scholar] [CrossRef]
- Varghese, J.N.; McKimm-Breschkin, J.L.; Caldwell, J.B.; Kortt, A.A.; Colman, P.M. The structure of the complex between influenza virus neuraminidase and sialic acid, the viral receptor. Proteins 1992, 14, 327–332. [Google Scholar] [CrossRef]
- McAuley, J.L.; Gilbertson, B.P.; Trifkovic, S.; Brown, L.E.; McKimm-Breschkin, J.L. Influenza Virus Neuraminidase Structure and Functions. Front Microbiol. 2019, 10, 39. [Google Scholar] [CrossRef] [Green Version]
- Harding, M.M. Geometry of metal-ligand interactions in proteins. Acta Crystallogr. Sect. D Biol. Crystallogr. 2001, 57, 401–411. [Google Scholar] [CrossRef]
- Russell, R.J.; Haire, L.F.; Stevens, D.J.; Collins, P.J.; Lin, Y.P.; Blackburn, G.M.; Hay, A.J.; Gamblin, S.J.; Skehel, J.J. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature 2006, 443, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Amaro, R.E.; Minh, D.D.L.; Cheng, L.S.; Lindstrom, W.M.; Olson, A.J.; Lin, J.-H.; Li, W.W.; McCammon, J.A. Remarkable Loop Flexibility in Avian Influenza N1 and Its Implications for Antiviral Drug Design. J. Am. Chem. Soc. 2007, 129, 7764–7765. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Qi, J.; Liu, Y.; Vavricka, C.J.; Wu, Y.; Li, Q.; Gao, G.F. Influenza A virus N5 neuraminidase has an extended 150-cavity. J. Virol. 2011, 85, 8431–8435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, B.J.; Colman, P.M.; Von Itzstein, M.; Danylec, B.; Varghese, J.N. Analysis of inhibitor binding in influenza virus neuraminidase. Protein Sci. A Publ. Protein Soc. 2001, 10, 689–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vavricka, C.J.; Li, Q.; Wu, Y.; Qi, J.; Wang, M.; Liu, Y.; Gao, F.; Liu, J.; Feng, E.; He, J.; et al. Structural and functional analysis of laninamivir and its octanoate prodrug reveals group specific mechanisms for influenza NA inhibition. PLoS Pathog. 2011, 7, e1002249. [Google Scholar] [CrossRef] [PubMed]
- Kerry, P.S.; Mohan, S.; Russell, R.J.; Bance, N.; Niikura, M.; Pinto, B.M. Structural basis for a class of nanomolar influenza A neuraminidase inhibitors. Sci. Rep. 2013, 3, 2871. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Qi, J.; Zhang, W.; Vavricka, C.J.; Shi, Y.; Wei, J.; Feng, E.; Shen, J.; Chen, J.; Liu, D.; et al. The 2009 pandemic H1N1 neuraminidase N1 lacks the 150-cavity in its active site. Nat. Struct. Mol. Biol. 2010, 17, 1266–1268. [Google Scholar] [CrossRef]
- Shtyrya, Y.A.; Mochalova, L.V.; Bovin, N.V. Influenza Virus Neuraminidase: Structure and Function. Acta Nat. 2009, 1, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Webster, R.G.; Webby, R.J. Influenza Virus: Dealing with a Drifting and Shifting Pathogen. Viral Immunol. 2018, 31, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Antiviral drugs for influenza for 2020–2021. Med. Lett. Drugs Ther. 2020, 62, 169–172.
- Hayden, F.G.; Sugaya, N.; Hirotsu, N.; Lee, N.; de Jong, M.D.; Hurt, A.C.; Ishida, T.; Sekino, H.; Yamada, K.; Portsmouth, S.; et al. Baloxavir Marboxil for Uncomplicated Influenza in Adults and Adolescents. N. Engl. J. Med. 2018, 379, 913–923. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, B.; Antigua, K.J.C.; Jeong, J.H.; Kim, C.I.; Choi, W.S.; Oh, S.; Kim, C.H.; Kim, E.G.; Choi, Y.K.; et al. Baloxavir-oseltamivir combination therapy inhibits the emergence of resistant substitutions in influenza A virus PA gene in a mouse model. Antivir. Res. 2021, 193, 105126. [Google Scholar] [CrossRef]
- Mayer, M.P.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005, 62, 670–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Östbye, H.; Gao, J.; Martinez, M.R.; Wang, H.; de Gier, J.W.; Daniels, R. N-Linked Glycan Sites on the Influenza A Virus Neuraminidase Head Domain Are Required for Efficient Viral Incorporation and Replication. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Bao, D.; Xue, R.; Zhang, M.; Lu, C.; Ma, T.; Ren, C.; Zhang, T.; Yang, J.; Teng, Q.; Li, X.; et al. N-Linked Glycosylation Plays an Important Role in Budding of Neuraminidase Protein and Virulence of Influenza Viruses. J. Virol. 2021, 95. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.; Jang, Y.H.; Kwon, S.B.; Lee, C.M.; Han, G.; Seong, B.L. Glycosylation of Hemagglutinin and Neuraminidase of Influenza A Virus as Signature for Ecological Spillover and Adaptation among Influenza Reservoirs. Viruses 2018, 10, 183. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.L.; Zhou, H.; Ethen, C.M.; Reinhold, V.N. Core-6 fucose and the oligomerization of the 1918 pandemic influenza viral neuraminidase. Biochem. Biophys. Res. Commun. 2016, 473, 524–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.L.; Ethen, C.; Hickey, G.E.; Jiang, W. Active 1918 pandemic flu viral neuraminidase has distinct N-glycan profile and is resistant to trypsin digestion. Biochem. Biophys. Res. Commun. 2009, 379, 749–753. [Google Scholar] [CrossRef]
