
Natural Phytochemicals, Luteolin and Isoginkgetin, Inhibit 3C Protease and Infection of FMDV, In Silico and In Vitro

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Structure Modeling of FMDV 3C Protease and Validation
2.2. Virtual Screening of Phytochemical Flavonoids
2.3. Cells, Viruses, and Compounds
2.4. Cytotoxicity Assay
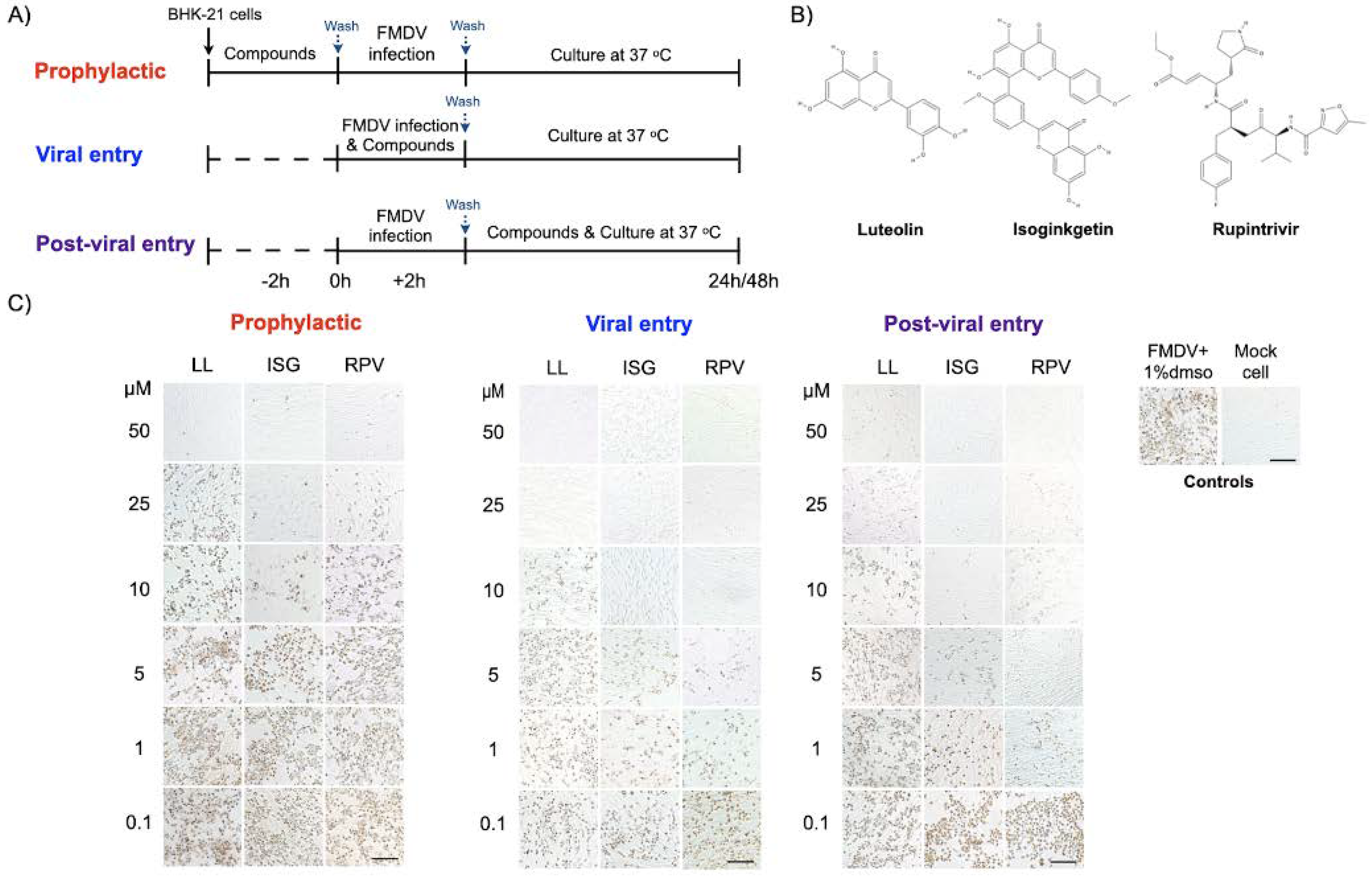
2.5. Antiviral Activity Assays (Prophylaxis, Viral Entry, and Post-Viral Entry)
2.6. Immunoperoxidase Monolayer Assay (IPMA) for FMDV Antigen Detection
2.7. RT Real-Time PCR (RT-qPCR) for Viral Load Quantification
2.8. Construction of Plasmids for Intracellular Protease Assay
2.9. Protease Inhibition Activity Using Cell-Based Protease Assay
2.10. Expression and Purification of Recombinant FMDV 3C Protease
2.11. In Vitro Protease Inhibition Using FRET Assay
2.12. Selectivity Index
3. Results
3.1. Homology Modeling and Virtual Screening of FMDV 3Cpro
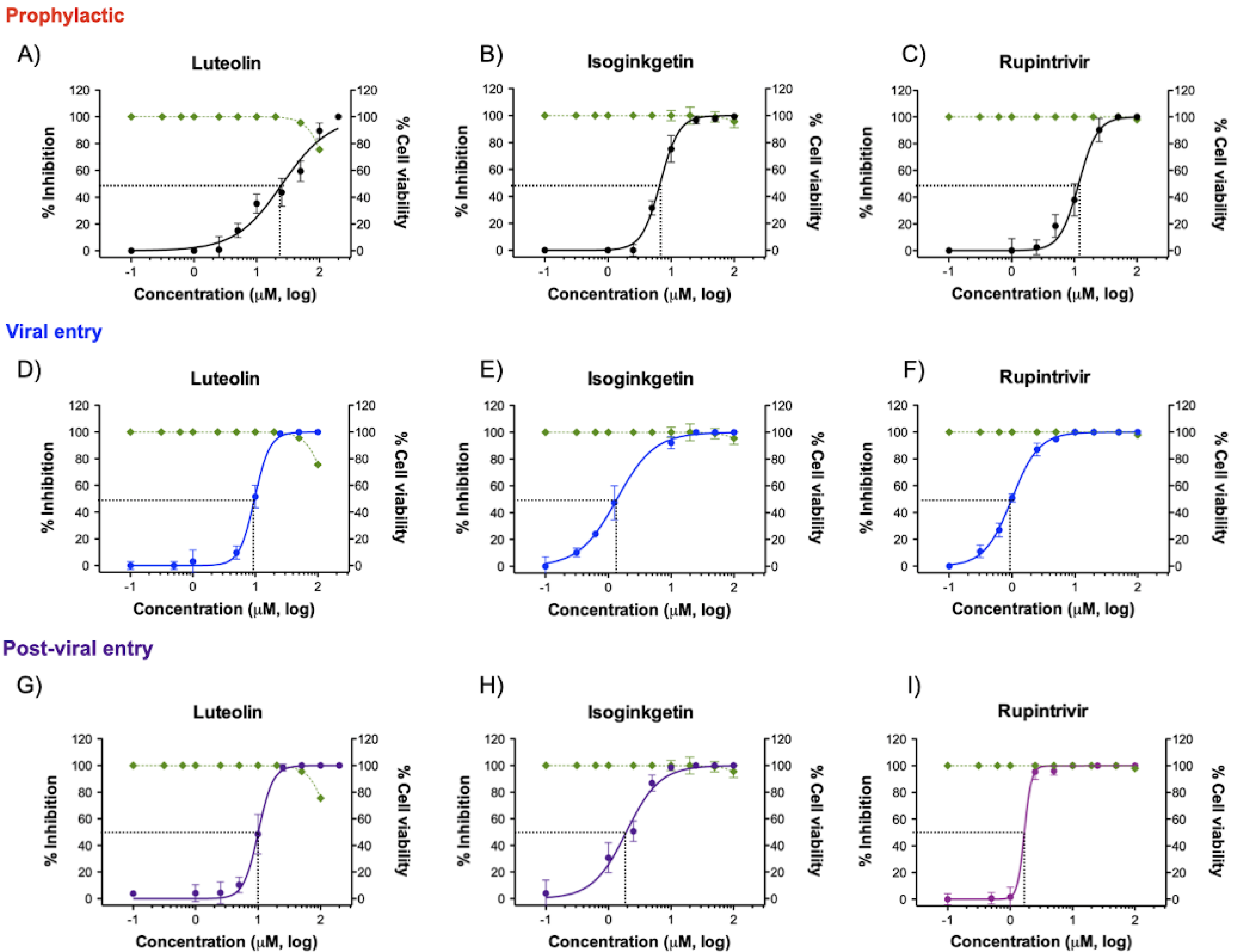
3.2. Cytotoxicity and Antiviral Activity of the Phytochemical Compounds
3.3. Viral Quantification by RT-qPCR
3.4. Evaluation of FMDV 3Cpro Inhibitors Using Cell-Based Protease Assay
3.5. Protease Inhibition Using FRET Assay
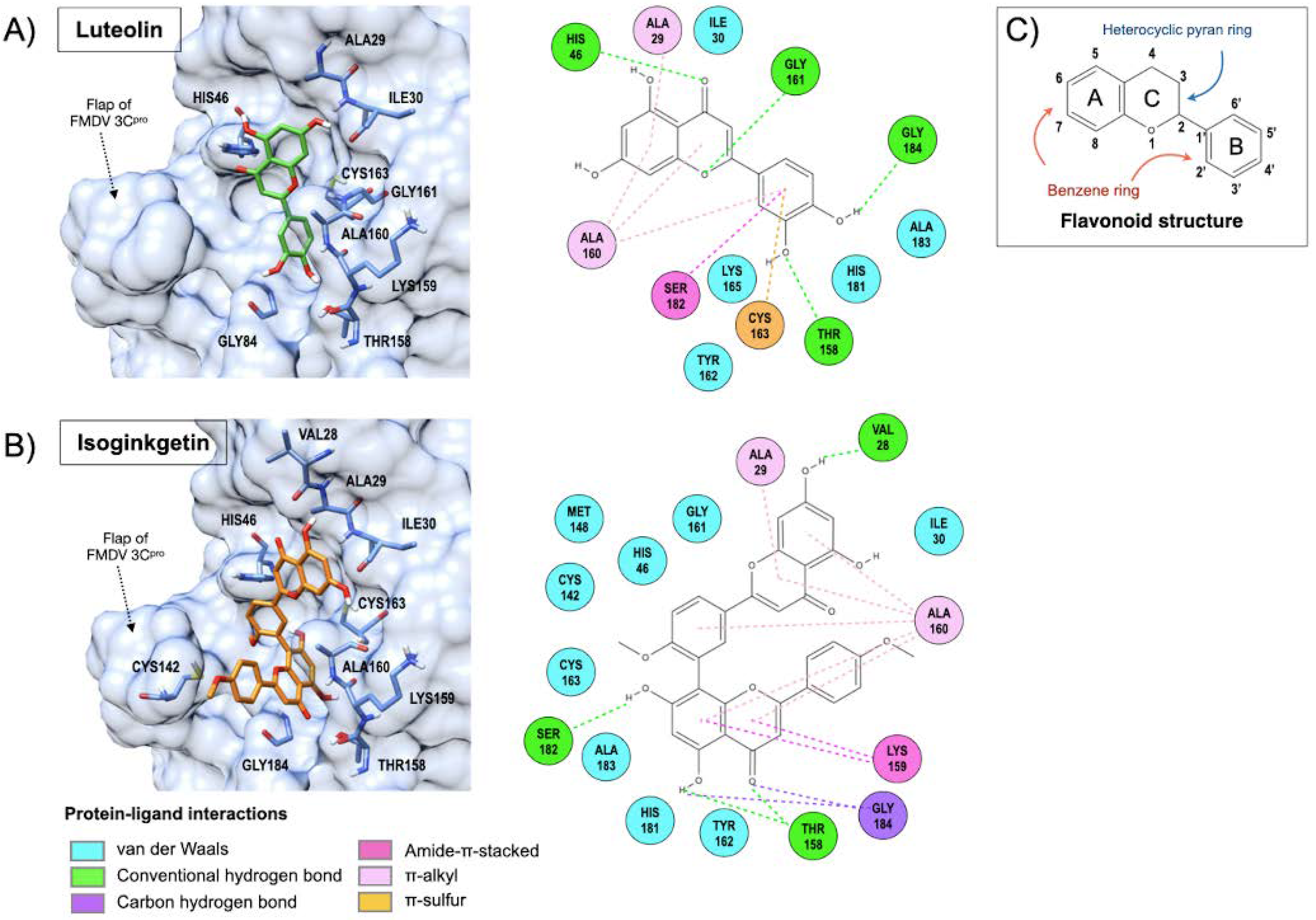
3.6. Interaction of Luteolin and Isoginkgetin with FMDV 3Cpro
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Office International des Epizooties (OIE)—Terrestrial Animal Health Code: Chapter 8.8—Infection with Foot and Mouth Disease Virus. Paris: World Organization for Animal Health; 2019. Available online: https://www.oie.int/fileadmin/Home/eng/Health_standards/tahm/3.01.08_FMD.pdf (accessed on 1 August 2021).
- Office International des Epizooties (OIE)—Terrestrial Manual: Chapter 3.1.8—Foot and Mouth Disease (Infection with Foot and Mouth Disease Virus. Paris: World Organization for Animal Health; 2021. Available online: http://www.oie.int/index.php?id=169&L=0&htmfile=chapitre_fmd.htm (accessed on 1 August 2021).
- Gao, Y.; Sun, S.Q.; Guo, H.C. Biological function of Foot-and-mouth disease virus non-structural proteins and non-coding elements. Virol. J. 2016, 13, 1–17. [Google Scholar] [CrossRef] [Green Version]
