
Genotypic Variants of Pandemic H1N1 Influenza A Viruses Isolated from Severe Acute Respiratory Infections in Ukraine during the 2015/16 Influenza Season

 ,
,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Epidemiology of Seasonal Influenza in Ukraine, 2015/16
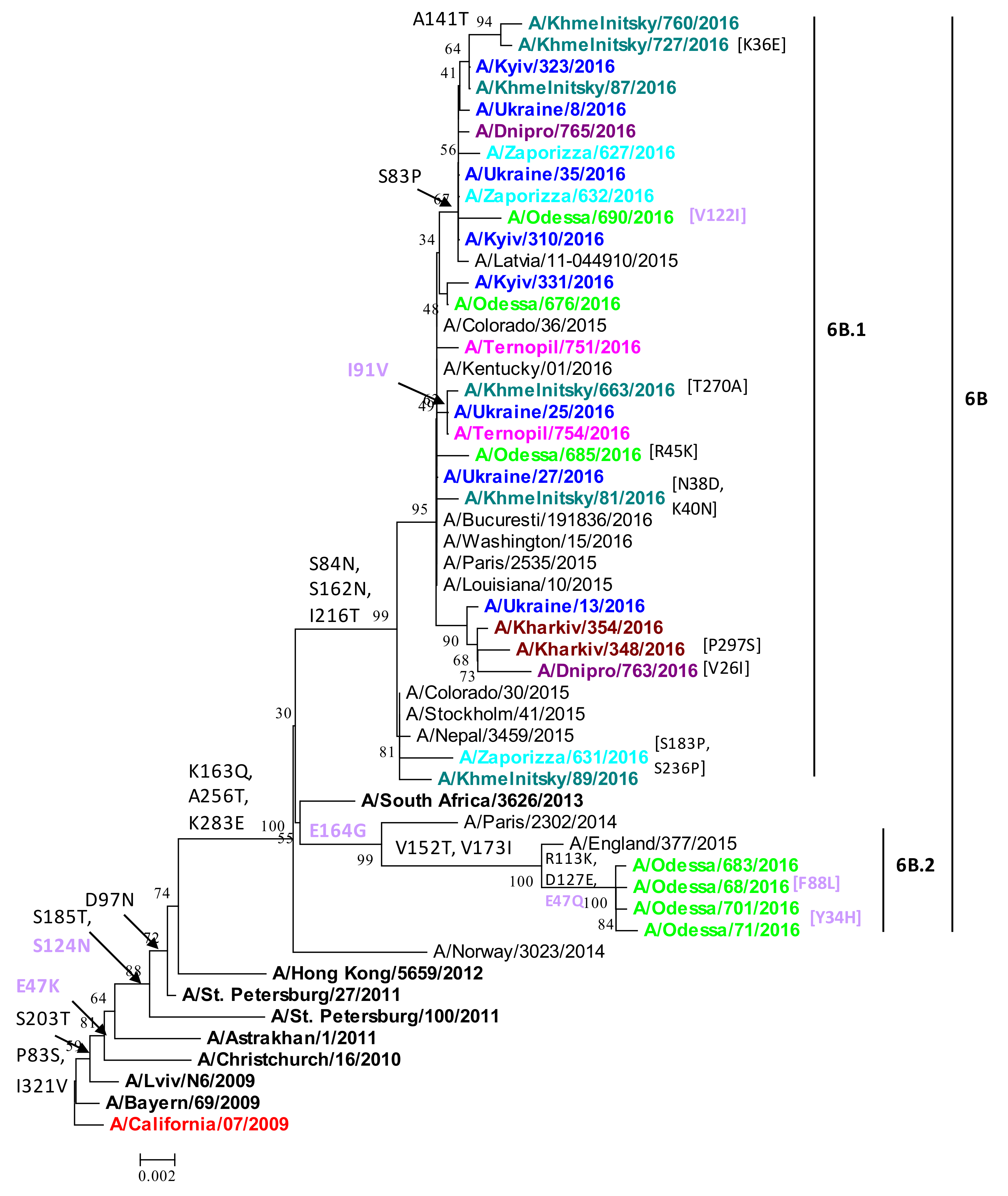
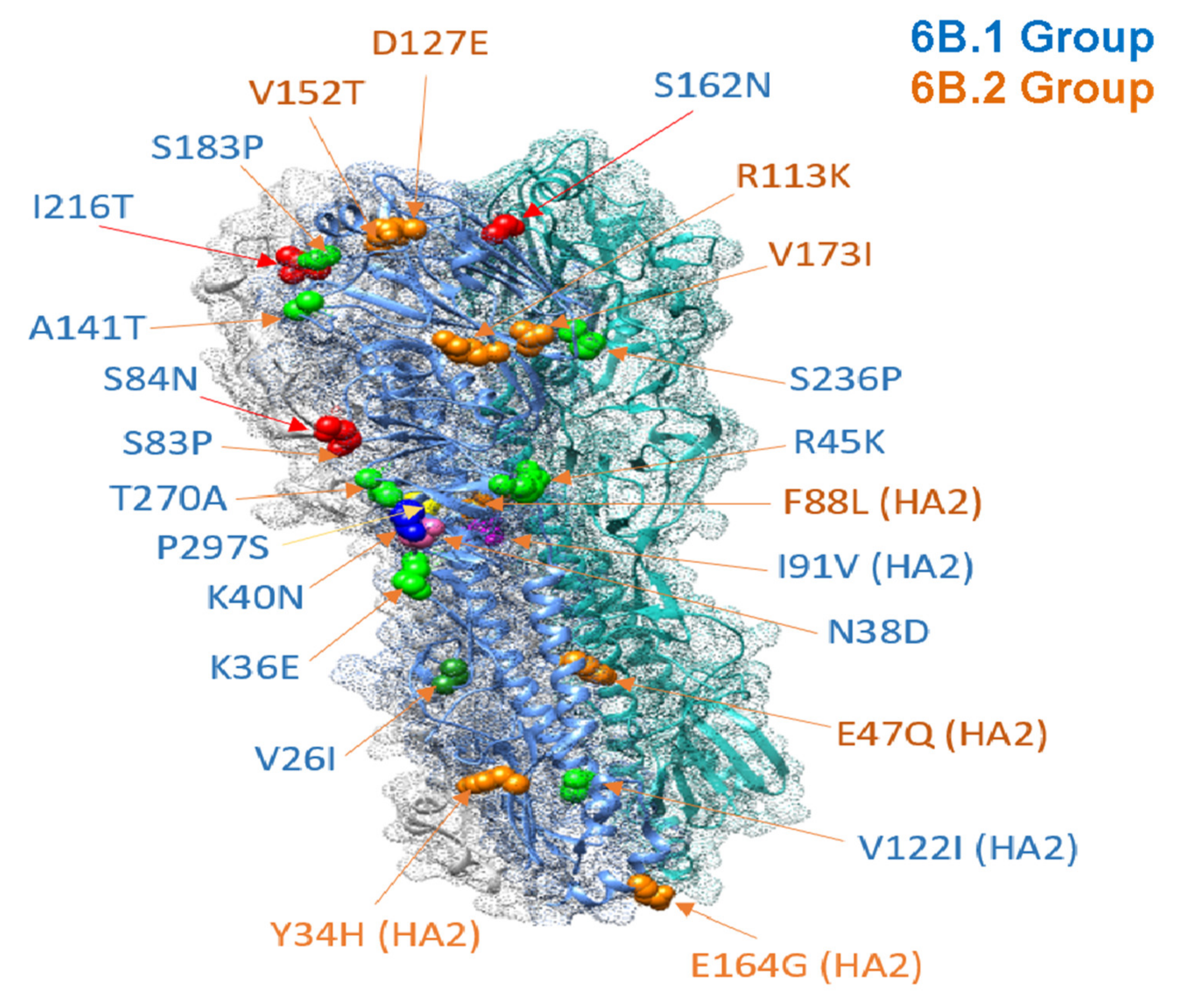
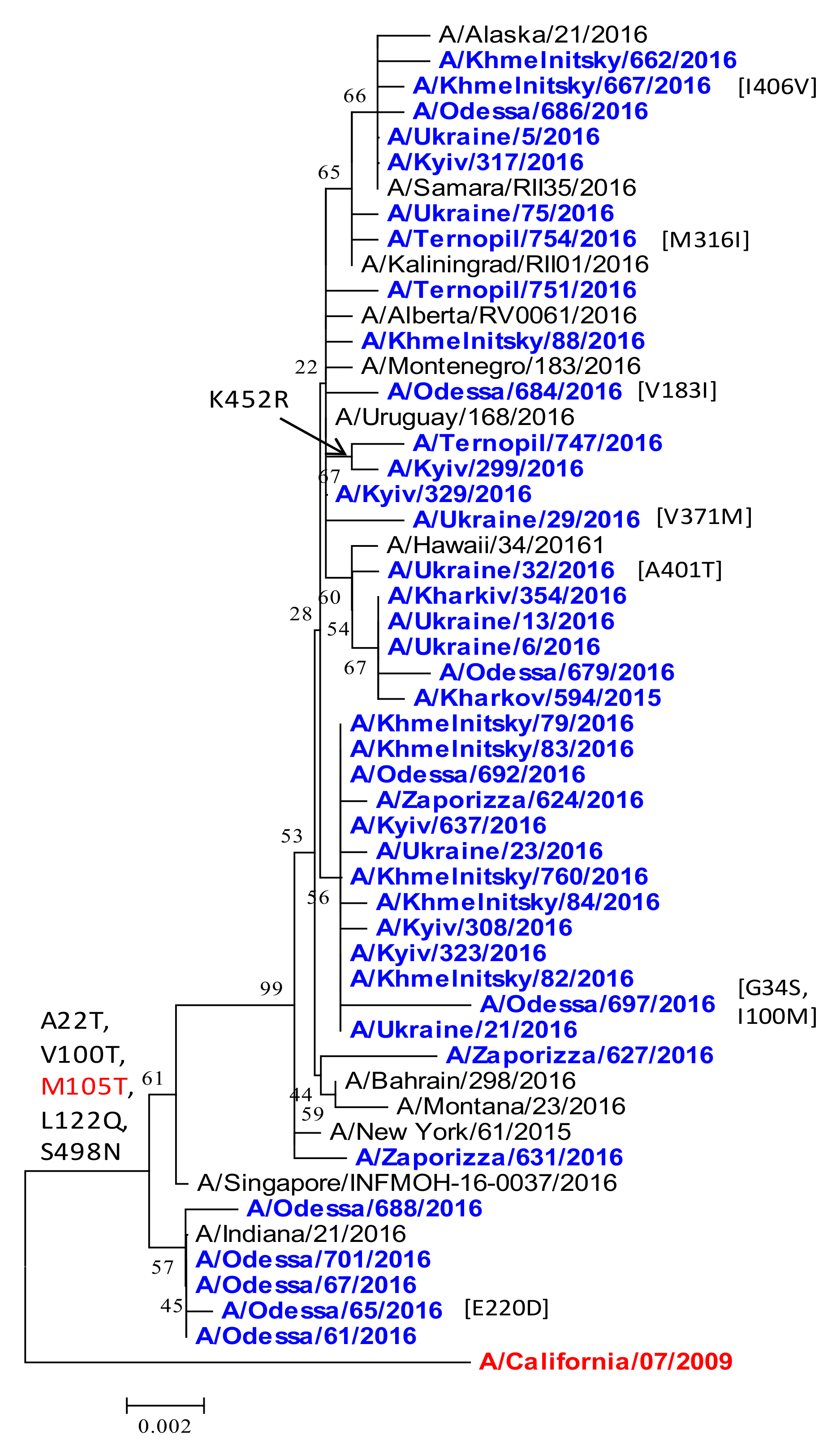
2.2. Phylogenetic Analysis of HA Protein
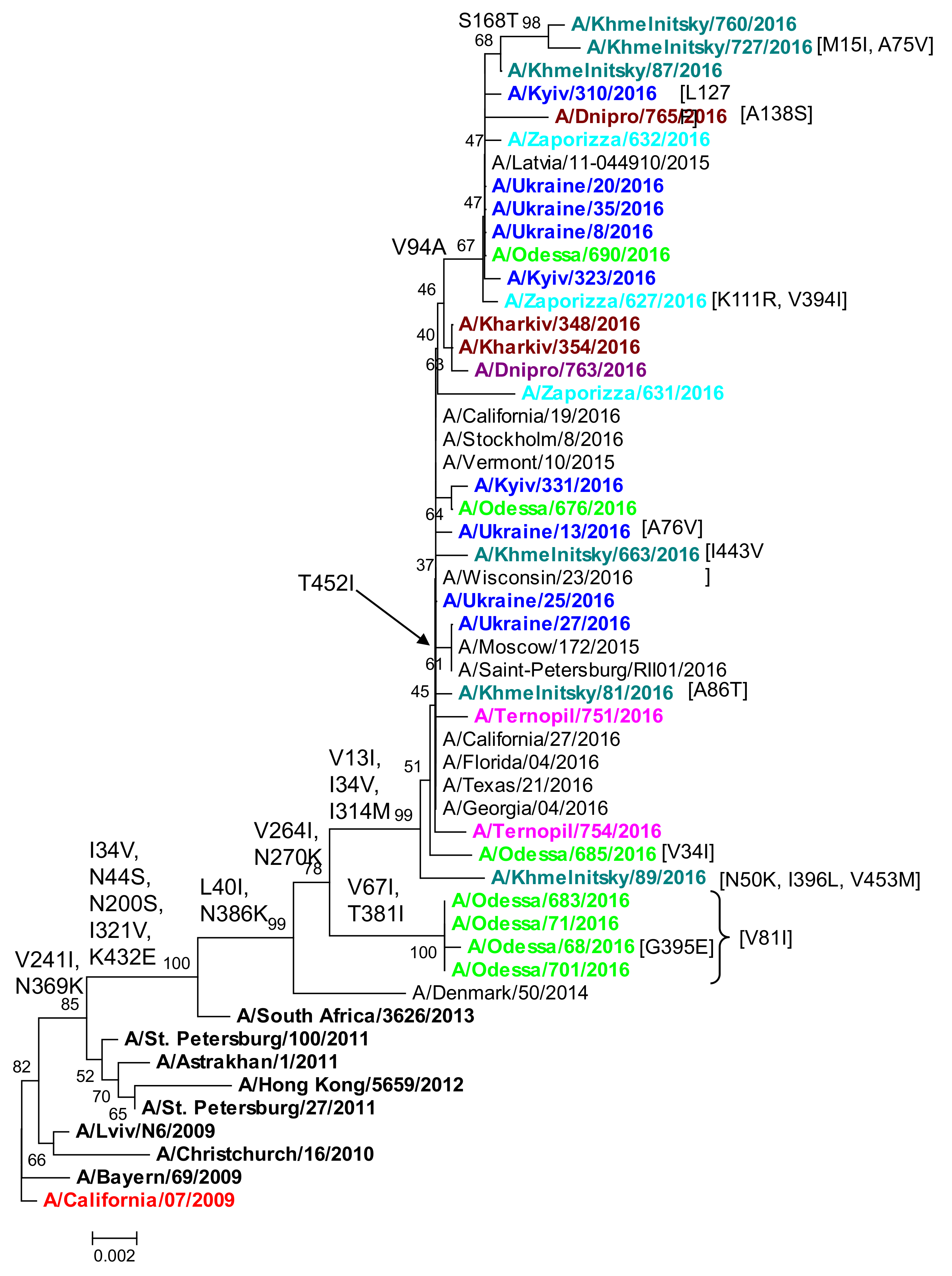
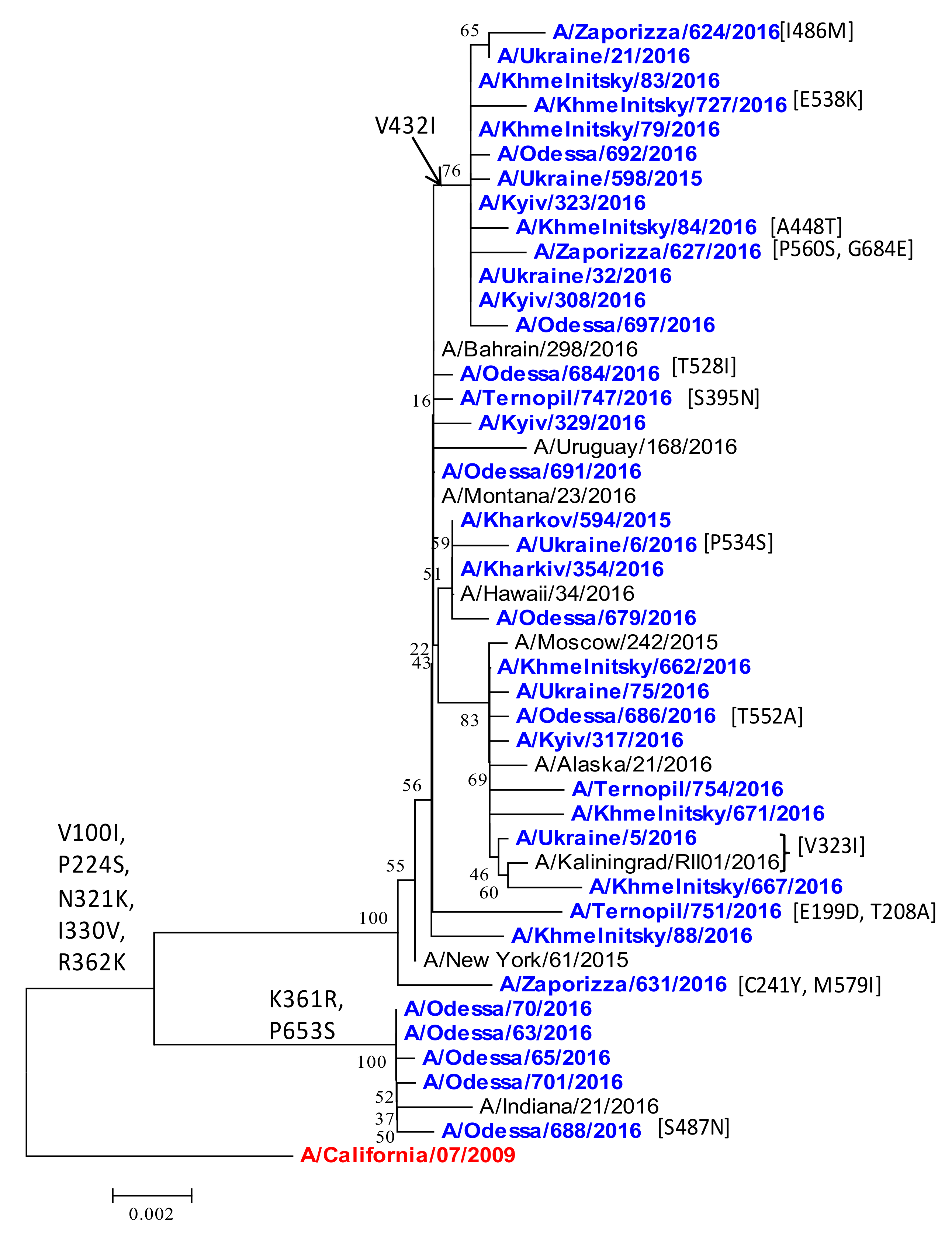
2.3. Phylogenetic Analysis of NA Protein
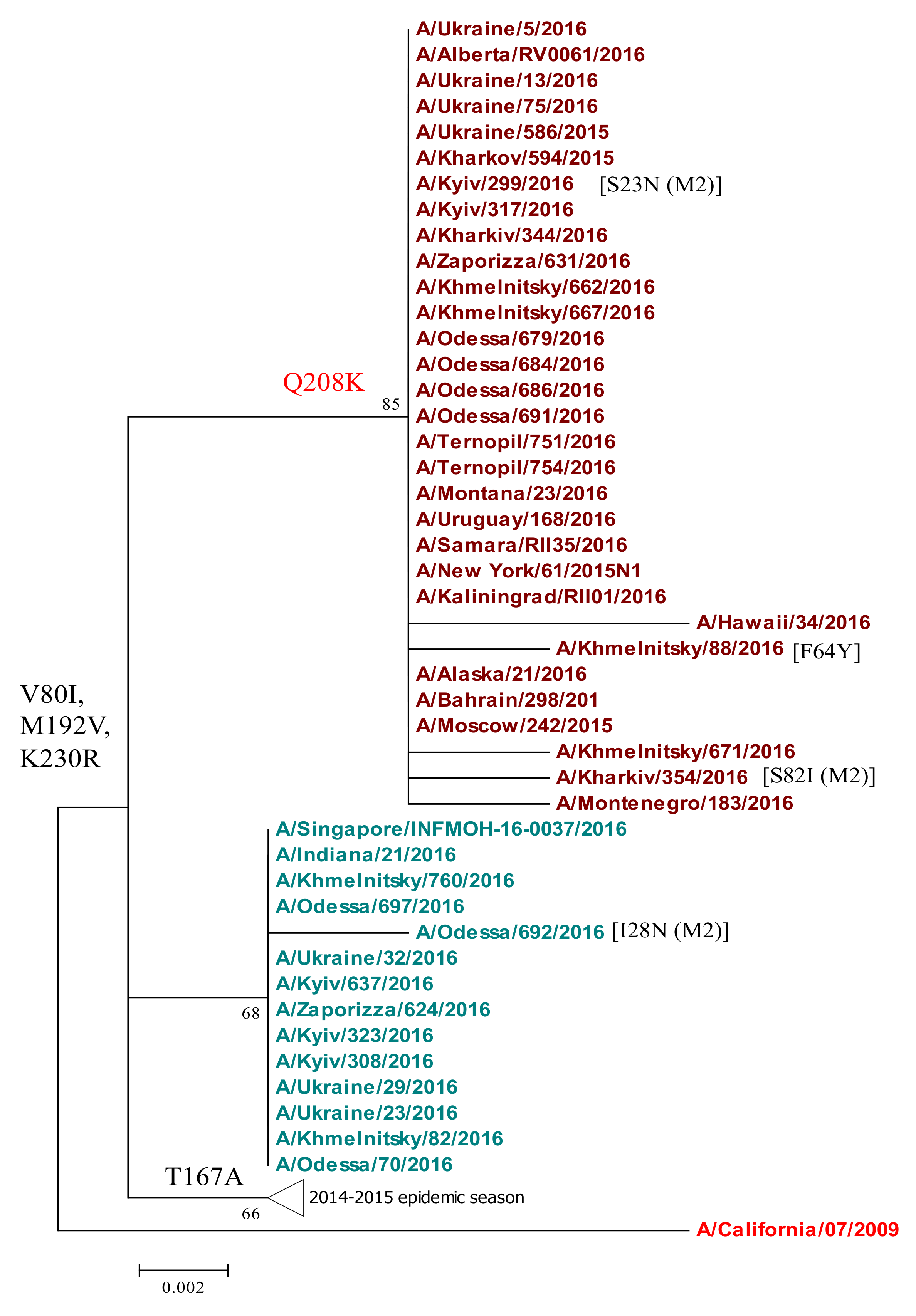
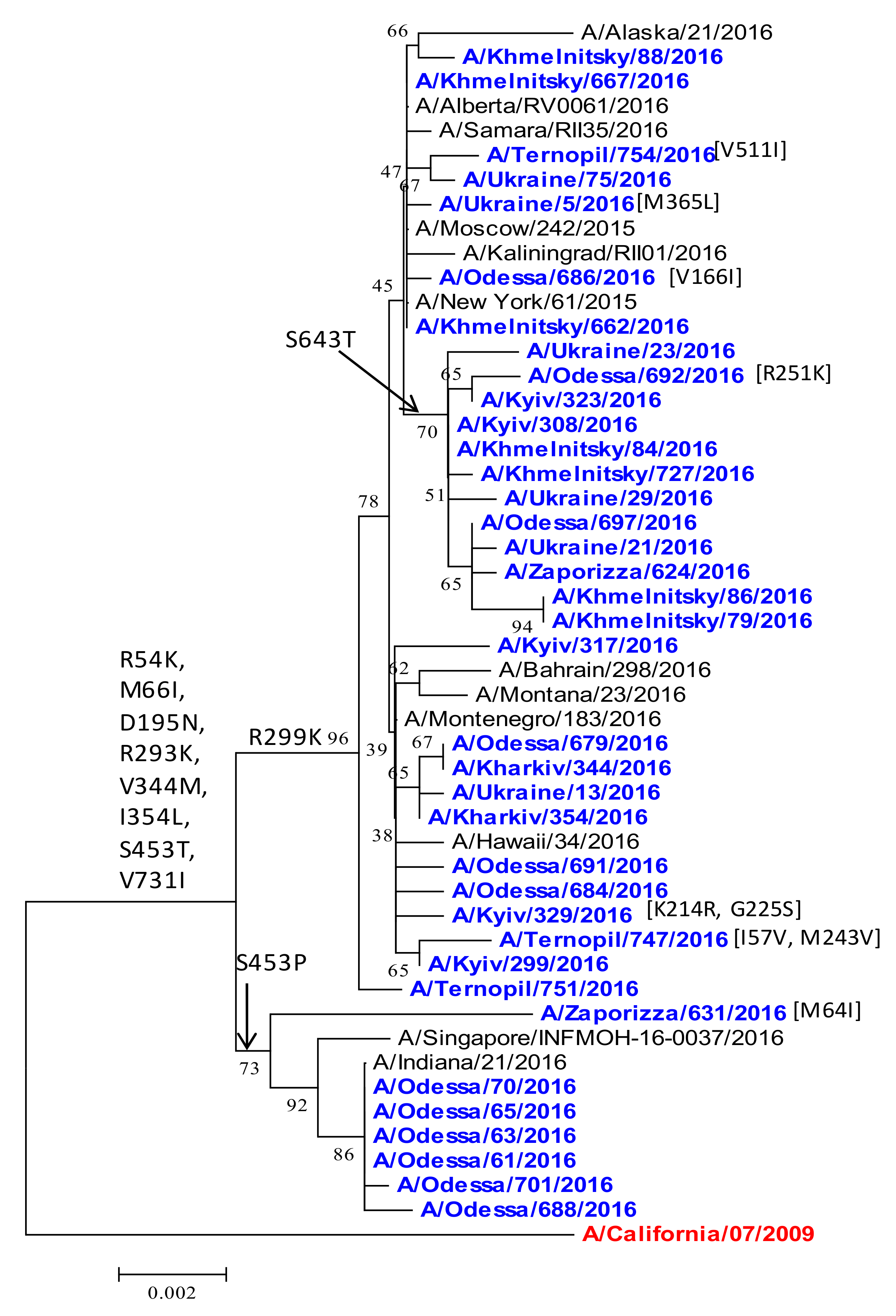
2.4. Phylogenetic Analyses of Internal Genes: M1, M2, and NP
2.5. Phylogenetic and Structural Analysis of Non-Structural (NS) Proteins
2.6. Phylogenetic Analysis of Polymerase Complex Genes
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Diagnostics
4.2. Phylogenetic Analysis
4.3. Protein Structural Modeling
