
Novel Cyclovirus Species in Dogs with Hemorrhagic Gastroenteritis




Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sampling
2.3. Amplification of Viral DNA
2.4. Nucleotide Sequencing
2.5. Sequence Analysis
2.6. GenBank Accession Numbers
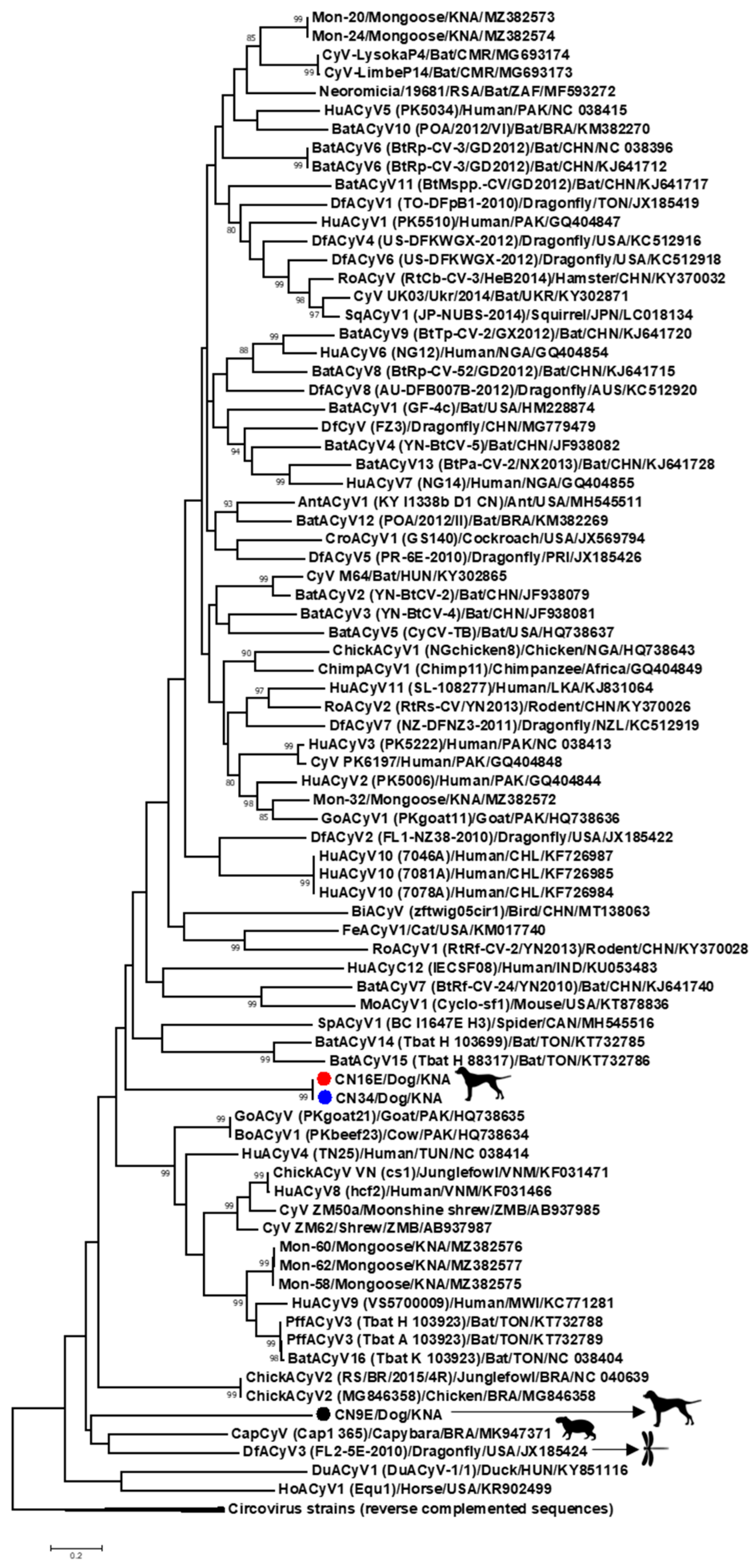
3. Results and Discussion
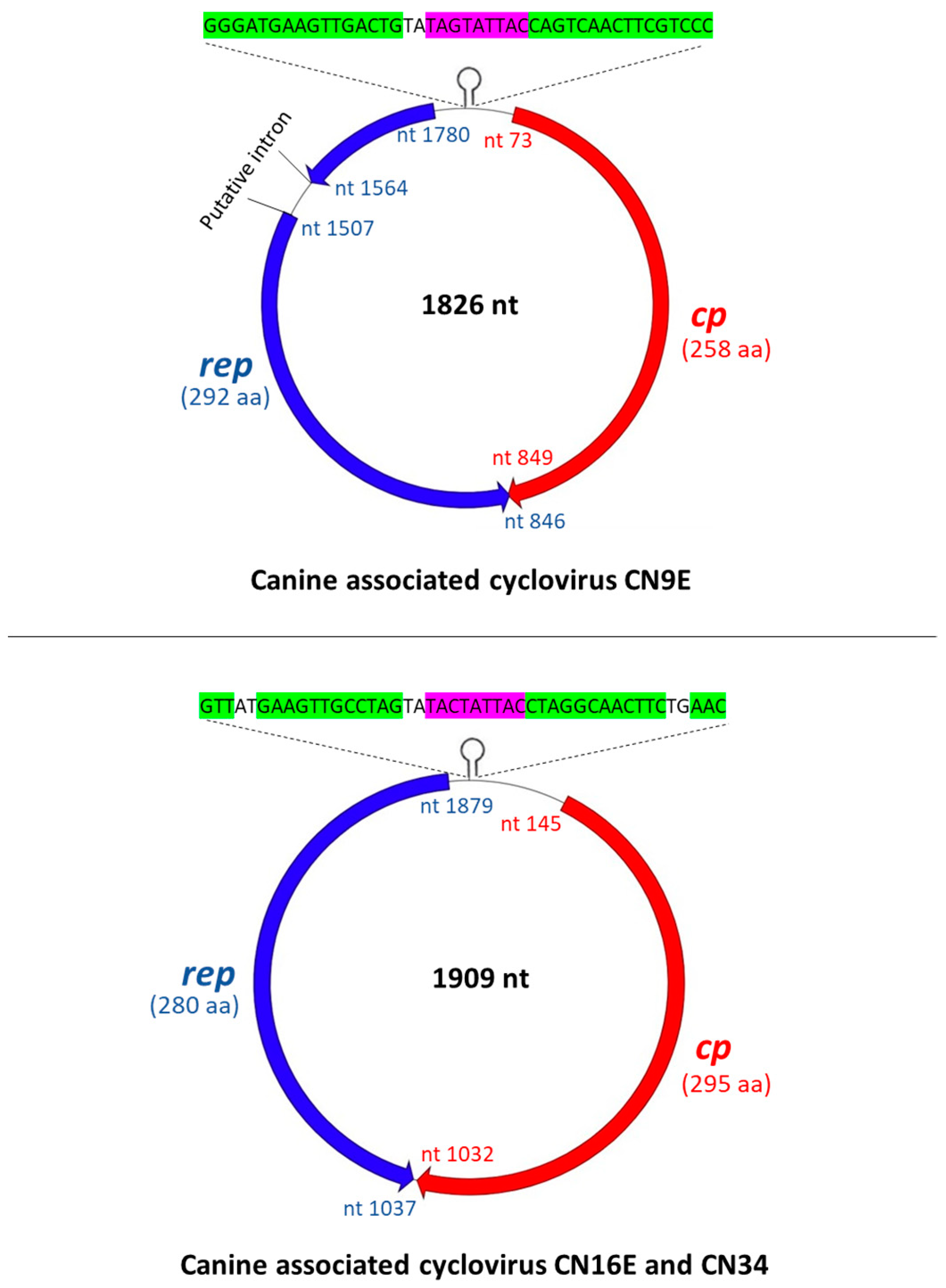
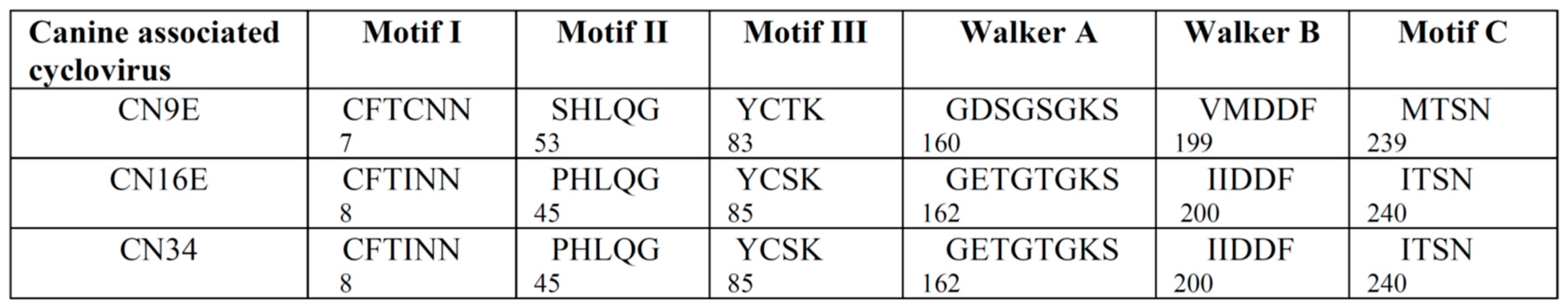
3.1. Complete Genome Analysis of Canine-Associated Cyclovirus CN9E
3.2. Complete Genome Analysis of Canine-Associated Cyclovirus CN16E and CN34
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Breitbart, M.; Delwart, E.; Rosario, K.; Segalés, J.; Varsani, A. ICTV virus taxonomy profile: Circoviridae. J. Gen. Virol. 2017, 98, 1997–1998. [Google Scholar] [CrossRef]
- Rosario, K.; Breitbart, M.; Harrach, B.; Segalés, J.; Delwart, E.; Biagini, P.; Varsani, A. Revisiting the taxonomy of the family Circoviridae: Establishment of the genus Cyclovirus and removal of the genus Gyrovirus. Arch. Virol. 2017, 162, 1447–1463. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Kapoor, A.; Slikas, B.; Bamidele, O.S.; Wang, C.; Shaukat, S.; Alam Masroor, M.; Wilson, M.L.; Ndjango, J.-B.N.; Peeters, M.; et al. Multiple diverse circoviruses infect farm animals and are commonly found in human and chimpanzee feces. J. Virol. 2010, 84, 1674–1682. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Rosario, K.; Breitbart, M.; Duffy, S. Eukaryotic circular rep-encoding single-stranded DNA (CRESS DNA) viruses: Ubiquitous viruses with small genomes and a diverse host range. In Advances in Clinical Chemistry; Elsevier: Amsterdam, The Netherlands, 2019; Volume 103, pp. 71–133. [Google Scholar]
- Delwart, E.; Li, L. Rapidly expanding genetic diversity and host range of the Circoviridae viral family and other Rep encoding small circular ssDNA genomes. Virus Res. 2012, 164, 114–121. [Google Scholar] [CrossRef] [Green Version]
- Steinfeldt, T.; Finsterbusch, T.; Mankertz, A. Demonstration of nicking/joining activity at the origin of DNA replication associated with the rep and rep’ proteins of porcine circovirus type 1. J. Virol. 2006, 80, 6225–6234. [Google Scholar] [CrossRef] [Green Version]