- King, A.M.Q.; Adams, M.J.; Carstens, E.B.; Lefkowitz, E.J. (Eds.) Picornavirales. In Virus Taxonomy; Elsevier: San Diego, CA, USA, 2012; pp. 835–839. [Google Scholar] [CrossRef]
- Jamal, S.M.; Belsham, G.J. Foot-and-mouth disease: Past, present and future. Vet. Res. 2013, 44, 116. [Google Scholar] [CrossRef] [Green Version]
- Kitching, P.; Hammond, J.; Jeggo, M.; Charleston, B.; Paton, D.; Rodriguez, L.; Heckert, R. Global FMD control-Is it an option? Vaccine 2007, 25, 5660–5664. [Google Scholar] [CrossRef] [PubMed]
- Goris, N.; Vandenbussche, F.; De Clercq, K. Potential of antiviral therapy and prophylaxis for controlling RNA viral infections of livestock. Antiviral Res. 2008, 78, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Mason, P.W.; Grubman, M.J.; Baxt, B. Molecular basis of pathogenesis of FMDV. Virus Res. 2003, 91, 9–32. [Google Scholar] [CrossRef]
- Birtley, J.R.; Knox, S.R.; Jaulent, A.M.; Brick, P.; Leatherbarrow, R.J.; Curry, S. Crystal structure of foot-and-mouth disease virus 3C protease: New insights into catalytic mechanism and cleavage specificity. J. Biol. Chem. 2005, 280, 11520–11527. [Google Scholar] [CrossRef] [Green Version]
- Carrillo, C.; Tulman, E.R.; Delhon, G.; Lu, Z.; Carreno, A.; Vagnozzi, A.; Kutish, G.F.; Rock, D.L. High throughput sequencing and comparative genomics of foot-and-mouth disease virus. Dev. Biol. 2006, 126, 23–30. [Google Scholar] [CrossRef]
- Curry, S.; Roqué-Rosell, N.; Zunszain, P.A.; Leatherbarrow, R.J. Foot-and-mouth disease virus 3C protease: Recent structural and functional insights into an antiviral target. Int. J. Biochem. Cell Biol. 2007, 39, 1–6. [Google Scholar] [CrossRef]
- Zunszain, P.A.; Knox, S.R.; Sweeney, T.R.; Yang, J.; Roqué-Rosell, N.; Belsham, G.J.; Leatherbarrow, R.J.; Curry, S. Insights into cleavage specificity from the crystal structure of foot-and-mouth disease virus 3C protease complexed with a peptide substrate. J. Mol. Biol. 2010, 395, 375–389. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Bi, J.; Liu, J.; Liu, X.; Wu, X.; Jiang, P.; Yoo, D.; Zhang, Y.; Wu, J.; Wan, R.; et al. 3Cpro of foot-and-mouth disease virus antagonizes the interferon signaling pathway by blocking STAT1/STAT2 nuclear translocation. J. Virol. 2014, 88, 4908–4920. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Chen, S.; Cheng, A.; Wang, M. Roles of the picornaviral 3C proteinase in the viral life cycle and host cells. Viruses 2016, 8, 82. [Google Scholar] [CrossRef] [Green Version]
- Patick, A.K.; Binford, S.L.; Brothers, M.A.; Jackson, R.L.; Ford, C.E.; Diem, M.D.; Maldonado, F.; Dragovich, P.S.; Zhou, R.; Prins, T.J.; et al. In vitro antiviral activity of AG7088, a potent inhibitor of human rhinovirus 3C protease. Antimicrob. Agents Chemother. 1999, 43, 2444–2450. [Google Scholar] [CrossRef] [Green Version]
- Binford, S.L.; Maldonado, F.; Brothers, M.A.; Weady, P.T.; Zalman, L.S.; Meador, J.W.; Matthews, D.A.; Patick, A.K. Conservation of amino acids in human rhinovirus 3C protease correlates with broad-spectrum antiviral activity of rupintrivir, a novel human rhinovirus 3C protease inhibitor. Antimicrob. Agents Chemother. 2005, 49, 619–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patick, A.K. Rhinovirus chemotherapy. Antiviral Res. 2006, 71, 391–396. [Google Scholar] [CrossRef]
- Ghildiyal, R.; Prakash, V.; Chaudhary, V.K.; Gupta, V.; Gabrani, R. Phytochemicals as antiviral agents: Recent updates. In Plant-derived Bioactives: Production, Properties and Therapeutic Applications; Swamy, M.K., Ed.; Springer: Singapore, 2020; pp. 279–295. [Google Scholar] [CrossRef]
- Mani, J.S.; Johnson, J.B.; Steel, J.C.; Broszczak, D.A.; Neilsen, P.M.; Walsh, K.B.; Naiker, M. Natural product-derived phytochemicals as potential agents against coronaviruses: A review. Virus Res. 2020, 284, 197989. [Google Scholar] [CrossRef]
- Zakaryan, H.; Arabyan, E.; Oo, A.; Zandi, K. Flavonoids: Promising natural compounds against viral infections. Arch. Virol. 2017, 162, 2539–2551. [Google Scholar] [CrossRef]
