
Genomic Signatures in HPV-Associated Tumors


Abstract
:1. Introduction
2. Radiosensitivity Differences between HPV+ and HPV− Tumors
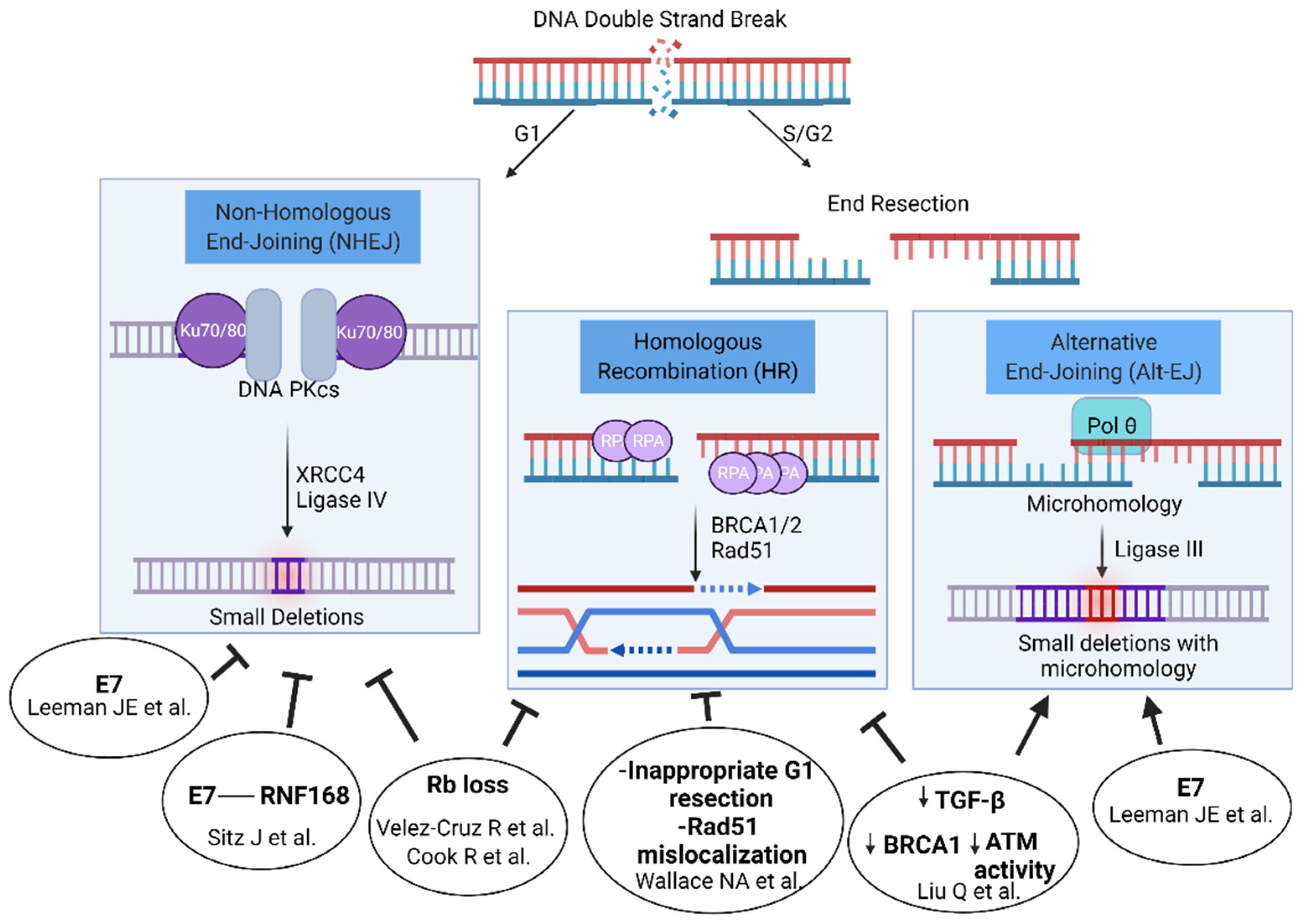
3. Proposed Mechanisms for Increased Radiosensitivity in HPV+ Tumors
4. Genomics Signatures in HPV Cancers
5. Clinical Responses to Platinum in HPV+ Cancer
6. Summary and Discussions
7. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Pei, X.F.; Meck, J.M.; Greenhalgh, D.; Schlegel, R. Cotransfection of HPV-18 and v-fos DNA induces tumorigenicity of primary human keratinocytes. Virology 1993, 196, 855–860. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helen, F.; Cancer Genome Atlas Research Network; Albert Einstein College of Medicine; Analytical Biological Services; Barretos Cancer Hospital; Baylor College of Medicine; Beckman Research Institute of City of Hope; Buck Institute for Research on Aging; Canada’s Michael Smith Genome Sciences Centre; Harvard Medical School; et al. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar] [CrossRef]
- Chung, J.H.; Sanford, E.; Johnson, A.; Klempner, S.J.; Schrock, A.B.; Palma, N.A.; Erlich, R.L.; Frampton, G.M.; Chalmers, Z.R.; Vergilio, J.; et al. Comprehensive genomic profiling of anal squamous cell carcinoma reveals distinct genomically defined classes. Ann. Oncol. 2016, 27, 1336–1341. [Google Scholar] [CrossRef]
- Ma, J.; Setton, J.; Lee, N.Y.; Riaz, N.; Powell, S.N. The therapeutic significance of mutational signatures from DNA repair deficiency in cancer. Nat. Commun. 2018, 9, 3292. [Google Scholar] [CrossRef] [PubMed]
- Setton, J.; Reis-Filho, J.S.; Powell, S.N. Homologous recombination deficiency: How genomic signatures are generated. Curr. Opin. Genet. Dev. 2021, 66, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Adelstein, D.J.; Li, Y.; Adams, G.L.; Wagner, H., Jr.; Kish, J.A.; Ensley, J.F.; Schuller, D.E.; Forastiere, A.A. An intergroup phase III comparison of standard radiation therapy and two schedules of concurrent chemoradiotherapy in patients with unresectable squamous cell head and neck cancer. J. Clin. Oncol. 2003, 21, 92–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forastiere, A.A.; Zhang, Q.; Weber, R.S.; Maor, M.H.; Goepfert, H.; Pajak, T.F.; Morrison, W.; Glisson, B.; Trotti, A.; Ridge, J.A.; et al. Long-term results of RTOG 91-11: A comparison of three nonsurgical treatment strategies to preserve the larynx in patients with locally advanced larynx cancer. J. Clin. Oncol. 2013, 31, 845–852. [Google Scholar] [CrossRef]
- Gillison, M.L.; Trotti, A.M.; Harris, J.; Eisbruch, A.; Harari, P.M.; Adelstein, D.J.; Jordan, R.C.K.; Zhao, W.; Sturgis, E.M.; Burtness, B.; et al. Radiotherapy plus cetuximab or cisplatin in human papillomavirus-positive oropharyngeal cancer (NRG Oncology RTOG 1016): A randomised, multicentre, non-inferiority trial. Lancet 2019, 393, 40–50. [Google Scholar] [CrossRef]
- Mehanna, H.; Robinson, M.; Hartley, A.; Kong, A.; Foran, B.; Fulton-Lieuw, T.; Dalby, M.; Mistry, P.; Sen, M.; O’Toole, L.; et al. Radiotherapy plus cisplatin or cetuximab in low-risk human papillomavirus-positive oropharyngeal cancer (De-ESCALaTE HPV): An open-label randomised controlled phase 3 trial. Lancet 2019, 393, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Rose, P.G.; Bundy, B.N.; Watkins, E.B.; Thigpen, J.T.; Deppe, G.; Maiman, M.A.; Clarke-Pearson, D.L.; Insalaco, S. Concurrent cisplatin-based radiotherapy and chemotherapy for locally advanced cervical cancer. N. Engl. J. Med. 1999, 340, 1144–1153. [Google Scholar] [CrossRef]
