
Phylogenetic Analysis of European Brown Hare Syndrome Virus Strains from Poland (1992–2004)


Abstract
:1. Introduction
2. Materials and Methods
2.1. EBHSV Strains and Lagovirus Sequences
2.2. Amplification of the EBHSV Genome
2.3. Sequencing and Phylogenetic Analyzis
3. Results
3.1. Sequence Analyzis of EBHSV Strains from Poland
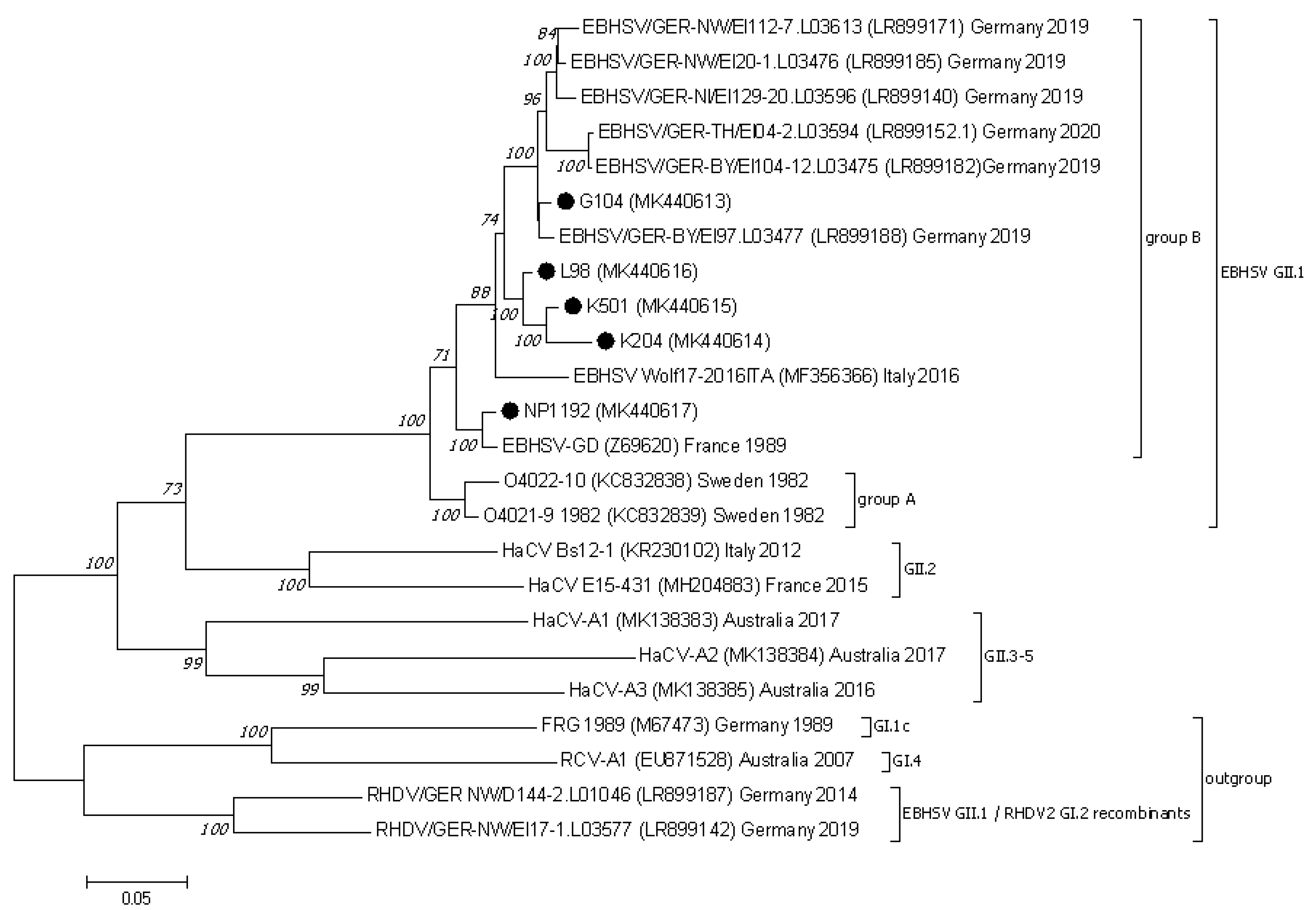
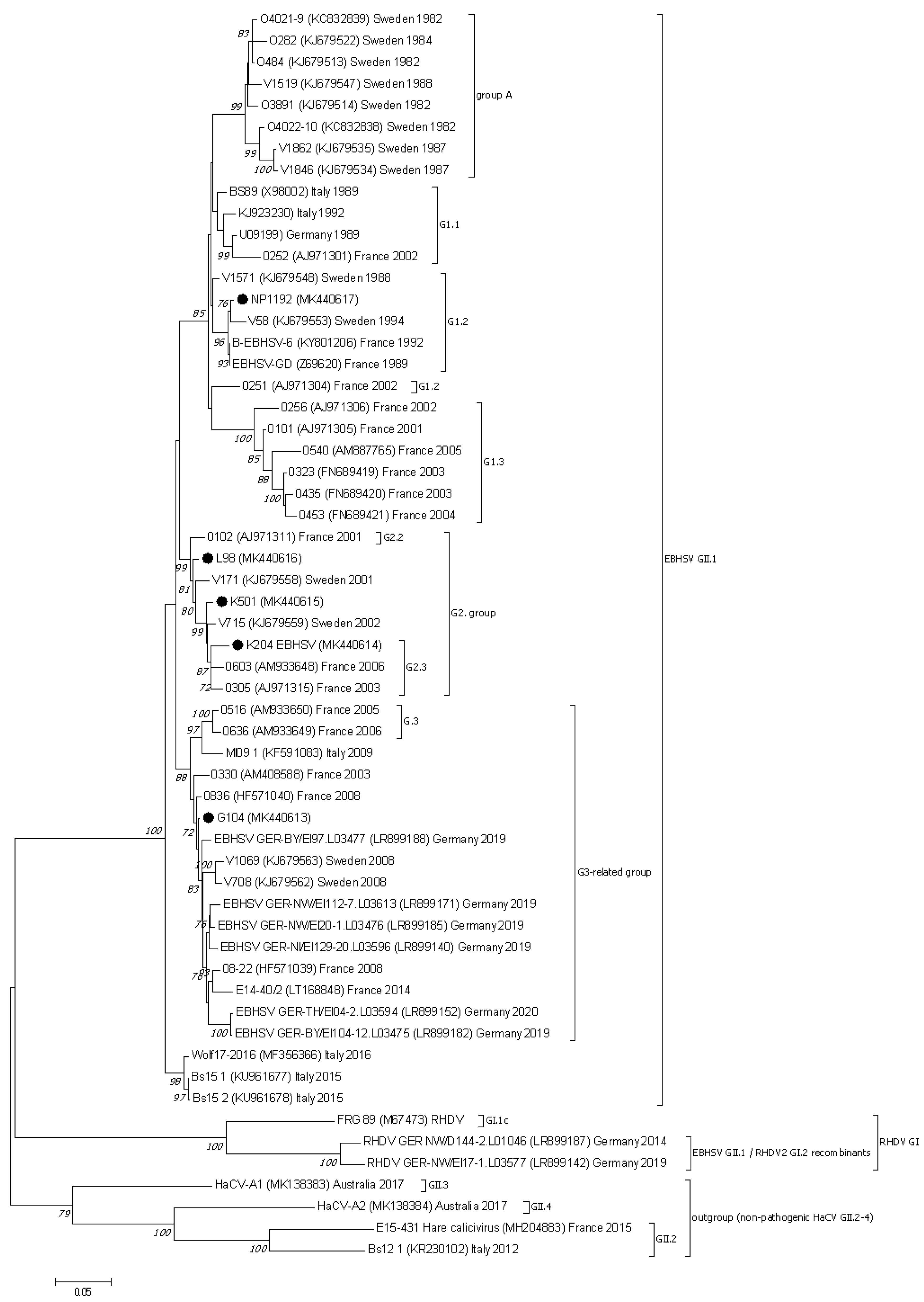
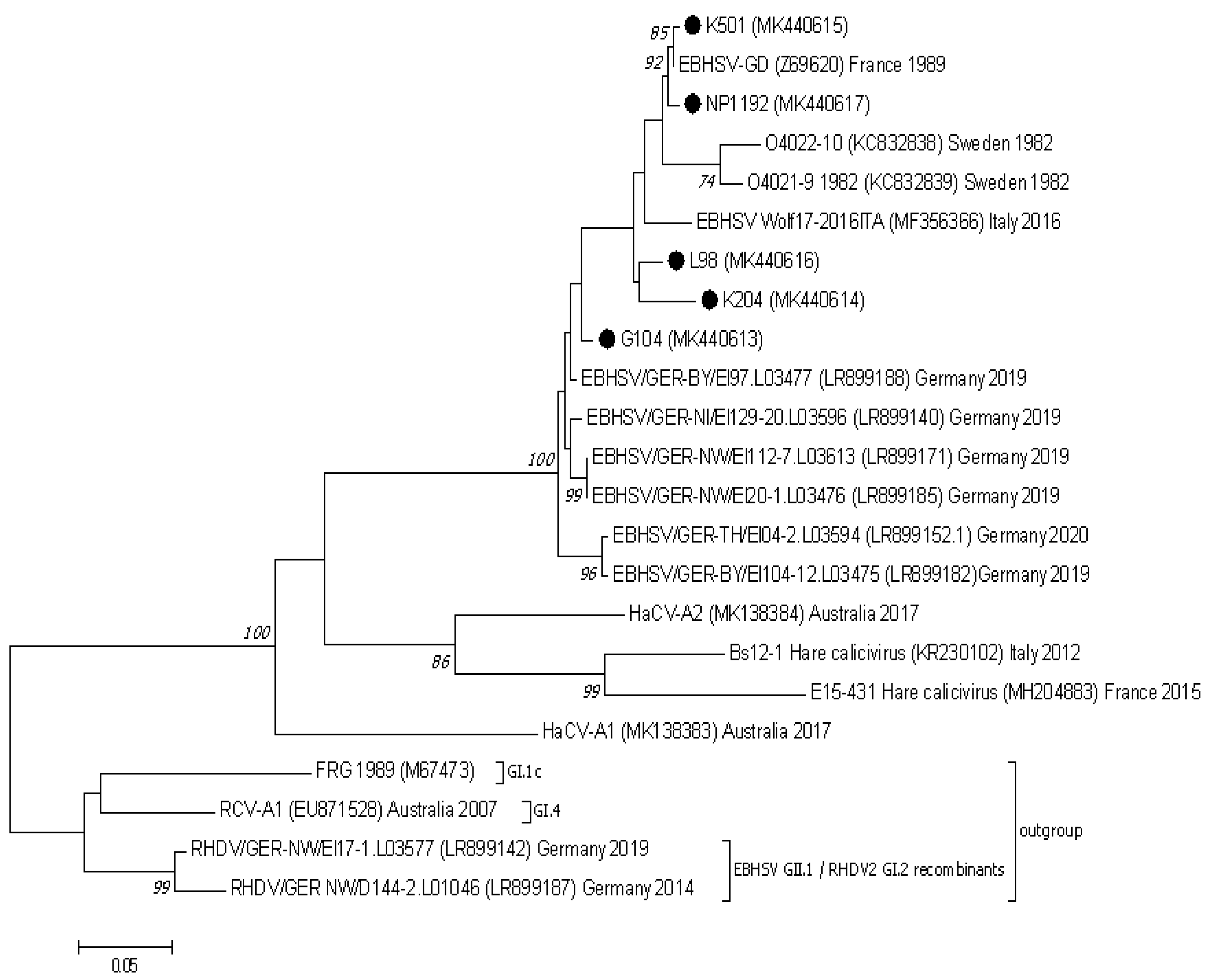
3.2. Analysis of the Lagovirus Phylogenetic Relationships Based on ORF1–ORF2 Genome Sequences
3.3. Nucletide and Amino Acid Sequence Analysis of the Structural Genes of EBHSV Strains from Poland and Elsewhere in Europe
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gavier-Widén, D.; Mörner, T. Epidemiology and diagnosis of the European brown hare syndrome in Scandinavian countries: A review. Rev. Sci. Tech. 1991, 10, 453–458. [Google Scholar] [CrossRef] [Green Version]
- Bascunana, R.C.; Novotny, N.; Belak, S. Detection and differentiation of rabbit hemorrhagic disease and European brown hare syndrome viruses by amplification of VP60 genomic sequences from fresh and fixed tissue specimens. J. Clin. Microbiol. 1997, 35, 2492–2495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duff, J.P.; Chasey, D.; Munro, R.; Wooldridge, M. European brown hare syndrome in England. Vet. Rec. 1994, 134, 669–673. [Google Scholar] [CrossRef]
- Syrjälä, P.; Nylund, M.; Heinikainen, S. European brown hare syndrome in free-living mountain hares (Lepus timidus) and European brown hares (Lepus europaeus) in Finland 1990–2002. J. Wildl. Dis. 2005, 41, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Cancellotti, F.M.; Renzi, M. Epidemiology and current situation of viral haemorrhagic disease of rabbits and the European brown hare syndrome in Italy. Rev. Sci. Tech. 1991, 10, 409–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capucci, L.; Scicluna, M.T.; Lavazza, A. Diagnosis of viral haemorrhagic disease of rabbits and the European brown hare syndrome. Rev. Sci. Tech. 1991, 10, 347–370. [Google Scholar] [CrossRef] [PubMed]
- Chasey, D.; Duff, P. European brown hare syndrome and associated virus particles in the UK. Vet. Rec. 1990, 126, 623–624. [Google Scholar] [PubMed]
- Frölich, K.; Fickel, J.; Ludwig, A.; Lieckfeldt, D.; Streich, W.J.; Jurčik, R.; Slamečka, J.; Wibbelt, G. New variants of European brown hare syndrome virus strains in free-ranging European brown hares (Lepus Europaeus) from Slovakia. J. Wildl. Dis. 2007, 43, 89–96. [Google Scholar] [CrossRef] [Green Version]