- Min, N.; Leong, P.T.; Lee, R.C.H.; Khuan, J.S.E.; Chu, J.J.H. A flavonoid compound library screen revealed potent antiviral activity of plant-derived flavonoids on human enterovirus A71 replication. Antivir. Res. 2018, 150, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Lalani, S.; Poh, C.L. Flavonoids as antiviral agents for enterovirus A71 (EV-A71). Viruses 2020, 12, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Su, W.; Jin, J.; Chen, J.; Li, X.; Zhang, X.; Sun, M.; Sun, S.; Fan, P.; An, D.; et al. Identification of luteolin as enterovirus 71 and coxsackievirus A16 inhibitors through reporter viruses and cell viability-based screening. Viruses 2014, 6, 2778–2795. [Google Scholar] [CrossRef] [Green Version]
- Kwon, B.E.; Song, J.H.; Song, H.H.; Kang, J.W.; Hwang, S.N.; Rhee, K.J.; Shim, A.; Hong, E.H.; Kim, Y.J.; Jeon, S.M.; et al. Antiviral activity of oroxylin A against coxsackievirus B3 alleviates virus-induced acute pancreatic damage in mice. PLoS ONE 2016, 11, e0155784. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Wang, H.Q.; Guo, T.T.; Li, Y.H. Luteolin inhibits CVB3 replication through inhibiting inflammation. J. Asian Nat. Prod. Res. 2020, 22, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Salvati, A.L.; De Dominicis, A.; Tait, S.; Canitano, A.; Lahm, A.; Fiore, L. Mechanism of action at the molecular level of the antiviral drug 3(2H)-isoflavene against type 2 poliovirus. Antimicrob. Agents Chemother. 2004, 48, 2233–2243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.J.; Chang, Y.C.; Hsiao, N.W.; Hsieh, J.L.; Wang, C.Y.; Kung, S.H.; Tsai, F.J.; Lan, Y.C.; Lin, C.W. Fisetin and rutin as 3C protease inhibitors of enterovirus A71. J. Virol. Methods 2012, 182, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Xi, C.; Hu, K.; Gao, W.; Cai, X.; Qin, J.; Lv, S.; Du, C.; Wei, Y. Inhibition of enterovirus 71 replication and viral 3C protease by quercetin. Virol. J. 2018, 15, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef]