- Morris, M.; Eifel, P.J.; Lu, J.; Grigsby, P.W.; Levenback, C.; Stevens, R.E.; Rotman, M.; Gershenson, D.M.; Mutch, D.G. Pelvic radiation with concurrent chemotherapy compared with pelvic and para-aortic radiation for high-risk cervical cancer. N. Engl. J. Med. 1999, 340, 1137–1143. [Google Scholar] [CrossRef]
- Green, J.A.; Kirwan, J.M.; Tierney, J.F.; Symonds, P.; Fresco, L.; Collingwood, M.; Williams, C.J. Survival and recurrence after concomitant chemotherapy and radiotherapy for cancer of the uterine cervix: A systematic review and meta-analysis. Lancet 2001, 358, 781–786. [Google Scholar] [CrossRef]
- Bartelink, H.; Roelofsen, F.; Eschwege, F.; Rougier, P.; Bosset, J.F.; Gonzalez, D.G.; Peiffert, D.; van Glabbeke, M.; Pierart, M. Concomitant radiotherapy and chemotherapy is superior to radiotherapy alone in the treatment of locally advanced anal cancer: Results of a phase III randomized trial of the European Organization for Research and Treatment of Cancer Radiotherapy and Gastrointestinal Cooperative Groups. J. Clin. Oncol. 1997, 15, 2040–2049. [Google Scholar] [CrossRef] [PubMed]
- Northover, J.; Glynne-Jones, R.; Sebag-Montefiore, D.; James, R.; Meadows, H.; Wan, S.; Jitlal, M.; Ledermann, J. Chemoradiation for the treatment of epidermoid anal cancer: 13-year follow-up of the first randomised UKCCCR Anal Cancer Trial (ACT I). Br. J. Cancer 2010, 102, 1123–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakhry, C.; Westra, W.H.; Li, S.; Cmelak, A.; Ridge, J.A.; Pinto, H.; Forastiere, A.; Gillison, M.L. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J. Natl. Cancer Inst. 2008, 100, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tan, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Rischin, D.; Young, R.J.; Fisher, R.; Fox, S.B.; Le, Q.T.; Peters, L.J.; Solomon, B.; Choi, J.; O’Sullivan, B.; Kenny, L.M.; et al. Prognostic significance of p16INK4A and human papillomavirus in patients with oropharyngeal cancer treated on TROG 02.02 phase III trial. J. Clin. Oncol. 2010, 28, 4142–4148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Carunchio, L.; Soveral, I.; Steenbergen, R.D.; Torne, A.; Martinez, S.; Fuste, P.; Pahisa, J.; Marimon, L.; Ordi, J.; del Pino, M. HPV-negative carcinoma of the uterine cervix: A distinct type of cervical cancer with poor prognosis. BJOG 2015, 122, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Wakeham, K.; Kavanagh, K.; Cuschieri, K.; Millan, D.; Pollock, K.G.; Bell, S.; Burton, K.; Reed, N.S.; Graham, S.V. HPV status and favourable outcome in vulvar squamous cancer. Int. J. Cancer 2017, 140, 1134–1146. [Google Scholar] [CrossRef]
- Hay, C.M.; Lachance, J.A.; Lucas, F.L.; Smith, K.A.; Jones, M.A. Biomarkers p16, Human Papillomavirus and p53 Predict Recurrence and Survival in Early Stage Squamous Cell Carcinoma of the Vulva. J. Low Genit. Tract. Dis. 2016, 20, 252–256. [Google Scholar] [CrossRef]
- Sand, F.L.; Nielsen, D.M.B.; Frederiksen, M.H.; Rasmussen, C.L.; Kjaer, S.K. The prognostic value of p16 and p53 expression for survival after vulvar cancer: A systematic review and meta-analysis. Gynecol. Oncol. 2018. [Google Scholar] [CrossRef]