- Cheung, A.K. Identification of an octanucleotide motif sequence essential for viral protein, DNA, and progeny virus biosynthesis at the origin of DNA replication of porcine circovirus type 2. Virology 2004, 324, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Nebbak, A.; Monteil-Bouchard, S.; Berenger, J.-M.; Almeras, L.; Parola, P.; Desnues, C. Virome diversity among mosquito populations in a sub-urban region of Marseille, France. Viruses 2021, 13, 768. [Google Scholar] [CrossRef]
- Wu, Z.; Lu, L.; Du, J.; Yang, L.; Ren, X.; Liu, B.; Jiang, J.; Yang, J.; Dong, J.; Sun, L.; et al. Comparative analysis of rodent and small mammal viromes to better understand the wildlife origin of emerging infectious diseases. Microbiome 2018, 6, 1–14. [Google Scholar] [CrossRef]
- Prades, Y.; Pizarro, R.; Ruiz, M.; Moreno, C.; Avendaño, L.F.; Luchsinger, V. Cyclovirus detection in Chilean adults with and without community-acquired pneumonia. J. Med. Virol. 2021, 93, 4786–4793. [Google Scholar] [CrossRef]
- Sasaki, M.; Orba, Y.; Ueno, K.; Ishii, A.; Moonga, L.; Hang’ombe, B.M.; Mweene, A.S.; Ito, K.; Sawa, H. Metagenomic analysis of the shrew enteric virome reveals novel viruses related to human stool-associated viruses. J. Gen. Virol. 2015, 96, 440–452. [Google Scholar] [CrossRef]
- Rosario, K.; Mettel, K.A.; Benner, B.E.; Johnson, R.; Scott, C.; Yusseff-Vanegas, S.Z.; Baker, C.C.; Cassill, D.L.; Storer, C.; Varsani, A.; et al. Virus discovery in all three major lineages of terrestrial arthropods highlights the diversity of single-stranded DNA viruses associated with invertebrates. PeerJ 2018, 6, e5761. [Google Scholar] [CrossRef]
- Dennis, T.P.W.; Flynn, P.J.; de Souza, W.M.; Singer, J.B.; Moreau, C.S.; Wilson, S.J.; Gifford, R.J. Insights into circovirus host range from the genomic fossil record. J. Virol. 2018, 92, 00145-18. [Google Scholar] [CrossRef] [Green Version]
- Smits, S.L.; Zijlstra, E.E.; van Hellemond, J.J.; Schapendonk, C.M.; Bodewes, R.; Schürch, A.; Haagmans, B.L.; Osterhaus, A. Novel cyclovirus in human cerebrospinal fluid, Malawi, 2010–2011. Emerg. Infect. Dis. 2013, 19, 1511–1513. [Google Scholar] [CrossRef]