- Von Grafenstein, S.; Wallnoefer, H.G.; Kirchmair, J.; Fuchs, J.E.; Huber, R.G.; Schmidtke, M.; Sauerbrei, A.; Rollinger, J.M.; Liedl, K.R. Interface dynamics explain assembly dependency of influenza neuraminidase catalytic activity. J. Biomol. Struct. Dyn. 2015, 33, 104–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Taylor, G.; Webster, R.G. Steps in maturation of influenza A virus neuraminidase. J. Virol. 1995, 69, 5011–5017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruswitz, F.; Chaudhary, S.; Ho, J.D.; Schlessinger, A.; Pezeshki, B.; Ho, C.-M.; Sali, A.; Westhoff, C.M.; Stroud, R.M. Function of human Rh based on structure of RhCG at 2.1 Å. Proc. Natl. Acad. Sci. USA 2010, 107, 9638–9643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Standfuss, J.; Xie, G.; Edwards, P.C.; Burghammer, M.; Oprian, D.D.; Schertler, G.F.X. Crystal Structure of a Thermally Stable Rhodopsin Mutant. J. Mol. Biol. 2007, 372, 1179–1188. [Google Scholar] [CrossRef] [Green Version]
- Penmatsa, A.; Wang, K.H.; Gouaux, E. X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 2013, 503, 85–90. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; zu Dohna, H.; Cardona, C.J.; Miller, J.; Carpenter, T.E. Emergence and Genetic Variation of Neuraminidase Stalk Deletions in Avian Influenza Viruses. PLoS ONE 2011, 6, e14722. [Google Scholar] [CrossRef]
- Li, Y.; Chen, S.; Zhang, X.; Fu, Q.; Zhang, Z.; Shi, S.; Zhu, Y.; Gu, M.; Peng, D.; Liu, X. A 20-amino-acid deletion in the neuraminidase stalk and a five-amino-acid deletion in the NS1 protein both contribute to the pathogenicity of H5N1 avian influenza viruses in mallard ducks. PLoS ONE 2014, 9, e95539. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection | |
|---|---|
| Space group | P 21 |
| Cell dimensions | |
| a, b, c (Å) | 122.7, 148.2, 127.2 |
| α, β, γ (°) | 90.0, 94.8, 90.0 |
| Wavelength (Å) | 0.9790 |
| Resolution range (Å) | 2.15–48.15 (2.15–2.19) |
| Observations | 914,067 (45,188) |
| Unique reflections | 242,924 (11,945) |
| Rmerge (I) | 0.241 (1.183) |
| Rmeas(I) | 0.281 (1.38) |
| Rpim (I) | 0.144 (0.703) |
| Mean I/σ | 6.1 (1.5) |
| Mean CC1/2 | 0.977 (0.412) |
| Completeness (%) | 99.1 (98.4) |
| Multiplicity | 3.8 (3.8) |
| No. of atoms | 25,641 |
| Protein | 23,774 |
| Carbohydrate | 476 |
| Ca2+ | 18 |
| Glycerol | 36 |
| Water | 1345 |
| Rwork | 0.1858 |
| Rfree b | 0.2151 |
| RMSD from ideal | |
| Bond length (Å) | 0.009 |
| Bond angle (°) | 0.970 |
| Ramachandran Plot | |
| Favoured (%) | 95.3 |
| Outliers (%) | 0.07 |
| Clash score (%-tile) c | 100 |
| MolProbity score (%-tile) c | 100 |
| B-factors (Å2) | |
| Average | 26.8 |
| Protein | 25.8 |
| Carbohydrate | 65.2 |
| Ca2+ | 29.8 |
| Glycerol | 45.5 |
| Water | 21.1 |
| Coordinate error (Å) d | 0.27 |
| PDB ID | 6D96 |
| Step | Volume (mL) | Purity (BIORAD) | [Protein] (μg/mL) | Total Protein (μg) | Total NA (μg) | Yield (%) a | NA Activity (EU/mL) | Total EU | Specific Activity (EU/μg) b | Fold Purification c |
|---|---|---|---|---|---|---|---|---|---|---|
| Supernatant | 1000 | 14 | 199 | 198,700 | 27,818 | 100 | 3.7 × 103 | 3.7 × 106 | 18 | 1 |
| His-Trap | 66 | 47 | 260 | 17,173 | 8071 | 29 | 1.9 × 104 | 1.2 × 106 | 75 | 4 |
| HIC | 9 | 100 | 80 | 727 | 727 | 2.6 | 2.7 × 104 | 2.4 × 105 | 336 | 18 |
| SEC | 1.5 | 100 | 269 | 403 | 403 | 1.4 | 1.9 × 105 | 2.8 × 105 | 710 | 38 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campbell, A.C.; Tanner, J.J.; Krause, K.L. Optimisation of Neuraminidase Expression for Use in Drug Discovery by Using HEK293-6E Cells. Viruses 2021, 13, 1893. https://doi.org/10.3390/v13101893
Campbell AC, Tanner JJ, Krause KL. Optimisation of Neuraminidase Expression for Use in Drug Discovery by Using HEK293-6E Cells. Viruses. 2021; 13(10):1893. https://doi.org/10.3390/v13101893
Chicago/Turabian StyleCampbell, Ashley C., John J. Tanner, and Kurt L. Krause. 2021. "Optimisation of Neuraminidase Expression for Use in Drug Discovery by Using HEK293-6E Cells" Viruses 13, no. 10: 1893. https://doi.org/10.3390/v13101893
APA StyleCampbell, A. C., Tanner, J. J., & Krause, K. L. (2021). Optimisation of Neuraminidase Expression for Use in Drug Discovery by Using HEK293-6E Cells. Viruses, 13(10), 1893. https://doi.org/10.3390/v13101893