- Horne, Z.D.; Dohopolski, M.J.; Pradhan, D.; Bhargava, R.; Edwards, R.P.; Kelley, J.L.; Comerci, J.T.; Olawaiye, A.B.; Courtney-Brooks, M.B.; Bockmeier, M.M.; et al. Human papillomavirus infection mediates response and outcome of vulvar squamous cell carcinomas treated with radiation therapy. Gynecol. Oncol. 2018, 151, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; Welzel, G.; Ottstadt, M.; Lohr, F.; Severa, S.; Prigge, E.S.; Wentzensen, N.; Trunk, M.J.; Wenz, F.; von Knebel-Doeberitz, M.; et al. Prognostic Relevance of HPV Infection and p16 Overexpression in Squamous Cell Anal Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2015, 93, 819–827. [Google Scholar] [CrossRef]
- Meulendijks, D.; Tomasoa, N.B.; Dewit, L.; Smits, P.H.; Bakker, R.; van Velthuysen, M.L.; Rosenberg, E.H.; Beijnen, J.H.; Schellens, J.H.; Cats, A. HPV-negative squamous cell carcinoma of the anal canal is unresponsive to standard treatment and frequently carries disruptive mutations in TP53. Br. J. Cancer 2015, 112, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Serup-Hansen, E.; Linnemann, D.; Skovrider-Ruminski, W.; Hogdall, E.; Geertsen, P.F.; Havsteen, H. Human papillomavirus genotyping and p16 expression as prognostic factors for patients with American Joint Committee on Cancer stages I to III carcinoma of the anal canal. J. Clin. Oncol. 2014, 32, 1812–1817. [Google Scholar] [CrossRef] [Green Version]
- Chera, B.S.; Amdur, R.J.; Green, R.; Shen, C.; Gupta, G.; Tan, X.; Knowles, M.; Fried, D.; Hayes, N.; Weiss, J.; et al. Phase II Trial of De-Intensified Chemoradiotherapy for Human Papillomavirus-Associated Oropharyngeal Squamous Cell Carcinoma. J. Clin. Oncol. 2019, 37, 2661–2669. [Google Scholar] [CrossRef] [PubMed]
- Yom, S.S.; Torres-Saavedra, P.; Caudell, J.J.; Waldron, J.N.; Gillison, M.L.; Xia, P.; Truong, M.T.; Kong, C.; Jordan, R.; Subramaniam, R.M.; et al. Reduced-Dose Radiation Therapy for HPV-Associated Oropharyngeal Carcinoma (NRG Oncology HN002). J. Clin. Oncol. 2021, 39, 956–965. [Google Scholar] [CrossRef]
- Marur, S.; Li, S.; Cmelak, A.J.; Gillison, M.L.; Zhao, W.J.; Ferris, R.L.; Westra, W.H.; Gilbert, J.; Bauman, J.E.; Wagner, L.I.; et al. E1308: Phase II Trial of Induction Chemotherapy Followed by Reduced-Dose Radiation and Weekly Cetuximab in Patients With HPV-Associated Resectable Squamous Cell Carcinoma of the Oropharynx-ECOG-ACRIN Cancer Research Group. J. Clin. Oncol. 2017, 35, 490–497. [Google Scholar] [CrossRef]
- Chen, A.M.; Felix, C.; Wang, P.C.; Hsu, S.; Basehart, V.; Garst, J.; Beron, P.; Wong, D.; Rosove, M.H.; Rao, S.; et al. Reduced-dose radiotherapy for human papillomavirus-associated squamous-cell carcinoma of the oropharynx: A single-arm, phase 2 study. Lancet Oncol. 2017, 18, 803–811. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.J.; Price, K.A.; Moore, E.J.; Patel, S.H.; Hinni, M.L.; Garcia, J.J.; Graner, D.E.; Foster, N.R.; Ginos, B.; Neben-Wittich, M.; et al. Phase II Evaluation of Aggressive Dose De-Escalation for Adjuvant Chemoradiotherapy in Human Papillomavirus-Associated Oropharynx Squamous Cell Carcinoma. J. Clin. Oncol. 2019, 37, 1909–1918. [Google Scholar] [CrossRef]