- Frölich, K.; Haerer, G.; Bacciarini, L.; Janovsky, M.; Rudolph, M.; Giacometti, M. European brown hare syndrome in free-ranging European brown and mountains hares from Switzerland. J. Wildl. Dis. 2001, 37, 803–807. [Google Scholar] [CrossRef] [Green Version]
- Frölich, K.; Meyer, H.; Pielowski, Z.; Ronsholt, L.; von Seck-Lanzendorf, S.; Stolte, M. European brown hare syndrome in free-ranging hares in Poland. J. Wildl. Dis. 1996, 32, 280–285. [Google Scholar] [CrossRef] [Green Version]
- Löliger, H.C.; Eskens, U. Incidence, epizootiology and control of viral haemorrhagic disease of rabbits and the European brown hare syndrome in Germany. Rev. Sci. Tech. 1991, 10, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Marcato, P.S.; Benazzi, C.; Vecchi, G.; Galeotti, M.; Della Salda, L.; Sarli, G.; Lucidi, P. Clinical and pathological features of rabbit hemorrhagic disease virus and of European brown hare syndrome. Rev. Sci. Tech. 1991, 10, 371–392. [Google Scholar] [CrossRef] [PubMed]
- Morisse, J.P.; Le Gall, G.; Boilletot, E. Hepatitis of viral origin in Leporidae: Introduction and aetiological hypotheses. Rev. Sci. Tech. 1991, 10, 283–295. [Google Scholar]
- Nauwynck, H.; Callebaut, P.; Peeters, J.; Ducatelle, R.; Uyttebroek, E. Susceptibility of hares and rabbits to a Belgian isolate of European brown hare syndrome virus. J. Wildl. Dis. 1993, 29, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Zanni, M.L.; Benassi, M.C.; Scicluna, M.T.; Lavazza, A.; Capucci, L. Clinical evolution and diagnosis of an outbreak of European brown hare syndrome in hares reared in captivity. Rev. Sci. Tech. 1993, 12, 931–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chrobocińska, M.; Górski, J. Prevalence of infection with EBHS (European Brown Hare Syndrome) virus in hares in Poland. Bull. Vet. I Pulawy 1995, 39, 17–21. [Google Scholar]
- Chrobocińska, M. The case of infection of hare with European brown hare syndrome virus. Med. Weter. 1999, 55, 750–752. [Google Scholar]
- Kwit, E.; Chrobocińska, M.; Grądzki, Z.; Jarosz, Ł.; Majer-Dziedzic, B.; Bigoraj, E. The genetic analysis of new Polish strains of European brown hare syndrome virus. Pol. J. Vet. Sci. 2014, 17, 353–355. [Google Scholar] [CrossRef] [Green Version]