- Gainor, K.; Becker, A.A.M.J.; Malik, Y.S.; Ghosh, S. Detection and complete genome analysis of circoviruses and cycloviruses in the small indian mongoose (Urva auropunctata): Identification of novel species. Viruses 2021, 13, 1700. [Google Scholar] [CrossRef]
- Kaszab, E.; Lengyel, G.; Marton, S.; Dán, Á.; Bányai, K.; Fehér, E. Occurrence and genetic diversity of CRESS DNA viruses in wild birds: A Hungarian study. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Male, M.F.; Kraberger, S.; Stainton, D.; Kami, V.; Varsani, A. Cycloviruses, gemycircularviruses and other novel replication-associated protein encoding circular viruses in Pacific flying fox (Pteropus tonganus) faeces. Infect. Genet. Evol. 2016, 39, 279–292. [Google Scholar] [CrossRef]
- Yinda, C.K.; Ghogomu, S.M.; Conceição-Neto, N.; Beller, L.; Deboutte, W.; Vanhulle, E.; Maes, P.; van Ranst, M.; Matthijnssens, J. Cameroonian fruit bats harbor divergent viruses, including rotavirus H, bastroviruses, and picobirnaviruses using an alternative genetic code. Virus Evol. 2018, 4, vey008. [Google Scholar] [CrossRef] [Green Version]
- Patterson, Q.M.; Kraberger, S.; Martin, D.P.; Shero, M.R.; Beltran, R.S.; Kirkham, A.L.; Aleamotu’a, M.; Ainley, D.G.; Kim, S.; Burns, J.M.; et al. Circoviruses and cycloviruses identified in Weddell seal fecal samples from McMurdo Sound, Antarctica. Infect. Genet. Evol. 2021, 95, 105070. [Google Scholar] [CrossRef]
- Yan, T.; Li, G.; Zhou, D.; Yang, X.; Hu, L.; Cheng, Z. Novel cyclovirus identified in broiler chickens with transmissible viral proventriculitis in China. Front. Vet. Sci. 2020, 7, 569098. [Google Scholar] [CrossRef]
- Phan, T.; Mori, D.; Deng, X.; Rajindrajith, S.; Ranawaka, U.; Fan Ng, T.F.; Bucardo-Rivera, F.; Orlandi, P.; Ahmed, K.; Delwart, E. Small circular single stranded DNA viral genomes in unexplained cases of human encephalitis, diarrhea, and in untreated sewage. Virology 2015, 482, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Li, Y.; Guo, Z.; Yi, Y.; Zhang, H.; Shangguan, H.; Huang, C.; Ge, J. Genetic changes and evolutionary analysis of canine circovirus. Arch. Virol. 2021, 166, 2235–2247. [Google Scholar] [CrossRef]