- Nulton, T.J.; Olex, A.L.; Dozmorov, M.; Morgan, I.M.; Windle, B. Analysis of The Cancer Genome Atlas sequencing data reveals novel properties of the human papillomavirus 16 genome in head and neck squamous cell carcinoma. Oncotarget 2017, 8, 17684–17699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nulton, T.J.; Kim, N.K.; DiNardo, L.J.; Morgan, I.M.; Windle, B. Patients with integrated HPV16 in head and neck cancer show poor survival. Oral Oncol. 2018, 80, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Kimple, R.J.; Smith, M.A.; Blitzer, G.C.; Torres, A.D.; Martin, J.A.; Yang, R.Z.; Peet, C.R.; Lorenz, L.D.; Nickel, K.P.; Klingelhutz, A.J.; et al. Enhanced radiation sensitivity in HPV-positive head and neck cancer. Cancer Res. 2013, 73, 4791–4800. [Google Scholar] [CrossRef] [Green Version]
- Rieckmann, T.; Tribius, S.; Grob, T.J.; Meyer, F.; Busch, C.J.; Petersen, C.; Dikomey, E.; Kriegs, M. HNSCC cell lines positive for HPV and p16 possess higher cellular radiosensitivity due to an impaired DSB repair capacity. Radiother. Oncol. 2013, 107, 242–246. [Google Scholar] [CrossRef]
- Lilja-Fischer, J.K.; Ulhoi, B.P.; Alsner, J.; Stougaard, M.; Thomsen, M.S.; Busk, M.; Lassen, P.; Steiniche, T.; Nielsen, V.E.; Overgaard, J. Characterization and radiosensitivity of HPV-related oropharyngeal squamous cell carcinoma patient-derived xenografts. Acta Oncol. 2019, 58, 1489–1494. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Nickel, K.P.; Torres, A.D.; Lee, D.; Lambert, P.F.; Kimple, R.J. Human papillomavirus type 16 E7 oncoprotein causes a delay in repair of DNA damage. Radiother. Oncol. 2014, 113, 337–344. [Google Scholar] [CrossRef] [Green Version]
- Kranjc Brezar, S.; Prevc, A.; Niksic Zakelj, M.; Brozic, A.; Cemazar, M.; Strojan, P.; Sersa, G. Synergistic effect of cisplatin chemotherapy combined with fractionated radiotherapy regimen in HPV-positive and HPV-negative experimental pharyngeal squamous cell carcinoma. Sci. Rep. 2020, 10, 1563. [Google Scholar] [CrossRef] [PubMed]
- Spiotto, M.T.; Taniguchi, C.M.; Klopp, A.H.; Colbert, L.E.; Lin, S.H.; Wang, L.; Frederick, M.J.; Osman, A.A.; Pickering, C.R.; Frank, S.J. Biology of the Radio- and Chemo-Responsiveness in HPV Malignancies. Semin. Radiat. Oncol. 2021, 31, 274–285. [Google Scholar] [CrossRef]
- Wallace, N.A.; Khanal, S.; Robinson, K.L.; Wendel, S.O.; Messer, J.J.; Galloway, D.A. High-Risk Alphapapillomavirus Oncogenes Impair the Homologous Recombination Pathway. J. Virol. 2017, 91, e01084-17. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Ma, L.; Jones, T.; Palomero, L.; Pujana, M.A.; Martinez-Ruiz, H.; Ha, P.K.; Murnane, J.; Cuartas, I.; Seoane, J.; et al. Subjugation of TGFbeta Signaling by Human Papilloma Virus in Head and Neck Squamous Cell Carcinoma Shifts DNA Repair from Homologous Recombination to Alternative End Joining. Clin. Cancer Res. 2018, 24, 6001–6014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leeman, J.E.; Li, Y.; Bell, A.; Hussain, S.S.; Majumdar, R.; Rong-Mullins, X.; Blecua, P.; Damerla, R.; Narang, H.; Ravindran, P.T.; et al. Human papillomavirus 16 promotes microhomology-mediated end-joining. Proc. Natl. Acad. Sci. USA 2019, 116, 21573–21579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitz, J.; Blanchet, S.A.; Gameiro, S.F.; Biquand, E.; Morgan, T.M.; Galloy, M.; Dessapt, J.; Lavoie, E.G.; Blondeau, A.; Smith, B.C.; et al. Human papillomavirus E7 oncoprotein targets RNF168 to hijack the host DNA damage response. Proc. Natl. Acad. Sci. USA 2019, 116, 19552–19562. [Google Scholar] [CrossRef] [Green Version]