- Lavazza, A.; Vecchi, G. Osservazioni su alcuni episodi di mortalita nella leper: Evidenziazione al microscopio elettronico di una particella virale. Selez. Vet. 1989, 30, 461–468. [Google Scholar]
- Le Pendu, J.; Abrantes, J.; Bertagnoli, S.; Guitton, J.S.; Le Gall-Reculé, G.; Lopes, A.M.; Marchandeau, S.; Alda, F.; Almeida, T.; Celio, A.P.; et al. Proposal for a unified classification system and nomenclature of lagoviruses. J. Gen. Virol. 2017, 98, 1658–1666. [Google Scholar] [CrossRef]
- Droillard, C.; Lemaitre, E.; Chatel, M.; Guitton, J.S.; Marchandeau, S.; Eterradossi, N.; Le Gall-Reculé, G. First complete genome sequence of a hare calicivirus strain isolated from Lepus europaeus. Microb. Res. Ann. 2018, 7, e01224-18. [Google Scholar] [CrossRef] [Green Version]
- Forrester, N.L.; Moss, S.R.; Turner, S.L.; Schirrmeier, H.; Gould, E.A. Recombination in rabbit haemorrhagic disease virus: Possible impact on evolution and epidemiology. Virology 2008, 376, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.M.; Dalton, K.P.; Magalhaes, M.J.; Parra, F.; Esteves, P.J.; Holmes, E.C.; Abrantes, J. Full genomic analysis of new variant rabbit hemorrhagic disease virus (RHDVb) revealed multiple recombination events. J. Gen. Virol. 2015, 96, 1309–1319. [Google Scholar] [CrossRef] [PubMed]
- Silvério, D.; Lopes, A.M.; Mel-Ferreira, J.; Magalhaes, M.J.; Monterosso, P.; Serronha, A.; Maio, E.; Alves, P.C.; Esteves, P.J. Insights into the evolution of the new variant rabbit haemorrhagic disease virus (GI.2) and the identification of novel recombinant strains. Transbound Emerg. Dis. 2018, 65, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, G.; Huguet, S.; Vende, P.; Vautherot, J.F.; Rasschaert, D. European brown hare syndrome virus: Molecular cloning and sequencing of the genome. J. Gen. Virol. 1996, 77, 1693–1697. [Google Scholar] [CrossRef]
- Wirblich, C.; Meyers, G.; Ohlinger, V.F.; Capucci, L.; Eskens, U.; Haas, B.; Thiel, H.J. European brown hare syndrome virus—Relationship to rabbit hemorrhagic disease virus and other caliciviruses. J. Virol. 1994, 68, 5164–5173. [Google Scholar] [CrossRef] [Green Version]
- Wirblich, C.; Thiel, H.J.; Meyers, G. Genetic map of the calicivirus rabbit hemorrhagic disease virus as deduced from in vitro translation studies. J. Virol. 1996, 70, 7974–7983. [Google Scholar] [CrossRef] [Green Version]
- Billinis, C.; Knowles, N.J.; Spyrou, V.; Sofianidis, G.; Psychas, V.; Birtsas, P.K.; Sofia, M.; Maslarinou, O.M.; Tontis, D.K.; Kanteres, D. Genetic analysis of the first European brown hare syndrome virus isolates from Greece. Wildl. Biol. Pract. 2005, 1, 118–127. [Google Scholar] [CrossRef]