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nelson, M.; Spiro, D.; Wentworth, D.; Beck, E.; Fan, J.; Ghedin, E.; Halpin, R.; Bera, J.; Hine, E.; Proudfoot, K.; et al. The early diversification of influenza A/H1N1pdm. PLoS Curr. Influenza 2009, 1, RRN1126. [Google Scholar] [CrossRef]
- Schweiger, B.; Zadow, I.; Heckler, R. Antigenic drift and variability of influenza viruses. Med. Microbiol. Immunol. 2002, 191, 133–138. [Google Scholar] [CrossRef]
- Smith, D.; Lapedes, A.; de Jong, J.; Bestebroer, T.; Rimmelzwaan, G.; Osterhaus, A.; Fouchier, R. Mapping the antigenic and genetic evolution of influenza virus. Science 2004, 305, 371–376. [Google Scholar] [CrossRef] [Green Version]
- Erbelding, E.; Post, D.; Stemmy, E.; Roberts, P.; Augustine, A.; Ferguson, S.; Paules, C.; Graham, B.; Fauci, A. A Universal Influenza Vaccine: The Strategic Plan for the National Institute of Allergy and Infectious Diseases. J. Infect. Dis. 2018, 218, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Belanov, S.; Bychkov, D.; Benner, C.; Ripatti, S.; Ojala, T.; Kankainen, M.; Lee, H.; Tang, J.; Kainov, D. Genome-Wide Analysis of Evolutionary Markers of Human Influenza A(H1N1)pdm09 and A(H3N2) Viruses May Guide Selection of Vaccine Strain Candidates. Genome Biol. Evol. 2015, 7, 3472–3483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elderfield, R.; Watson, S.; Godlee, A.; Adamson, W.; Thompson, C.; Dunning, J.; Fernandez-Alonso, M.; Blumenkrantz, D.; Hussell, T.; Zambon, M.; et al. Accumulation of human-adapting mutations during circulation of A(H1N1)pdm09 influenza virus in humans in the United Kingdom. J. Virol. 2014, 88, 13269–13283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, Y.; Tsao, K.; Huang, C.; Chang, K.; Huang, Y.; Gong, Y. Clinical characteristics of patients with laboratory-confirmed influenza A(H1N1)pdm09 during the 2013/2014 and 2015/2016 clade 6B/6B.1/6B.2-predominant outbreaks. Sci. Rep. 2018, 8, 15636. [Google Scholar] [CrossRef]