- Prevc, A.; Kranjc, S.; Cemazar, M.; Todorovic, V.; Zegura, B.; Novak, M.; Filipic, M.; Flezar, M.S.; Kirbis, I.S.; Rotter, A.; et al. Dose-Modifying Factor of Radiation Therapy with Concurrent Cisplatin Treatment in HPV-Positive Squamous Cell Carcinoma: A Preclinical Study. Radiat. Res. 2018, 189, 644–651. [Google Scholar] [CrossRef]
- Jha, S.; Vande Pol, S.; Banerjee, N.S.; Dutta, A.B.; Chow, L.T.; Dutta, A. Destabilization of TIP60 by human papillomavirus E6 results in attenuation of TIP60-dependent transcriptional regulation and apoptotic pathway. Mol. Cell 2010, 38, 700–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.; Cheng, S.; Iovane, A.; Laimins, L.A. STAT-5 Regulates Transcription of the Topoisomerase IIbeta-Binding Protein 1 (TopBP1) Gene to Activate the ATR Pathway and Promote Human Papillomavirus Replication. mBio 2015, 6, e02006-15. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Laimins, L.A. The JAK-STAT transcriptional regulator, STAT-5, activates the ATM DNA damage pathway to induce HPV 31 genome amplification upon epithelial differentiation. PLoS Pathog. 2013, 9, e1003295. [Google Scholar] [CrossRef] [Green Version]
- Morgan, E.L.; Macdonald, A. Manipulation of JAK/STAT Signalling by High-Risk HPVs: Potential Therapeutic Targets for HPV-Associated Malignancies. Viruses 2020, 12, 977. [Google Scholar] [CrossRef]
- Dok, R.; Kalev, P.; Van Limbergen, E.J.; Asbagh, L.A.; Vazquez, I.; Hauben, E.; Sablina, A.; Nuyts, S. p16INK4a impairs homologous recombination-mediated DNA repair in human papillomavirus-positive head and neck tumors. Cancer Res. 2014, 74, 1739–1751. [Google Scholar] [CrossRef] [Green Version]
- Jirawatnotai, S.; Hu, Y.; Michowski, W.; Elias, J.E.; Becks, L.; Bienvenu, F.; Zagozdzon, A.; Goswami, T.; Wang, Y.E.; Clark, A.B.; et al. A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature 2011, 474, 230–234. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhang, P.; Molkentine, D.P.; Chen, C.; Molkentine, J.M.; Piao, H.; Raju, U.; Zhang, J.; Valdecanas, D.R.; Tailor, R.C.; et al. TRIP12 as a mediator of human papillomavirus/p16-related radiation enhancement effects. Oncogene 2017, 36, 820–828. [Google Scholar] [CrossRef] [Green Version]
- Velez-Cruz, R.; Manickavinayaham, S.; Biswas, A.K.; Clary, R.W.; Premkumar, T.; Cole, F.; Johnson, D.G. RB localizes to DNA double-strand breaks and promotes DNA end resection and homologous recombination through the recruitment of BRG1. Genes Dev. 2016, 30, 2500–2512. [Google Scholar] [CrossRef] [Green Version]
- Busch, C.J.; Becker, B.; Kriegs, M.; Gatzemeier, F.; Kruger, K.; Mockelmann, N.; Fritz, G.; Petersen, C.; Knecht, R.; Rothkamm, K.; et al. Similar cisplatin sensitivity of HPV-positive and -negative HNSCC cell lines. Oncotarget 2016, 7, 35832–35842. [Google Scholar] [CrossRef]
- Hussain, S.S.; Majumdar, R.; Moore, G.M.; Narang, H.; Buechelmaier, E.S.; Bazil, M.J.; Ravindran, P.T.; Leeman, J.E.; Li, Y.; Jalan, M.; et al. Measuring nonhomologous end-joining, homologous recombination and alternative end-joining simultaneously at an endogenous locus in any transfectable human cell. Nucleic Acids Res. 2021, 49, e74. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, K.A.; Mehta, K.P.; Laimins, L.A.; Moody, C.A. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J. Virol. 2012, 86, 9520–9526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moody, C.A.; Laimins, L.A. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef] [Green Version]