- Novotny, N.; Bascunana, C.R.; Uhlén, M.; Belák, S. Phylogenetic analysis of rabbit haemorrhagic disease and European brown hare syndrome viruses by comparison of sequences from the capsid protein gene. Arch. Virol. 1997, 142, 657–673. [Google Scholar] [CrossRef]
- Lopes, A.M.; Capucci, L.; Gaviér-Widen, D.; Le Gall-Reculé, G.; Brocchi, E.; Barbieri, I.; Quéméner, A.; Le Pendu, J.; Geoghegan, J.L.; Holmes, E.C.; et al. Molecular evolution and antigenic variation of European brown hare syndrome virus (EBHSV). Virology 2014, 468–470, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Drews, B.; Szentiks, C.A.; Roellig, K.; Fickel, J.; Schroeder, K.; Duff, J.P.; Lavazza, A.; Hildebrandt, T.B.; Goeritz, F. Epidemiology, control and management of an EBHS outbreak in captive hares. Vet. Microbiol. 2011, 154, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Le Gall-Reculé, G.; Zwingelstein, F.; Laurent, S.; Portejoie, Y.; Rasschaert, D. Molecular epidemiology of European brown hare syndrome virus in France between 1989 and 2003. Arch. Virol. 2006, 151, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Szillat, K.P.; Höper, D.; Beer, M.; König, P. Full-genome sequencing of German rabbit haemorrhagic disease virus uncovers recombination between RHDV (GI.2) and EBHSV (GII.1). Virus Evol. 2020, 6, veaa080. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Hall, R.N.; Mahar, J.E.; Read, A.J.; Mourant, R.; Piper, M.; Huang, N.; Strive, T. A strain-specific multiplex RT-PCR for Australian Rabbit haemorrhagic disease viruses uncovers a new recombinant virus variant in rabbits and hares. Transbound Emerg. Dis. 2018, 65, e444–e456. [Google Scholar] [CrossRef]
- Mahar, J.E.; Hall, R.N.; Shi, M.; Mourant, R.; Huang, N.; Strive, T.; Holmes, E.C. The discover of three new hare lagoviruses reveals unexplored viral diversity in this genus. Virus Evol. 2019, 5, vez005. [Google Scholar] [CrossRef] [Green Version]
- Panek, M. Current situation of hares and partridges and the management of their populations. In Animal Population Management; Polski Związek Łowiecki: Nowy Świat, Poland, 2016; pp. 99–109. Available online: www.czempin.pzlow.pl (accessed on 29 January 2021). (In Polish)