- Broberg, E.; Melidou, A.; Prosenc, K.; Bragstad, K.; Hungnes, O.; WHO European Region and the European Influenza Surveillance Network members of the reporting countries. Predominance of influenza A(H1N1)pdm09 virus genetic subclade 6B.1 and influenza B/Victoria lineage viruses at the start of the 2015/16 influenza season in Europe. Eurosurveillance 2016, 21. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.A.; Huang, Y.C.; Chiu, C.H.; Tsao, K.C.; Lin, T.Y. Impaired Vaccine-Induced Antibody Response Against Clade 6B H1N1 Viruses in Individuals Before Viral Emergence. Open Forum Infect Dis. 2020, 7, ofz513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Gong, Y.-N.; Shaw-Saliba, K.; Mehoke, T.; Evans, J.; Liu, Z.-Y.; Lewis, M.; Sauer, L.; Thielen, P.; Rothman, R.; et al. Differential Disease Severity and Whole Genome Sequence Analysis for Human Influenza A/H1N1pdm Virus in 2015-2016 Influenza Season. BioRxiv 2020, 02.20.957068. Available online: https://www.biorxiv.org/content/10.1101/2020.02.20.957068v2.full (accessed on 26 September 2021). [CrossRef] [Green Version]
- Newitt, S.; Mironenko, A.; Holubka, O.; Zaika, O.; Gubar, O.; Jalava, K.; Brown, C.; Demchshyna, I.; Dykhanovska, T. Rapid risk assessment during the early weeks of the 2015-2016 influenza season in Ukraine. Influenza. Other Respir. Viruses 2018, 12, 241–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogales, A.; Martinez-Sobrido, L.; Chiem, K.; Topham, D.; DeDiego, M. Functional evolution of the pandemic H1N1 influenza virus NS1 and PA in humans. J. Virol. 2018, 92, e01206-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization Influenza Centre. NIMR Interim Report February 2016. Available online: https://www.crick.ac.uk/media/286458/crick_feb2016_vcm_report_to_post.pdf (accessed on 26 September 2021).