- Studer, G.; Rempfer, C.; Waterhouse, A.M.; Gumienny, R.; Haas, J.; Schwede, T. QMEANDisCo—distance constraints applied on model quality estimation. Bioinformatics 2020, 36, 1765–1771. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theerawatanasirikul, S.; Lekcharoensuk, P. Virtual Screening of Natural Compounds Targeting Proteases of Coronaviruses and Picornaviruses. In Methods in Pharmacology and Toxicology; Roy, K., Ed.; Springer USA: New York, NY, USA, 2021; pp. 661–681. [Google Scholar] [CrossRef]
- Sayers, E.W.; Barrett, T.; Benson, D.A.; Bolton, E.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Federhen, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2011, 39, D38–D51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neveu, V.; Perez-Jiménez, J.; Vos, F.; Crespy, V.; du Chaffaut, L.; Mennen, L.; Knox, C.; Eisner, R.; Cruz, J.; Wishart, D.; et al. Phenol-Explorer: An online comprehensive database on polyphenol contents in foods. Database 2010, 2010, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Erehman, J.; Gohlke, B.-O.; Wilhelm, T.; Preissner, R.; Dunkel, M. Super Natural II—a database of natural products. Nucleic Acids Res. 2014, 43, D935–D939. [Google Scholar] [CrossRef] [Green Version]
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [CrossRef]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Semkum, P.; Kaewborisuth, C.; Thangthamniyom, N.; Theerawatanasirikul, S.; Lekcharoensuk, C.; Hansoongnern, P.; Ramasoota, P.; Lekcharoensuk, P. A novel plasmid DNA-based foot and mouth disease virus minigenome for intracytoplasmic mRNA production. Viruses 2021, 13, 1047. [Google Scholar] [CrossRef]
- Lekcharoensuk, P.; Wiriyarat, W.; Petcharat, N.; Lekcharoensuk, C.; Auewarakul, P.; Richt, J.A. Cloned cDNA of A/swine/Iowa/15/1930 internal genes as a candidate backbone for reverse genetics vaccine against influenza A viruses. Vaccine 2012, 30, 1453–1459. [Google Scholar] [CrossRef] [Green Version]
- Theerawatanasirikul, S.; Kuo, C.J.; Phecharat, N.; Chootip, J.; Lekcharoensuk, C.; Lekcharoensuk, P. Structural-based virtual screening and in vitro assays for small molecules inhibiting the feline coronavirus 3CL protease as a surrogate platform for coronaviruses. Antiviral Res. 2020, 182, 104927. [Google Scholar] [CrossRef]