- Camarda, A.; Pugliese, N.; Cavadini, P.; Circella, E.; Capucci, L.; Caroli, A.; Legretto, M.; Malia, E.; Lavazza, A. Detection of the new emerging rabbit haemorrhagic disease type 2 virus (RHDV2) in Sicily from rabbit (Oryctolagus cuniculus) and Italian hare (Lepus corsicanus). Res. Vet. Sci. 2014, 97, 642–645. [Google Scholar] [CrossRef]
- Le Gall-Reculé, G.; Lemaitre, E.; Bertagnoli, S.; Hubert, C.; Top, S.; Decors, A.; Marchandeau, S.; Guitton, J.S. Large-scale lagovirus disease outbreaks in European brown hares (Lepus europaeus) in France caused by RHDV2 strains spatially shared with rabbits (Oryctolagus cuniculus). Vet. Res. 2017, 48, 70. [Google Scholar] [CrossRef] [Green Version]
- Lopes, A.M.; Marques, S.; Silva, E.; Magalhães, M.J.; Pinheiro, A.; Alves, P.C.; Le Pendu, J.; Esteves, P.J.; Thompson, G.; Abrantes, J. Detection of RHDV strains in the Iberian hare (Lepus granatensis): Earliest evidence of rabbit lagovirus cross-species infection. Vet. Res. 2014, 45, 94–101. [Google Scholar] [PubMed] [Green Version]
- Neimanis, A.; Ahola, H.; Larsson Petersson, U.; Lopes, A.M.; Abrantes, J.; Zohari, S.; Esteves, P.J.; Gavier-Widen, D. Overcoming species barriers: An outbreak of Lagovirus europaeus GI.2/RHDV2 in an isolated population of mountain hares (Lepus timidus). BMC Vet. Res. 2018, 14, 367. [Google Scholar] [CrossRef] [Green Version]
- Puggioni, G.; Cavadini, P.; Maestrale, C.; Scivoli, R.; Botti, G.; Ligios, C.; Le Gall-Reculé, G.; Lavazza, A.; Capucci, L. The new French 2010 variant of the rabbit of the hemorrhagic disease virus causes an RHD-like disease in the Sardinian Cape hare (Lepus capensis mediterraneus). Vet. Res. 2013, 44, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velarde, R.; Cavadini, P.; Neimanis, A.; Cabezon, O.; Chiari, M.; Gaffuri, A.; Lavin, S.; Grilli, G.; Gavier-Widen, D.; Lavazza, A.; et al. Spillover events of infection of Brown hares (Lepus europaeus) with rabbit haemorrhagic disease type 2 virus (RHDV2) caused sporadic cases of an European Brown Hare Syndrome-like disease in Italy and Spain. Transbound Emerg. Dis. 2017, 64, 1750–1761. [Google Scholar] [CrossRef] [PubMed]
- Chrobocińska, M. Analysis of the fragment of capsid protein gene sequences of Polish strains of European brown hare syndrome virus. Bull. Vet. Inst. Pulawy 2002, 46, 213–222. [Google Scholar]