- Anacker, D.C.; Gautam, D.; Gillespie, K.A.; Chappell, W.H.; Moody, C.A. Productive replication of human papillomavirus 31 requires DNA repair factor Nbs1. J. Virol. 2014, 88, 8528–8544. [Google Scholar] [CrossRef] [Green Version]
- Mehta, K.; Laimins, L. Human Papillomaviruses Preferentially Recruit DNA Repair Factors to Viral Genomes for Rapid Repair and Amplification. mBio 2018, 9, e00064-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattiroli, F.; Vissers, J.H.; van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Alvarez-Quilon, A.; Moatti, N.; Zimmermann, M.; et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef]
- Mirman, Z.; Lottersberger, F.; Takai, H.; Kibe, T.; Gong, Y.; Takai, K.; Bianchi, A.; Zimmermann, M.; Durocher, D.; de Lange, T. 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature 2018, 560, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Dev, H.; Chiang, T.W.; Lescale, C.; de Krijger, I.; Martin, A.G.; Pilger, D.; Coates, J.; Sczaniecka-Clift, M.; Wei, W.; Ostermaier, M.; et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018, 20, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Somyajit, K.; Narita, T.; Maskey, E.; Stanlie, A.; Kremer, M.; Typas, D.; Lammers, M.; Mailand, N.; Nussenzweig, A.; et al. DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell 2018, 173, 972–988.e923. [Google Scholar] [CrossRef] [Green Version]
- Cook, R.; Zoumpoulidou, G.; Luczynski, M.T.; Rieger, S.; Moquet, J.; Spanswick, V.J.; Hartley, J.A.; Rothkamm, K.; Huang, P.H.; Mittnacht, S. Direct involvement of retinoblastoma family proteins in DNA repair by non-homologous end-joining. Cell Rep. 2015, 10, 2006–2018. [Google Scholar] [CrossRef] [Green Version]
- Chappell, W.H.; Gautam, D.; Ok, S.T.; Johnson, B.A.; Anacker, D.C.; Moody, C.A. Homologous Recombination Repair Factors Rad51 and BRCA1 Are Necessary for Productive Replication of Human Papillomavirus 31. J. Virol. 2015, 90, 2639–2652. [Google Scholar] [CrossRef] [Green Version]
- Anacker, D.C.; Aloor, H.L.; Shepard, C.N.; Lenzi, G.M.; Johnson, B.A.; Kim, B.; Moody, C.A. HPV31 utilizes the ATR-Chk1 pathway to maintain elevated RRM2 levels and a replication-competent environment in differentiating Keratinocytes. Virology 2016, 499, 383–396. [Google Scholar] [CrossRef]
- Reinson, T.; Toots, M.; Kadaja, M.; Pipitch, R.; Allik, M.; Ustav, E.; Ustav, M. Engagement of the ATR-dependent DNA damage response at the human papillomavirus 18 replication centers during the initial amplification. J. Virol. 2013, 87, 951–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauson, E.J.; Donaldson, M.M.; Dornan, E.S.; Wang, X.; Bristol, M.; Bodily, J.M.; Morgan, I.M. Evidence supporting a role for TopBP1 and Brd4 in the initiation but not continuation of human papillomavirus 16 E1/E2-mediated DNA replication. J. Virol. 2015, 89, 4980–4991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakakibara, N.; Mitra, R.; McBride, A.A. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J. Virol. 2011, 85, 8981–8995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, D.; Bristol, M.L.; Smith, N.W.; James, C.D.; Wang, X.; Pichierri, P.; Morgan, I.M. Werner Helicase Control of Human Papillomavirus 16 E1-E2 DNA Replication Is Regulated by SIRT1 Deacetylation. mBio 2019, 10, e00263-19. [Google Scholar] [CrossRef] [Green Version]