- Kapoor, A.; Dubovi, E.J.; Henriquez-Rivera, J.A.; Lipkin, W.I. Complete genome sequence of the first canine circovirus. J. Virol. 2012, 86, 7018. [Google Scholar] [CrossRef] [Green Version]
- Anderson, A.; Hartmann, K.; Leutenegger, C.M.; Proksch, A.L.; Mueller, R.S.; Unterer, S. Role of canine circovirus in dogs with acute haemorrhagic diarrhoea. Vet. Rec. 2017, 180, 542. [Google Scholar] [CrossRef]
- Thaiwong, T.; Wise, A.G.; Maes, R.K.; Mullaney, T.; Kiupel, M. Canine circovirus 1 (CaCV-1) and canine parvovirus 2 (CPV-2). Vet. Pathol. 2016, 53, 1204–1209. [Google Scholar] [CrossRef] [Green Version]
- Zaccaria, G.; Malatesta, D.; Scipioni, G.; di Felice, E.; Campolo, M.; Casaccia, C.; Savini, G.; di Sabatino, D.; Lorusso, A. Circovirus in domestic and wild carnivores: An important opportunistic agent? Virology 2016, 490, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; McGraw, S.; Zhu, K.; Leutenegger, C.M.; Marks, S.L.; Kubiski, S.; Gaffney, P.; dela Cruz, F.N.; Wang, C.; Delwart, E.; et al. Circovirus in tissues of dogs with vasculitis and hemorrhage. Emerg. Infect. Dis. 2013, 19, 534–541. [Google Scholar] [CrossRef]
- Bexton, S.; Wiersma, L.C.; Getu, S.; van Run, P.R.; Verjans, G.; Schipper, D.; Schapendonk, C.M.; Bodewes, R.; Oldroyd, L.; Haagmans, B.L.; et al. Detection of circovirus in foxes with meningoencephalitis, United Kingdom, 2009–2013. Emerg. Infect. Dis. 2015, 21, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Urbani, L.; Tryland, M.; Ehrich, D.; Fuglei, E.; Battilani, M.; Balboni, A. Ancient origin and genetic segregation of canine circovirus infecting arctic foxes (Vulpes lagopus) in Svalbard and red foxes (Vulpes vulpes) in northern Norway. Transbound. Emerg. Dis. 2021, 68, 1283–1293. [Google Scholar] [CrossRef]
- Gainor, K.; Bowen, A.; Bolfa, P.; Peda, A.; Malik, Y.; Ghosh, S. Molecular investigation of canine parvovirus-2 (CPV-2) outbreak in Nevis Island: Analysis of the nearly complete genomes of CPV-2 strains from the caribbean region. Viruses 2021, 13, 1083. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayaram, A.; Potter, K.A.; Moline, A.B.; Rosenstein, D.D.; Marinov, M.; Thomas, J.E.; Breitbart, M.; Rosario, K.; Argüello-Astorga, G.R.; Varsani, A. High global diversity of cycloviruses amongst dragonflies. J. Gen. Virol. 2013, 94, 1827–1840. [Google Scholar] [CrossRef] [Green Version]