- Igarashi, M.; Ito, K.; Yoshida, R.; Tomabechi, D.; Kida, H.; Takada, A. Predicting the Antigenic Structure of the Pandemic (H1N1) 2009 Influenza Virus Hemagglutinin. PLoS ONE. 2010, 5, e8553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlee, G.G.; Fodor, E. The predicted antigenicity of the haemagglutinin of the 1918 Spanish influenza pandemic suggests an avian origin. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2001, 356, 1871–1876. [Google Scholar] [CrossRef]
- Zolotarova, O.; Budzanivska, I.; Leibenko, L.; Radchenko, L.; Mironenko, A. Antigenic site variation in the hemagglutinin of pandemic influenza A(H1N1)pdm09 viruses between 2009–2017 in Ukraine. Pathogens 2019, 8, 194. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, P.; Henneberry, J.; Wilson, I.; Sambrook, J.; Gething, M. Addition of carbohydrate side chains at novel sites on influenza virus hemagglutinin can modulate the folding, transport, and activity of the molecule. J. Cell. Biol. 1988, 107, 2059–2073. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.; DeDiego, M.; Anderson, C.; Wang, J.; Yang, H.; Nogales, A.; Martinez-Sobrido, L.; Zand, M.; Sangster, M.; Topham, D. Antigenicity of the 2015-2016 seasonal H1N1 human influenza virus HA and NA proteins. PLoS ONE 2017, 12, e0188267. [Google Scholar] [CrossRef] [Green Version]
- Puig-Barberà, J.; Guglieri-López, B.; Tortajada-Girbés, M.; López-Labrador, F.X.; Carballido-Fernández, M.; Mollar-Maseres, J.; Schwarz-Chavarri, G.; Baselga-Moreno, V.; Mira-Iglesias, A.; Díez-Domingo, J. Valencia Hospital Network for the Study of Influenza, Respiratory Viruses Disease. Low influenza vaccine effectiveness and the effect of previous vaccination in preventing admission with A(H1N1)pdm09 or B/Victoria-Lineage in patients 60 years old or older during the 2015/2016 influenza season. Vaccine 2017, 35, 7331–7338. [Google Scholar] [CrossRef] [PubMed]
- Mohebbi, A.; Fotouhi, F.; Jamali, A.; Yaghobi, R.; Farahmand, B.; Mohebbi, R. Molecular epidemiology of the hemagglutinin gene of prevalent influenza virus A/H1N1/pdm09 among patient in Iran. Virus Res. 2019, 259, 38–45. [Google Scholar] [CrossRef]
- Nakamura, K.; Shirakura, M.; Fujisaki, S.; Kishida, N.; Burke, D.F.; Smith, D.J.; Kuwahara, T.; Takashita, E.; Takayama, I.; Nakauchi, M.; et al. Characterization of influenza A(H1N1)pdm09 viruses isolated from Nepalese and Indian outbreak patients in early 2015. Influenza Other Respir Viruses 2017, 11, 399–403. [Google Scholar] [CrossRef]
- Seok, J.H.; Kim, J.; Lee, D.B.; Cho, K.J.; Lee, J.-H.; Bae, G.; Chung, M.S.; Kim, K.H. Conformational modulation of influenza virus hemagglutinin: Characterization and in vivo efficacy of monomeric form. Sci. Rep. 2017, 7, 7540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapia, R.; Torremorell, M.; Culhane, M.; Medina, R.A.; Neira, V. Antigenic characterization of novel H1 influenza A viruses in swine. Sci. Rep. 2020, 10, 4510. [Google Scholar] [CrossRef] [PubMed]
- Opanda, S.; Bulimo, W.; Gachara, G.; Ekuttan, C.; Amukoye, E. Assessing antigenic drift and phylogeny of influenza A (H1N1) pdm09 virus in Kenya using HA1 sub-unit of the hemagglutinin gene. PLoS ONE 2020, 15, e0228029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath Neerukonda, S.; Vassell, R.; Weiss, C.D. Neutralizing Antibodies Targeting the Conserved Stem Region of Influenza Hemagglutinin. Vaccines 2020, 8, 382. [Google Scholar] [CrossRef]
- Puzelli, S.; Facchini, M.; Spagnolo, D.; De Marco, M.A.; Calzoletti, L.; Zanetti, A.; Fumagalli, R.; Tanzi, M.L.; Cassone, A.; Rezza, G.; et al. Surveillance Group for Pandemic A H1N1 2009 Influenza Virus in Italy. Transmission of hemagglutinin D222G mutant strain of pandemic (H1N1) 2009 virus. Emerg. Infect Dis. 2010, 16, 863–865. [Google Scholar] [CrossRef] [PubMed]
- Ilyushina, N.A.; Komatsu, T.E.; Ince, W.L.; Donaldson, E.F.; Lee, N.; O’Rear, J.J.; Donnelly, R.P. Influenza A virus hemagglutinin mutations associated with use of neuraminidase inhibitors correlate with decreased inhibition by anti-influenza antibodies. Virol. J. 2019, 16, 149. [Google Scholar] [CrossRef] [Green Version]
- Korsun, N.; Angelova, S.; Gregory, V.; Daniels, R.; Georgieva, I.; McCauley, J. Antigenic and genetic characterization of influenza viruses circulating in Bulgaria during the 2015/2016 season. Infect. Genet. Evol. 2017, 49, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Chutinimitkul, S.; Herfst, S.; Steel, J.; Lowen, A.C.; Ye, J.; van Riel, D.; Schrauwen, E.J.; Bestebroer, T.M.; Koel, B.; Burke, D.F.; et al. Virulence-associated substitution D222G in the hemagglutinin of 2009 pandemic influenza A(H1N1) virus affects receptor binding. J. Virol. 2010, 84, 11802–11813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houng, H.S.; Garner, J.; Zhou, Y.; Lyons, A.; Kuschner, R.; Deye, G.; St Clair, K.; Douce, R.W.; Chicaiza, W.; Blair, P.J.; et al. Emergent 2009 influenza A(H1N1) viruses containing HA D222N mutation associated with severe clinical outcomes in the Americas. J. Clin. Virol. 2012, 53, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, T.; De Rosa, F.; Cerutti, F.; Pagani, N.; Allice, T.; Stella, M.L.; Milia, M.G.; Calcagno, A.; Burdino, E.; Gregori, G.; et al. A(H1N1)pdm09 hemagglutinin D222G and D222N variants are frequently harbored by patients requiring extracorporeal membrane oxygenation and advanced respiratory assistance for severe A(H1N1)pdm09 infection. Influenza Other Respir Viruses 2013, 7, 1416–1426. [Google Scholar] [CrossRef] [Green Version]
- Doud, M.; Hensley, S.; Bloom, J. Complete mapping of viral escape from neutralizing antibodies. PLoS Pathog. 2017, 13, e1006271. [Google Scholar] [CrossRef] [Green Version]
- Childs, R.A.; Palma, A.S.; Wharton, S.; Matrosovich, T.; Liu, Y.; Chai, W.; Campanero-Rhodes, M.A.; Zhang, Y.; Eickmann, M.; Kiso, M.; et al. Receptor-binding specificity of pandemic influenza A (H1N1) 2009 virus determined by carbohydrate microarray. Nat. Biotechnol. 2009, 27, 797–799. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.Q.; Mozdzanowska, K.; Gerhard, W. Complement component C1q enhances the biological activity of influenza virus hemagglutinin-specific antibodies depending on their fine antigen specificity and heavy-chain isotype. J. Virol. 2002, 76, 1369–1378. [Google Scholar] [CrossRef] [Green Version]