- Sariya, L.; Thangthumniyom, N.; Wajjwalku, W.; Chumsing, W.; Ramasoota, P.; Lekcharoensuk, P. Expression of foot and mouth disease virus nonstructural polyprotein 3ABC with inactive 3C pro in Escherichia coli. Protein Expr. Purif. 2011, 80, 17–21. [Google Scholar] [CrossRef]
- Van Der Linden, L.; Ulferts, R.; Nabuurs, S.B.; Kusov, Y.; Liu, H.; George, S.; Lacroix, C.; Goris, N.; Lefebvre, D.; Lanke, K.H.W.; et al. Application of a cell-based protease assay for testing inhibitors of picornavirus 3C proteases. Antivir. Res. 2014, 103, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.J.; Chi, Y.H.; Hsu, J.T.A.; Liang, P.H. Characterization of SARS main protease and inhibitor assay using a fluorogenic substrate. Biochem. Biophys. Res. Commun. 2004, 318, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Hiyama, Y.; Itoh, K.; Hashimoto, Y.; Shimizu, M.; Morita, N.; Nakayama, M.; Horie, T. Antiviral activity of natural occurring flavonoids in vitro. Chem. Pharm. Bull. 1985, 33, 3881–3886. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Pandey, A.K. Chemistry and biological activities of flavonoids: An overview. Sci. World J. 2013, 2013, 162750. [Google Scholar] [CrossRef] [Green Version]
- Manzoor, M.F.; Ahmad, N.; Ahmed, Z.; Siddique, R.; Zeng, X.A.; Rahaman, A.; Muhammad Aadil, R.; Wahab, A. Novel extraction techniques and pharmaceutical activities of luteolin and its derivatives. J. Food Biochem. 2019, 43, e12974. [Google Scholar] [CrossRef]
- Fan, W.; Qian, S.; Qian, P.; Li, X. Antiviral activity of luteolin against Japanese encephalitis virus. Virus Res. 2016, 220, 112–116. [Google Scholar] [CrossRef]
- Wu, C.C.; Fang, C.Y.; Hsu, H.Y.; Chen, Y.J.; Chou, S.P.; Huang, S.Y.; Cheng, Y.J.; Lin, S.F.; Chang, Y.; Tsai, C.H.; et al. Luteolin inhibits Epstein-Barr virus lytic reactivation by repressing the promoter activities of immediate-early genes. Antivir. Res. 2016, 132, 99–110. [Google Scholar] [CrossRef]