- Van Tan, L.; van Doorn, H.R.; Nghia, H.D.T.; Chau, T.T.H.; Tu, L.T.P.; de Vries, M.; Canuti, M.; Deijs, M.; Jebbink, M.F.; Baker, S.; et al. Identification of a new cyclovirus in cerebrospinal fluid of patients with acute central nervous system infections. mBio 2013, 4, e00231-13. [Google Scholar] [CrossRef] [Green Version]
- Fontenele, R.S.; Lacorte, C.; Lamas, N.S.; Schmidlin, K.; Varsani, A.; Ribeiro, S.G. Single stranded DNA viruses associated with capybara faeces sampled in Brazil. Viruses 2019, 11, 710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosario, K.; Dayaram, A.; Marinov, M.; Ware, J.; Kraberger, S.; Stainton, D.; Breitbart, M.; Varsani, A. Diverse circular ssDNA viruses discovered in dragonflies (Odonata: Epiprocta). J. Gen. Virol. 2012, 93, 2668–2681. [Google Scholar] [CrossRef] [PubMed]
- Nibert, M.L.; Debat, H.J.; Manny, A.R.; Grigoriev, I.V.; de Fine Licht, H.H. Mitovirus and mitochondrial coding sequences from basal fungus Entomophthora muscae. Viruses 2019, 11, 351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shackelton, L.A.; Holmes, E.C. The role of alternative genetic codes in viral evolution and emergence. J. Theor. Biol. 2008, 254, 128–134. [Google Scholar] [CrossRef]
- Yinda, C.K.; Vanhulle, E.; Conceição-Neto, N.; Beller, L.; Deboutte, W.; Shi, C.; Ghogomu, S.M.; Maes, P.; van Ranst, M.; Matthijnssens, J. Gut virome analysis of cameroonians reveals high diversity of enteric viruses, including potential interspecies transmitted viruses. mSphere 2019, 4, e00585-18. [Google Scholar] [CrossRef] [Green Version]
- Kleymann, A.; Becker, A.A.M.J.; Malik, Y.S.; Kobayashi, N.; Ghosh, S. Detection and molecular characterization of picobirnaviruses (PBVs) in the mongoose: Identification of a novel PBV using an alternative genetic code. Viruses 2020, 12, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Malik, Y.S. The true host/s of picobirnaviruses. Front. Vet. Sci. 2021, 7, 615293. [Google Scholar] [CrossRef] [PubMed]