- Chrobocińska, M. Charakterystyka Fenotypowa i Molekularna Krajowych Szczepów Wirusów Krwotocznej Choroby Zajęcy (EBHSV) i Krwotocznej Choroby Królików (RHDV) (Phenotypic and Molecular Characteristics of Native Strains of European Brown Hare Syndrome Virus (EBHSV) and Rabbit Haemorrhagic Disease Virus (RHDV) (In Polish)). In Habilitation Dissertation; The National Veterinary Research Institute: Puławy, Poland, 2007. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| EBHSV Strain/Acc. Number | NP1192 | L98 | K501 | G104 | K204 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ORF1-ORF2 | NSP | VP60 | VP10 | ORF1-ORF2 | NSP | VP60 | VP10 | ORF1-ORF2 | NSP | VP60 | VP10 | ORF1-ORF2 | NSP | VP60 | VP10 | ORF1-ORF2 | NSP | VP60 | VP10 | |
| * 21–7330 | 21–5282 | 5282–7013 | 7006–7330 | 21–7330 | 21–5282 | 5282–7013 | 7006–7330 | 21–7330 | 21–5282 | 5282–7013 | 7006–7330 | 21–7330 | 21–5282 | 5282–7013 | 7006–7330 | 21–7330 | 21–5282 | 5282–7013 | 7006–7330 | |
| NP1192/MK440617 | ||||||||||||||||||||
| L98/MK440616 | 94.7 | 94.6 | 94.8 | 96.5 | ||||||||||||||||
| K501/MK440615 | 94.1 | 93.7 | 94.3 | 98.8 | 97.6 | 97.6 | 97.8 | 95.9 | ||||||||||||
| G104/MK440613 | 94.5 | 94.4 | 94.7 | 94.2 | 96.5 | 96.6 | 96.4 | 95.4 | 95.5 | 95.5 | 95.7 | 94.8 | ||||||||
| K204/MK440614 | 93.0 | 92.8 | 93.4 | 94.8 | 96.0 | 95.8 | 96.6 | 95.9 | 97.0 | 96.9 | 97.5 | 94.8 | 94.5 | 94.4 | 95.2 | 93.6 | ||||
| EBHSV-GD **/Z69620 | 97.7 | 97.1 | 99.4 | 99.1 | 94.3 | 94.0 | 94.9 | 96.2 | 93.7 | 93.1 | 94.2 | 99.7 | 94.0 | 93.7 | 94.9 | 94.5 | 92.7 | 92.3 | 93.5 | 95.1 |
| O4022-10 **/KC832838 | 93.7 | 93.7 | 93.6 | 94.2 | 92.5 | 92.6 | 92.1 | 92.8 | 91.7 | 91.7 | 91.3 | 94.2 | 91.9 | 92.2 | 91.4 | 91.3 | 91.0 | 91.0 | 90.7 | 92.2 |
| WOLF17/MF356366 | 93.5 | 93.2 | 94.0 | 96.4 | 95.0 | 94.8 | 95.5 | 95.7 | 94.3 | 93.9 | 94.8 | 96.7 | 94.8 | 94.7 | 95.3 | 94.2 | 93.4 | 93.0 | 94.2 | 95.8 |
| L03596/2019/LR899140 | 93.5 | 93.2 | 94.3 | 94.2 | 95.2 | 95.3 | 95.4 | 94.2 | 94.3 | 94.2 | 94.5 | 94.2 | 97.5 | 97.3 | 98.3 | 97.7 | 93.4 | 93.1 | 94.6 | 92.5 |
| L03594/2020/LR899152 | 92.7 | 92.5 | 93.6 | 92.2 | 94.6 | 94.6 | 95.1 | 92.2 | 93.8 | 93.6 | 94.6 | 92.3 | 96.4 | 96.3 | 97.1 | 95.7 | 92.9 | 92.6 | 94.2 | 90.7 |
| L03613/2019/LR899171 | 93.6 | 93.4 | 94.0 | 93.6 | 95.4 | 95.4 | 95.6 | 93.9 | 94.5 | 94.3 | 95.2 | 93.6 | 97.4 | 97.3 | 98.0 | 97.4 | 93.4 | 93.1 | 94.5 | 92.8 |
| L03475/2019/LR899182 | 92.8 | 92.6 | 93.7 | 92.2 | 94.6 | 94.6 | 95.3 | 92.2 | 93.8 | 93.6 | 94.8 | 92.3 | 96.5 | 96.3 | 97.2 | 95.7 | 92.9 | 92.5 | 94.3 | 90.7 |
| L03476/2019/LR899185 | 93.8 | 93.6 | 94.3 | 93.6 | 95.6 | 95.8 | 95.8 | 93.9 | 94.6 | 94.5 | 95.4 | 93.6 | 97.8 | 97.5 | 98.5 | 97.4 | 93.6 | 93.3 | 94.9 | 92.8 |