- Nogales, A.; Martinez-Sorbido, L.; Topham, D.; DeDiego, M. Modulation of innate immune responses by the influenza A NS1 and PA-X proteins. Viruses 2018, 10, E708. [Google Scholar] [CrossRef] [Green Version]
- Hale, B.; Steel, J.; Medina, R.; Manicassamy, B.; Ye, J.; Hickman, D.; Hai, R.; Schmolke, M.; Lowen, A.; Perez, D.; et al. Inefficient control of host gene expression by the 2009 pandemic H1N1 influenza A virus NS1 protein. J. Virol. 2010, 84, 6909–6922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, A.M.; Nogales, A.; Martinez-Sobrido, L.; Topham, D.J.; DeDiego, M.L. Functional Evolution of Influenza Virus NS1 Protein in Currently Circulating Human 2009 Pandemic H1N1 Viruses. J. Virol. 2017, 91, e00721-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komissarov, A.; Fadeev, A.; Sergeeva, M.; Petrov, S.; Sintsova, K.; Egorova, A.; Pisareva, M.; Buzitskaya, Z.; Musaeva, T.; Danilenko, D.; et al. Rapid spread of influenza A(H1N1)pdm09 viruses with a new set of specific mutations in the internal genes in the beginning of 2015/2016 epidemic season in Moscow and Saint Petersburg (Russian Federation). Influenza. Other. Respir. Viruses 2016, 5, 247–253. [Google Scholar] [CrossRef]
- Kainov, D.E.; Müller, K.H.; Theisen, L.L.; Anastasina, M.; Kaloinen, M.; Muller, C.P. Differential effects of NS1 proteins of human pandemic H1N1/2009, avian highly pathogenic H5N1, and low pathogenic H5N2 influenza A viruses on cellular pre-mRNA polyadenylation and mRNA translation. J. Biol. Chem. 2011, 286, 7239–7247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, J.Y.; Li, S.; Sen, G.C.; Krug, R.M. A site on the influenza A virus NS1 protein mediates both inhibition of PKR activation and temporal regulation of viral RNA synthesis. Virology 2007, 363, 236–243. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Zeng, W.; Hao, X.; Huang, J.; Cai, M.; Zhou, P.; Zhang, G. Continuous evolution of influenza A viruses of swine from 2013 to 2015 in Guangdong, China. PLoS ONE 2019, 14, e0217607. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.S.; Noh, J.Y.; Song, J.Y.; Cheong, H.J.; Choi, W.S.; Jeong, H.W.; Wie, S.-H.; Kim, W.J. Molecular genetic characteristics of influenza A virus clinically isolated during 2011–2016 influenza seasons in Korea. Influenza Other Respir Viruses 2018, 12, 497–507. [Google Scholar] [CrossRef]
- Pinilla, L.T.; Holder, B.P.; Abed, Y.; Boivin, G.; Beauchemin, C.A. The H275Y neuraminidase mutation of the pandemic A/H1N1 influenza virus lengthens the eclipse phase and reduces viral output of infected cells, potentially compromising fitness in ferrets. J. Virol. 2012, 86, 10651–10660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potdar, V.A.; Padbidri, V.V.; Chadha, M.S. Oseltamivir-resistant influenza A(H1N1) pdm09 virus: First reported case from India. WHO South. East. Asia J. Public Health 2013, 2, 181–183. [Google Scholar] [CrossRef]
- Bialas, K.M.; Desmet, E.A.; Takimoto, T. Specific residues in the 2009 H1N1 swine-origin influenza matrix protein influence virion morphology and efficiency of viral spread in vitro. PLoS ONE 2012, 7, e50595. [Google Scholar] [CrossRef] [Green Version]
- Dong, G.; Peng, C.; Luo, J.; Wang, C.; Han, L.; Wu, B.; Ji, G.; He, H. Adamantane-resistant influenza a viruses in the world (1902–2013): Frequency and distribution of M2 gene mutations. PLoS ONE 2015, 10, e0119115. [Google Scholar] [CrossRef] [Green Version]
- Ashenberg, O.; Padmakumar, J.; Doud, M.B.; Bloom, J.D. Deep mutational scanning identifies sites in influenza nucleoprotein that affect viral inhibition by MxA. PLoS Pathog. 2017, 13, e1006288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Xu, Q.; Shen, Y.; Liu, L.; Wei, K.; Sun, H.; Pu, J.; Chang, K.; Liu, J. Naturally occurring mutations in the PA gene are key contributors to increased virulence of pandemic H1N1/09 influenza virus in mice. J. Virol. 2014, 88, 4600–4604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bortz, E.; Westera, L.; Maamary, J.; Steel, J.; Albrecht, R.; Manicassamy, B.; Chase, G.; Martinez-Sorbido, L.; Schwemmle, M.; Garcia-Sastre, A. Host- and strain-specific regulation of influenza virus polymerase activity by interacting cellular proteins. mBio 2011, 2, e00151-11. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, G.; Fodor, E. Molecular determinants of pathogenicity in the polymerase complex. Curr. Top. Microbiol. Immunol. 2014, 385, 35–60. [Google Scholar] [CrossRef] [PubMed]
- Mishel, P.; Ojala, T.; Banner, C.; Lakspere, T.; Bychkov, D.; Jalovaara, P.; Kakkola, L.; Kallio-Kokko, H.; Kantele, A.; Kankainen, M.; et al. Comparative analysis of whole-genome sequences of influenza A(H1N1)pdm09 viruses isolated from hospitalized and non-hospitalized patients identifies missense mutations that might be associated with patient hospital admissions in Finland during 2009 to 2014. Genome. Announc. 2015, 3, e00676-15. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Zhao, X.; Hua, S.; Du, X.; Peng, Y.; Li, X.; Lan, Y.; Wang, D.; Wu, A.; Shu, Y.; et al. Antigenic patterns and evolution of the human influenza A(H1N1) virus. Sci. Rep. 2015, 5, 14171. [Google Scholar] [CrossRef] [PubMed]
- Schaduangrat, N.; Phanich, J.; Rungrotmongkol, T.; Lerdssamran, H.; Puthavathana, P.; Ubol, S. The significance of naturally occurring neuraminidase quasispecies of H5N1 avian influenza virus on resistance to oseltamivir: A point of concern. J. Gen. Virol. 2016, 97, 1311–1323. [Google Scholar] [CrossRef] [Green Version]
- Otte, A.; Marriott, A.; Dreier, C.; Dove, B.; Mooren, K.; Klingen, T.; Sauter, M.; Thompson, K.; Bennett, A.; Klingel, K.; et al. Evolution of 2009 H1N1 influenza viruses during the pandemic correlates with increased viral pathogenicity and transmissibility in the ferret model. Sci. Rep. 2016, 6, 28583. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization (WHO). CDC Protocol of Realtime RT-PCR for Influenza H1N1. World Health Organization, Geneva, Switzerland. 2009. Available online: https://www.who.int/csr/resources/publications/swineflu/CDCRealtimeRTPCR_SwineH1Assay-2009_20090430.pdf (accessed on 26 September 2021).
- WHO Global Influenza Surveillance Network Manual for the Laboratory Diagnosis and Virological Surveillance of Influenza. 2011, 153. Available online: https://apps.who.int/iris/bitstream/handle/10665/44518/9789241548090_eng.pdf;jsessionid=1E7BC05AD56E9F5C9C353368D811AE13?sequence=1 (accessed on 26 September 2021).