- Fradet-Turcotte, A.; Bergeron-Labrecque, F.; Moody, C.A.; Lehoux, M.; Laimins, L.A.; Archambault, J. Nuclear accumulation of the papillomavirus E1 helicase blocks S-phase progression and triggers an ATM-dependent DNA damage response. J. Virol. 2011, 85, 8996–9012. [Google Scholar] [CrossRef] [Green Version]
- Boner, W.; Taylor, E.R.; Tsirimonaki, E.; Yamane, K.; Campo, M.S.; Morgan, I.M. A Functional interaction between the human papillomavirus 16 transcription/replication factor E2 and the DNA damage response protein TopBP1. J. Biol. Chem. 2002, 277, 22297–22303. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.; Gaykalova, D.A.; Guo, T.; Favorov, A.V.; Fertig, E.J.; Tamayo, P.; Callejas-Valera, J.L.; Allevato, M.; Gilardi, M.; Santos, J.; et al. HPV E2, E4, E5 drive alternative carcinogenic pathways in HPV positive cancers. Oncogene 2020, 39, 6327–6339. [Google Scholar] [CrossRef] [PubMed]
- Pang, E.; Delic, N.C.; Hong, A.; Zhang, M.; Rose, B.R.; Lyons, J.G. Radiosensitization of oropharyngeal squamous cell carcinoma cells by human papillomavirus 16 oncoprotein E6 *I. Int. J. Radiat. Oncol. Biol. Phys. 2011, 79, 860–865. [Google Scholar] [CrossRef]
- Shamseddine, A.A.; Burman, B.; Lee, N.Y.; Zamarin, D.; Riaz, N. Tumor Immunity and Immunotherapy for HPV-Related Cancers. Cancer Discov. 2021, 11, 1896–1912. [Google Scholar] [CrossRef]
- Cleary, C.; Leeman, J.E.; Higginson, D.S.; Katabi, N.; Sherman, E.; Morris, L.; McBride, S.; Lee, N.; Riaz, N. Biological Features of Human Papillomavirus-related Head and Neck Cancers Contributing to Improved Response. Clin. Oncol. R Coll. Radiol. 2016, 28, 467–474. [Google Scholar] [CrossRef] [Green Version]
- Akagi, K.; Li, J.; Broutian, T.R.; Padilla-Nash, H.; Xiao, W.; Jiang, B.; Rocco, J.W.; Teknos, T.N.; Kumar, B.; Wangsa, D.; et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 2014, 24, 185–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duensing, S.; Munger, K. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 2002, 62, 7075–7082. [Google Scholar]
- White, A.E.; Livanos, E.M.; Tlsty, T.D. Differential disruption of genomic integrity and cell cycle regulation in normal human fibroblasts by the HPV oncoproteins. Genes Dev. 1994, 8, 666–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Gillison, M.L.; Akagi, K.; Xiao, W.; Jiang, B.; Pickard, R.K.L.; Li, J.; Swanson, B.J.; Agrawal, A.D.; Zucker, M.; Stache-Crain, B.; et al. Human papillomavirus and the landscape of secondary genetic alterations in oral cancers. Genome Res. 2019, 29, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Popova, T.; Manie, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Deriano, L.; Stracker, T.H.; Baker, A.; Petrini, J.H.; Roth, D.B. Roles for NBS1 in alternative nonhomologous end-joining of V(D)J recombination intermediates. Mol. Cell 2009, 34, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuler, W.; Weiler, I.J.; Schuler, A.; Phillips, R.A.; Rosenberg, N.; Mak, T.W.; Kearney, J.F.; Perry, R.P.; Bosma, M.J. Rearrangement of antigen receptor genes is defective in mice with severe combined immune deficiency. Cell 1986, 46, 963–972. [Google Scholar] [CrossRef]