- Mehla, R.; Bivalkar-Mehla, S.; Chauhan, A. A flavonoid, luteolin, cripples HIV-1 by abrogation of Tat function. PLoS ONE 2011, 6, e27915. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.; Bi, J.; Li, F.; Wang, S.; Huang, X.; Meng, X.; Sun, B.; Wang, D.; Kong, W.; Jiang, C.; et al. Antiviral efficacy of flavonoids against enterovirus 71 infection in vitro and in newborn mice. Viruses 2019, 11, 625. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Ding, Y.; Ke, Z.; Cao, L.; Li, N.; Ding, G.; Wang, Z.; Xiao, W. Luteoloside acts as 3C protease inhibitor of enterovirus 71 in vitro. PLoS ONE 2016, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Mallick, S.; Chakraborty, T.; Ghosh, N.; Singh, A.K.; Manna, S.; Majumdar, S. Synthesis, characterisation and antioxidant activity of luteolin-vanadium(II) complex. Food Chem. 2015, 173, 1172–1178. [Google Scholar] [CrossRef]
- Xue, G.; Gong, L.; Yuan, C.; Xu, M.; Wang, X.; Jiang, L.; Huang, M. A structural mechanism of flavonoids in inhibiting serine proteases. Food Funct. 2017, 8, 2437–2443. [Google Scholar] [CrossRef]
- Hayashi, K.; Hayashi, T.; Morita, N. Mechanism of action of the antiherpesvirus biflavone ginkgetin. Antimicrob. Agents Chemother. 1992, 36, 1890–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.R.; Wei, L.H.; Guan, X.Q.; Huang, C.; Liu, Z.Y.; Wang, F.J.; Hou, J.; Jin, Q.; Liu, Y.F.; Wen, P.H.; et al. Biflavones from Ginkgo biloba as inhibitors of human thrombin. Bioorganic Chem. 2019, 92, 103199. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.; Matlin, A.J.; Lowell, A.M.; Moore, M.J. The biflavonoid isoginkgetin is a general inhibitor of pre-mRNA splicing. J. Biol. Chem. 2008, 283, 33147–33154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Li, B.; Xia, Z.M.; Tian, Y.; Zhang, D.; Rui, W.J.; Dong, J.X.; Xiao, F.J. Anticancer effects of five biflavonoids from ginkgo biloba l. Male flowers in vitro. Molecules 2019, 24, 1496. [Google Scholar] [CrossRef] [Green Version]
- Tsalikis, J.; Abdel-Nour, M.; Farahvash, A.; Sorbara, M.T.; Poon, S.; Philpott, D.J.; Girardin, S.E. Isoginkgetin, a natural biflavonoid proteasome inhibitor, sensitizes cancer cells to apoptosis via disruption of lysosomal homeostasis and impaired protein clearance. Mol. Cell. Biol. 2019, 39, 1–17. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Binding Affinity (kcal/mol) | Cell-Based Assay | |||