| L03477/2019/LR899188 | 94.2 | 94.1 | 94.5 | 94.5 | 96.0 | 96.4 | 95.6 | 95.1 | 95.0 | 95.1 | 94.9 | 94.5 | 98.5 | 98.4 | 98.7 | 98.6 | 94.0 | 94.0 | 94.5 | 93.3 |
| EBHSV Strain | Accession Number | Origin | Genetic Group/Subgroup | Position of Amino Acids in Particular Regions of VP60 Gene | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| b | c | d | e | f | |||||||||||||||||||||
| 58 | 66 | 231 | 270 | 291 | 302 | 327 | 343 | 383 | 410 | 415 | 417 | 427 | 461 | 476 | 522 | 524 | 536 | 542 | 544 | 565 | 566 | ||||
| EBHSV-GD | Z69620 | France | B/G1.2 | A | V | D | S | I | S | I | T | S | T | I | L | V | A | A | V | M | A | D | T | L | A |
| NP1192 | MK440617 | Poland 1992 | B/G1.2 | A | V | D | S | I | S | I | T | S | T | I | L | V | A | A | V | M | A | D | T | L | A |
| L98 | MK440616 | Poland 1998 | B/G2.2 | V | A | D | S | F | T | V | T | N | A | I | L | T | A | A | I | L | A | E | A | L | T |
| K501 | MK440615 | Poland 2001 | B/G2.3 | V | A | D | S | I | T | V | S | N | A | I | L | T | A | S | I | L | A | D | T | L | A |
| K204 | MK440614 | Poland 2004 | B G2.3 | V | A | D | S | I | T | V | S | N | A | L | L | T | S | S | I | L | T | E | T | F | T |
| G104 | MK440613 | Poland 2004 | B/G3 | V | A | E | C | I | T | V | T | N | S | I | M | T | A | A | I | L | A | E | T | L | T |
| BS89 | X98002 | Italy 1989 | B/G1.1 | A | V | D | S | I | T | I | T | S | S | I | L | V | A | V | V | M | A | D | T | L | A |
| V58 | KJ679553 | Sweden 1994 | B/G1.2 | A | V | D | S | I | T | I | T | N | T | I | M | V | A | V | V | M | A | D | T | L | A |
| A0256 | AJ971306 | France 2002 | B/G1.3 | A | V | D | S | I | T | V | S | N | T | L | L | V | A | S | V | M | T | E | T | F | T |
| V171 | KJ679558 | Sweden 2001 | B/G2.2 | A | A | D | S | I | T | V | T | N | A | I | L | T | A | A | I | L | T | E | T | L | T |
| V715 | KJ679559 | Sweden 2002 | B/G2.3 | V | A | D | S | I | T | V | S | N | A | I | L | T | A | S | I | L | A | E | T | L | A |
| O516 | AM933650 | France 2005 | B/G3 | A | V | E | C | I | T | I | T | N | S | I | M | T | A | A | I | L | A | E | T | L | A |
| MI09 | KF591083 | Italy 2009 | B/G3 | A | V | E | C | I | T | I | T | N | S | L | L | T | A | A | I | L | A | E | T | L | T |
| BS15-1 | KU961677 | Italy 2015 | B/-undefined | A | A | D | S | I | T | V | S | N | S | I | M | V | A | A | I | L | A | E | T | L | T |