- Schmale, M.C.; Gibbs, P.D.; Rahn, J.J.; Vidal, D. 60 A parasite of mitochondria? A virus-like agent in neurogenic tumors of a tropical marine fish. Mitochondrion 2010, 10, 216. [Google Scholar] [CrossRef]
- Zhang, W.; Li, L.; Deng, X.; Kapusinszky, B.; Pesavento, P.A.; Delwart, E. Faecal virome of cats in an animal shelter. J. Gen. Virol. 2014, 95, 2553–2564. [Google Scholar] [CrossRef]
- Kotsias, F.; Bucafusco, D.; Nuñez, D.A.; Borisovsky, L.A.L.; Rodriguez, M.; Bratanich, A.C. Genomic characterization of canine circovirus associated with fatal disease in dogs in South America. PLoS ONE 2019, 14, e0218735. [Google Scholar] [CrossRef]
- Giraldo-Ramirez, S.; Rendon-Marin, S.; Vargas-Bermudez, D.S.; Jaime, J.; Ruiz-Saenz, J. First detection and full genomic analysis of canine circovirus in CPV-2 infected dogs in Colombia, South America. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Canine-Associated Cyclovirus (GenBank Accession Number) | ||||
|---|---|---|---|---|
| CN9E (OK148727) | CN16E (OK148728) | CN34 (OK148729) | ||
| Maximum/significant pairwise nucleotide sequence (%) identities of complete genome | Between canine-associated cycloviruses | 59.704% with CN16E 59.642% with CN34 | 99.790% between CN16E and CN34 | |
| With cyclovirus (Strain name/Detected in animal species/Country/Year/GenBank accession number) from other animal species | 61.646% with human-associated cyclovirus 8 isolate hcf2/Human/ Vietnam/2009/KF031466 61.337% with capybara-associated cyclovirus 1 isolate Cap1_365/ Capybara/Brazil/2016/MK947371 | 63.010% with feline-associated cyclovirus 1/Cat/USA/2013/ KM017740 62.910% with dragonfly-associated cyclovirus 1 isolate TODFpB12010/ Dragonfly/Tonga/2010/JX185419 | 63.040% with feline-associated cyclovirus 1/Cat/USA/2013/ KM017740 62.709% with dragonfly-associated cyclovirus 1 isolate TODFpB12010/ Dragonfly/Tonga/2010/JX185419 | |
| Maximum/significant pairwise deduced amino acid (aa) sequence (%) identities of putative Rep | Between canine-associated cycloviruses | 35.379% with CN34 35.018% with CN16E | 99.285% between CN16E and CN34 | |