- Riaz, N.; Sherman, E.; Pei, X.; Schoder, H.; Grkovski, M.; Paudyal, R.; Katabi, N.; Selenica, P.; Yamaguchi, T.N.; Ma, D.; et al. Precision Radiotherapy: Reduction in Radiation for Oropharyngeal Cancer in the 30 ROC Trial. J. Natl. Cancer Inst. 2021, 113, 742–751. [Google Scholar] [CrossRef]
- Park, W.; Chen, J.; Chou, J.F.; Varghese, A.M.; Yu, K.H.; Wong, W.; Capanu, M.; Balachandran, V.; McIntyre, C.A.; El Dika, I.; et al. Genomic Methods Identify Homologous Recombination Deficiency in Pancreas Adenocarcinoma and Optimize Treatment Selection. Clin. Cancer Res. 2020, 26, 3239–3247. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.M.; Lee, J.W.; Zalupski, M.; Capanu, M.; Park, J.; Golan, T.; Tahover, E.; Lowery, M.A.; Chou, J.F.; Sahai, V.; et al. Randomized, Multicenter, Phase II Trial of Gemcitabine and Cisplatin with or Without Veliparib in Patients with Pancreas Adenocarcinoma and a Germline BRCA/PALB2 Mutation. J. Clin. Oncol. 2020, 38, 1378–1388. [Google Scholar] [CrossRef] [PubMed]
- Pomerantz, M.M.; Spisak, S.; Jia, L.; Cronin, A.M.; Csabai, I.; Ledet, E.; Sartor, A.O.; Rainville, I.; O’Connor, E.P.; Herbert, Z.T.; et al. The association between germline BRCA2 variants and sensitivity to platinum-based chemotherapy among men with metastatic prostate cancer. Cancer 2017, 123, 3532–3539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.C.; Zhang, J.; Xu, B.H.; Cai, L.; Ragaz, J.; Wang, Z.H.; Wang, B.Y.; Teng, Y.E.; Tong, Z.S.; Pan, Y.Y.; et al. Cisplatin plus gemcitabine versus paclitaxel plus gemcitabine as first-line therapy for metastatic triple-negative breast cancer (CBCSG006): A randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2015, 16, 436–446. [Google Scholar] [CrossRef]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Z.; Xie, X. BRCA mutations in the manifestation and treatment of ovarian cancer. Oncotarget 2017, 8, 97657–97670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermorken, J.B.; Psyrri, A.; Mesia, R.; Peyrade, F.; Beier, F.; de Blas, B.; Celik, I.; Licitra, L. Impact of tumor HPV status on outcome in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck receiving chemotherapy with or without cetuximab: Retrospective analysis of the phase III EXTREME trial. Ann. Oncol. 2014, 25, 801–807. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Characteristic | HPV+ HNSCC | HPV− HNSCC |
|---|---|---|
| Dysregulated tumor suppressors | Rb (E7), p53 (E6) | p16(INK4A), p53 |
| Copy number alterations (median) | 113 | 136 |
| “M” class tumors | 58% | 27% |
| Somatic mutation frequency | Similar | Similar |
| Single base substitutions signature | APOBEC | Smoking |
| Alt-EJ genomic scars | Higher | Lower |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussain, S.S.; Lundine, D.; Leeman, J.E.; Higginson, D.S. Genomic Signatures in HPV-Associated Tumors. Viruses 2021, 13, 1998. https://doi.org/10.3390/v13101998
Hussain SS, Lundine D, Leeman JE, Higginson DS. Genomic Signatures in HPV-Associated Tumors. Viruses. 2021; 13(10):1998. https://doi.org/10.3390/v13101998
Chicago/Turabian StyleHussain, Suleman S., Devon Lundine, Jonathan E. Leeman, and Daniel S. Higginson. 2021. "Genomic Signatures in HPV-Associated Tumors" Viruses 13, no. 10: 1998. https://doi.org/10.3390/v13101998
APA StyleHussain, S. S., Lundine, D., Leeman, J. E., & Higginson, D. S. (2021). Genomic Signatures in HPV-Associated Tumors. Viruses, 13(10), 1998. https://doi.org/10.3390/v13101998