| WOLF | MF356366 | Italy 2016 | B/-undefined | A | A | D | S | I | T | V | S | N | S | I | M | V | A | A | I | L | A | E | T | L | T |
| 0330 | AM408588 | France 2003 | B/G3 | V | A | E | C | I | T | I | T | N | S | I | M | T | A | A | I | L | A | E | T | L | T |
| E14-40/2 | LT168848 | France 2014 | B/G3 | A | A | E | C | I | T | V | T | N | S | I | M | T | A | A | I | L | A | E | T | L | T |
| 08-36 | HF571040 | Sweden | B/G3 | V | A | E | C | I | T | V | T | N | S | I | M | T | A | A | I | L | A | E | T | L | T |
| L03596/2019 | LR899140 | Germany | B/G3 | A | A | E | C | I | T | V | S | N | S | I | M | T | A | A | I | L | A | E | T | L | T |
| L03594/2020 | LR899152 | Germany | B/G3 | A | A | E | C | I | T | V | T | N | S | I | M | T | A | A | I | L | A | E | T | L | T |
| L03613/2019 | LR899171 | Germany | B/G3 | A | A | E | C | I | T | V | S | N | S | I | M | T | A | A | I | L | T | E | T | L | T |
| L03475/2019 | LR899182 | Germany | B/G3 | A | A | E | C | I | T | V | T | N | S | I | M | T | A | A | I | L | A | E | T | L | T |
| L03476/2019 | LR899185 | Germany | B/G3 | A | A | E | C | I | T | V | S | N | S | I | M | T | A | A | I | L | A | E | T | L | T |
| L03477/2019 | LR899188 | Germany | B/G3 | A | A | E | C | I | T | V | T | N | S | I | M | T | A | A | I | L | A | E | T | L | T |
| O4021-9 | KC832839 | Sweden 1982 | A | A | M | D | S | I | N | I | T | N | S | I | L | V | A | D | V | M | A | D | T | L | A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fitzner, A.; Kwit, E.; Niedbalski, W.; Bigoraj, E.; Kęsy, A.; Rzeżutka, A. Phylogenetic Analysis of European Brown Hare Syndrome Virus Strains from Poland (1992–2004). Viruses 2021, 13, 1999. https://doi.org/10.3390/v13101999
Fitzner A, Kwit E, Niedbalski W, Bigoraj E, Kęsy A, Rzeżutka A. Phylogenetic Analysis of European Brown Hare Syndrome Virus Strains from Poland (1992–2004). Viruses. 2021; 13(10):1999. https://doi.org/10.3390/v13101999
Chicago/Turabian StyleFitzner, Andrzej, Ewa Kwit, Wiesław Niedbalski, Ewelina Bigoraj, Andrzej Kęsy, and Artur Rzeżutka. 2021. "Phylogenetic Analysis of European Brown Hare Syndrome Virus Strains from Poland (1992–2004)" Viruses 13, no. 10: 1999. https://doi.org/10.3390/v13101999
APA StyleFitzner, A., Kwit, E., Niedbalski, W., Bigoraj, E., Kęsy, A., & Rzeżutka, A. (2021). Phylogenetic Analysis of European Brown Hare Syndrome Virus Strains from Poland (1992–2004). Viruses, 13(10), 1999. https://doi.org/10.3390/v13101999