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.; Goddard, T.; Huang, C.; Couch, G.; Greenblatt, D.; Meng, E.; Ferrin, T. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Viral Protein | Substitutions | A(H1N1)pdm09 Strains | Potential Functions | Epidemiology | Refs. |
|---|---|---|---|---|---|
| HA | P83S, I321V, S203T, D97N, S185T, E47K, S124N | Defining group 6B.1, 6B.2 mutations in H1 HA in all Ukrainian strains reported herein. | Antigenic drift; resistance to neutralizing antibody generated by vaccine; S185T RBD mutation may impact receptor specificity. | Group 6B.1, 6B.2 dominated in many countries (2015/16 and 2017/18). Vaccine mismatch. | [7,8,9,10] |
| S84N, I216T, S162N, D127E | All group 6B.1 strains | Signature mutations in group 6B.1. Potential novel glycosylation in Sa antigenic site: S162N. | S84N appeared early 2015, a year prior to 6B.1 wave, in India. | [19,20,21] | |
| F88L | A/Odessa/68/2016 (6B.2) | F88L is on trimeric interaction surface in HA2. | [22] | ||
| T270A | A/Khmelnitsky/663/2016 (6B.1) | Functional significance unknown. | T270A found in 2 strains in swine, Chile (2015). | [23] | |
| R45K | A/Odessa/685/2016 | Antigenic epitope C. | R45K in strains circulating in Kenya in 2015, 2018. | [24] | |
| N38D, K40N | A/Khelmitsky/81/2016 | N38 highly conserved glycosylation site in H1 HA that prevents nAb binding. Loss possibly compensated by K40N. | [25] | ||
| P297S | A/Kharkiv/348/2016 | P297S possibly stabilizes HA1 in context of the D222G/E mutation that increases virulence. | [26] | ||
| S183P, S326P | A/Zaporizzia/631/2016 | S183P, S326P in H1 RBD; increases binding affinity to a-2,6 SA; S326P in Ca antigenic site. | S183P, S326P strongly selected for by 2018. | [27] | |
| I91V | A/Khmelnitsky/663/2016, A/Ukraine/25/2016, A/Ternopil/754/2016 | I91V is in HA2 domain (functional significance unknown). | I91V found in subset of strains in Bulgaria 2015/16. | [28] | |
| D222G, D222N | A/Dnipro/580/2016, A/Ukraine/7182/2016 | D222G/N increases bindiE17atory tract, increases severity of ILI; increased virulence in mice. | D222G/M mutations occurred in 32% of severe/lethal cases in Russia (2017/18) and elsewhere. | [16,29,30,31] | |
| A141T | A/Khmelnitsky/727/2016, A/Khmelnitsky/760/2016, A/Zaporizza/631/2016 | A141T mutation in Ca antigenic site in HA1. | A141T found in subset of strains in Bulgaria 2015/16. | [28] | |
| S83P | 11 Ukrainian strains (6B.1) * | S83P mutation in Cb antigenic site, epitope E nAb site. | S83P is a common mutation observed in H1N1 isolates and back transferred into swine. | [32,33,34] | |
| NS1 | E55K, L90I, I123V, K131E, N205S | All Ukrainian strains ** | Mutations inhibit host interferon response and gene expression. | Common to NS1 in group 6B.1. | [35,36,37] |
| D2E, E125D | 17 Ukrainian strains ** | D2E in RNA binding domain; E125D in effector domain (functional significance unknown). | Observed in Russia (2015/16) and China (2016–18). | [38] | |
| N48S, M124T | A/Ukraine/586/2015, A/Ukraine/5/2016, A/Ukraine75/2016 | N48S in RNA binding domain enhances viral mRNA translation; M124T inhibits antiviral PKR/RAP55 binding. | N48S present in H5 HPAI NS1 and swine H1 strains in China. | [6,39,40,41] | |
| I18V | A/Odessa/61/2016 | I18V in NS1 site of genetic instability, arises in MDCK passage. | Revertant to swine triple reassortant lineage residue, observed in China. | [41] | |
| V129I, I182V | A/Dnipro/439/2015 | Mutations in NS1 efector domain (functional significance unknown). | Sporadic occurrence, observed in Korea (2013). | [42] | |
| NA | H275Y | Not detected | Oseltamivir resistance; reduced fitness. | <2% prevalence in A(H1N1)pdm09 strains. | [43] |
| V453M | A/Khmelnitsky/89/2016 | V453 is a potentially stabilizing mutations that can co-occur with H275Y. | Sporadic co-occurrence with H275Y. | [43,44] | |
| M1 | V80I | All Ukrainian strains. | Increased replication in cell culture. | Worldwide. | [5] |
| Q208K | 62.5% of Ukrainian isolates sequenced in 2015/16. *** | M1 residues 207–209 (alpha helix #12) determine filmentous morphology and budding. | First occurred in Ukraine 2013, 2015, fixed by 2015/16; found in Russia and Bulgaria. | [28,38,45] | |
| M2 | D21G | All Ukrainian strains. | Resistance to amantadine. | Worldwide. | [38,46] |
| NP | M105T | All Ukrainian strains. | Residues 98–105 form variable motif in human influenza that affects sensitivity/resistance to antiviral protein MXA. | 86% of strains in Russia in subsequent year; worldwide. | [28,38,47] |
| PA | N321K | All Ukrainian strains. | Increased virulence. | All A(H1N1)pdm09 strains. | [48] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zolotarova, O.; Fesenko, A.; Holubka, O.; Radchenko, L.; Bortz, E.; Budzanivska, I.; Mironenko, A. Genotypic Variants of Pandemic H1N1 Influenza A Viruses Isolated from Severe Acute Respiratory Infections in Ukraine during the 2015/16 Influenza Season. Viruses 2021, 13, 2125. https://doi.org/10.3390/v13112125
Zolotarova O, Fesenko A, Holubka O, Radchenko L, Bortz E, Budzanivska I, Mironenko A. Genotypic Variants of Pandemic H1N1 Influenza A Viruses Isolated from Severe Acute Respiratory Infections in Ukraine during the 2015/16 Influenza Season. Viruses. 2021; 13(11):2125. https://doi.org/10.3390/v13112125
Chicago/Turabian StyleZolotarova, Oksana, Anna Fesenko, Olga Holubka, Larysa Radchenko, Eric Bortz, Iryna Budzanivska, and Alla Mironenko. 2021. "Genotypic Variants of Pandemic H1N1 Influenza A Viruses Isolated from Severe Acute Respiratory Infections in Ukraine during the 2015/16 Influenza Season" Viruses 13, no. 11: 2125. https://doi.org/10.3390/v13112125
APA StyleZolotarova, O., Fesenko, A., Holubka, O., Radchenko, L., Bortz, E., Budzanivska, I., & Mironenko, A. (2021). Genotypic Variants of Pandemic H1N1 Influenza A Viruses Isolated from Severe Acute Respiratory Infections in Ukraine during the 2015/16 Influenza Season. Viruses, 13(11), 2125. https://doi.org/10.3390/v13112125