|---|---|---|---|---|---|
| Cytotoxicity (CC50 a; μM) | Prophylaxis (EC50 b; μM) | Viral (EC50; μM) | Post-Viral (EC50; μM) | ||
|
Luteolin (ChemFaces) | −7.0 | >100 e | 25.83 ± 1.29 | 9.73 ± 0.94 | 10.00 ± 0.98 |
|
Isoginkgetin (TargetMol) | −7.2 | >100 | 6.76 ± 0.80 | 2.01 ± 0.07 | 1.93 ± 0.21 |
|
Apigenin (Vitas-M Laboratory) | −7.0 | 68.73 ± 4.10 | >100 | 75–100 | 75–100 |
|
Quercetin 7-rhamnoside (ChemFaces) | −6.8 | >100 | >100 | >100 | 75–100 |
|
7-O-Methyl luteolin (ChemFaces) | −6.3 | >100 | >100 | 75–100 | 75–100 |
|
Ribavirin (Sigma Aldrich) c | ND | >100 | 283.90 ± 2.30 | 41.80 ± 1.58 | 133.30 ± 2.03 |
|
Rupintrivir d (Sigma Aldrich) | ND | >100 | 11.79 ± 1.03 | 1.99 ± 0.01 | 1.688 ± 0.19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Theerawatanasirikul, S.; Thangthamniyom, N.; Kuo, C.-J.; Semkum, P.; Phecharat, N.; Chankeeree, P.; Lekcharoensuk, P. Natural Phytochemicals, Luteolin and Isoginkgetin, Inhibit 3C Protease and Infection of FMDV, In Silico and In Vitro. Viruses 2021, 13, 2118. https://doi.org/10.3390/v13112118
Theerawatanasirikul S, Thangthamniyom N, Kuo C-J, Semkum P, Phecharat N, Chankeeree P, Lekcharoensuk P. Natural Phytochemicals, Luteolin and Isoginkgetin, Inhibit 3C Protease and Infection of FMDV, In Silico and In Vitro. Viruses. 2021; 13(11):2118. https://doi.org/10.3390/v13112118
Chicago/Turabian StyleTheerawatanasirikul, Sirin, Nattarat Thangthamniyom, Chih-Jung Kuo, Ploypailin Semkum, Nantawan Phecharat, Penpitcha Chankeeree, and Porntippa Lekcharoensuk. 2021. "Natural Phytochemicals, Luteolin and Isoginkgetin, Inhibit 3C Protease and Infection of FMDV, In Silico and In Vitro" Viruses 13, no. 11: 2118. https://doi.org/10.3390/v13112118
APA StyleTheerawatanasirikul, S., Thangthamniyom, N., Kuo, C. -J., Semkum, P., Phecharat, N., Chankeeree, P., & Lekcharoensuk, P. (2021). Natural Phytochemicals, Luteolin and Isoginkgetin, Inhibit 3C Protease and Infection of FMDV, In Silico and In Vitro. Viruses, 13(11), 2118. https://doi.org/10.3390/v13112118