| With cyclovirus (Strain name/Detected in animal species/Country/Year/GenBank accession number) from other animal species | 47.272% with dragonfly cyclovirus 3 isolate FL2-5E-2010/Dragonfly/ USA/2010/JX185424 45.112% with human-associated cyclovirus 5 isolate PK5034/ Human/Pakistan/2007/NC_038415 | 49.462% with capybara-associated cyclovirus 1 isolate Cap1_365/ Capybara/Brazil/2016/MK947371 49.446% with dragonfly-associated cyclovirus isolate DfCyV-FZ3/ Dragonfly/China/2016/MG779479 | 49.820% with capybara-associated cyclovirus 1 isolate Cap1_365/ Capybara/Brazil/2016/MK947371 49.815% with dragonfly-associated cyclovirus isolate DfCyV-FZ3/ Dragonfly/China/2016/ MG779479 | |
| Maximum/significant pairwise deduced aa sequence (%) identities of putative Cp | Between canine-associated cycloviruses | 26.601% with CN16E and CN34 | 100% between CN16E and CN34 | |
| With cyclovirus/CRESS DNA virus (Strain name/Detected in animal species, or environmental sample/Country/Year/GenBank accession number) from other animal species/environmental sample | 38.201% with uncultured virus clone CG130/Wastewater/USA/ 2015/ KY487801 30.901% with cyclovirus strain ZM41/Red musk shrew/Zambia/ 2012/AB937984 | 30.401% with uncultured virus, clone CG130/Wastewater/USA/2015/ KY487801 28.601% with chicken-associated cyclovirus 2 strain RS/BR/2015/4R/ Brazil/2015/NC_040639 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gainor, K.; Malik, Y.S.; Ghosh, S. Novel Cyclovirus Species in Dogs with Hemorrhagic Gastroenteritis. Viruses 2021, 13, 2155. https://doi.org/10.3390/v13112155
Gainor K, Malik YS, Ghosh S. Novel Cyclovirus Species in Dogs with Hemorrhagic Gastroenteritis. Viruses. 2021; 13(11):2155. https://doi.org/10.3390/v13112155
Chicago/Turabian StyleGainor, Kerry, Yashpal S. Malik, and Souvik Ghosh. 2021. "Novel Cyclovirus Species in Dogs with Hemorrhagic Gastroenteritis" Viruses 13, no. 11: 2155. https://doi.org/10.3390/v13112155
APA StyleGainor, K., Malik, Y. S., & Ghosh, S. (2021). Novel Cyclovirus Species in Dogs with Hemorrhagic Gastroenteritis. Viruses, 13(11), 2155. https://doi.org/10.3390/v13112155