
Genetic Analysis of SARS-CoV-2 Variants in Mexico during the First Year of the COVID-19 Pandemic

 , , , , , , , , , , , , add
Show full author list
, , , , , , , , , , , , add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Statement and Sample Collection
2.2. Sample Processing, Sequencing, and Viral Genome Assembly
2.3. Sequence Data Collation
2.4. Genomic Characterization
2.5. Lineage Classification and Analysis
2.6. Phylogenetic Analyses
2.7. Statistical Analysis
3. Results
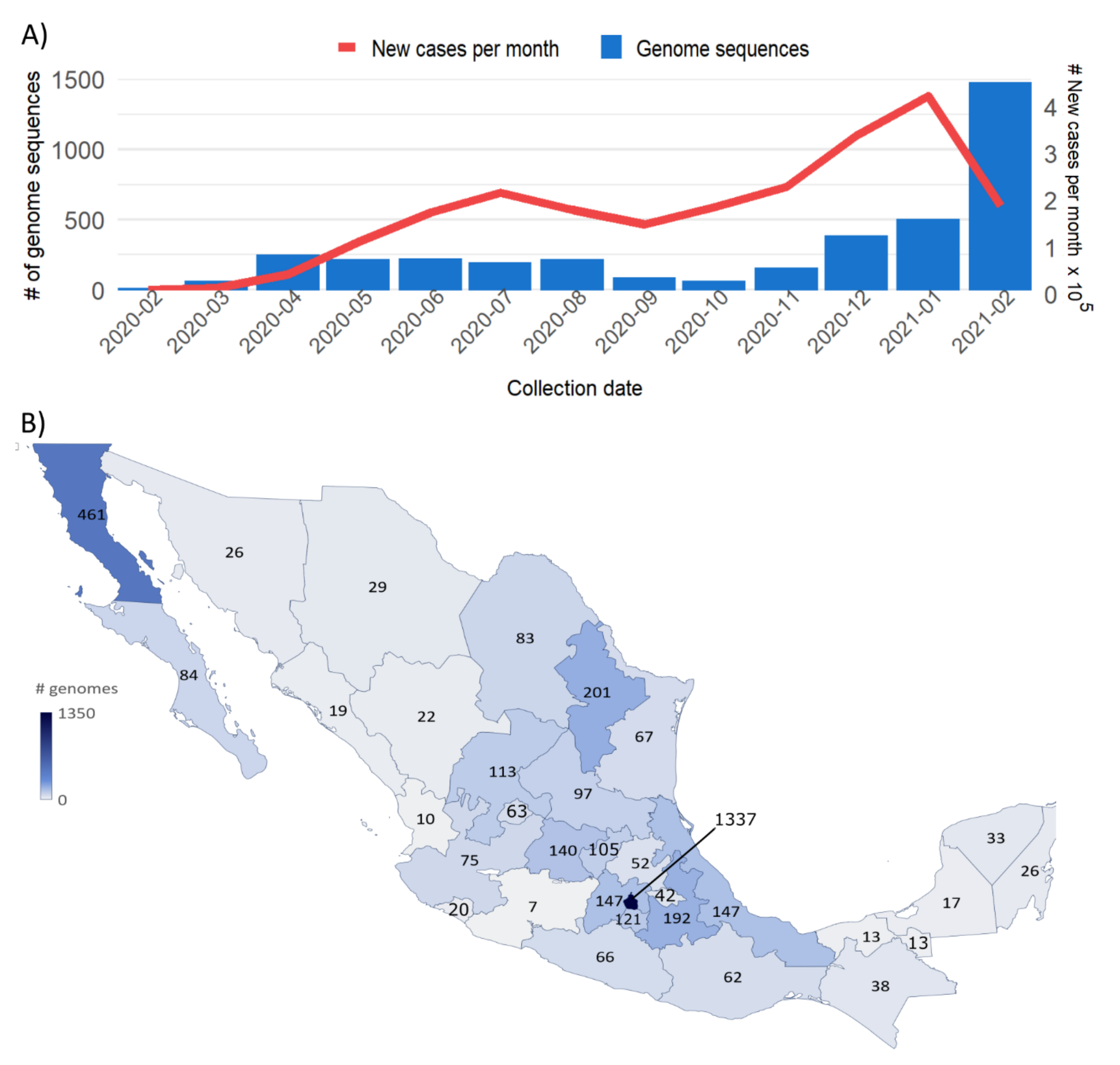
3.1. Sample Distribution
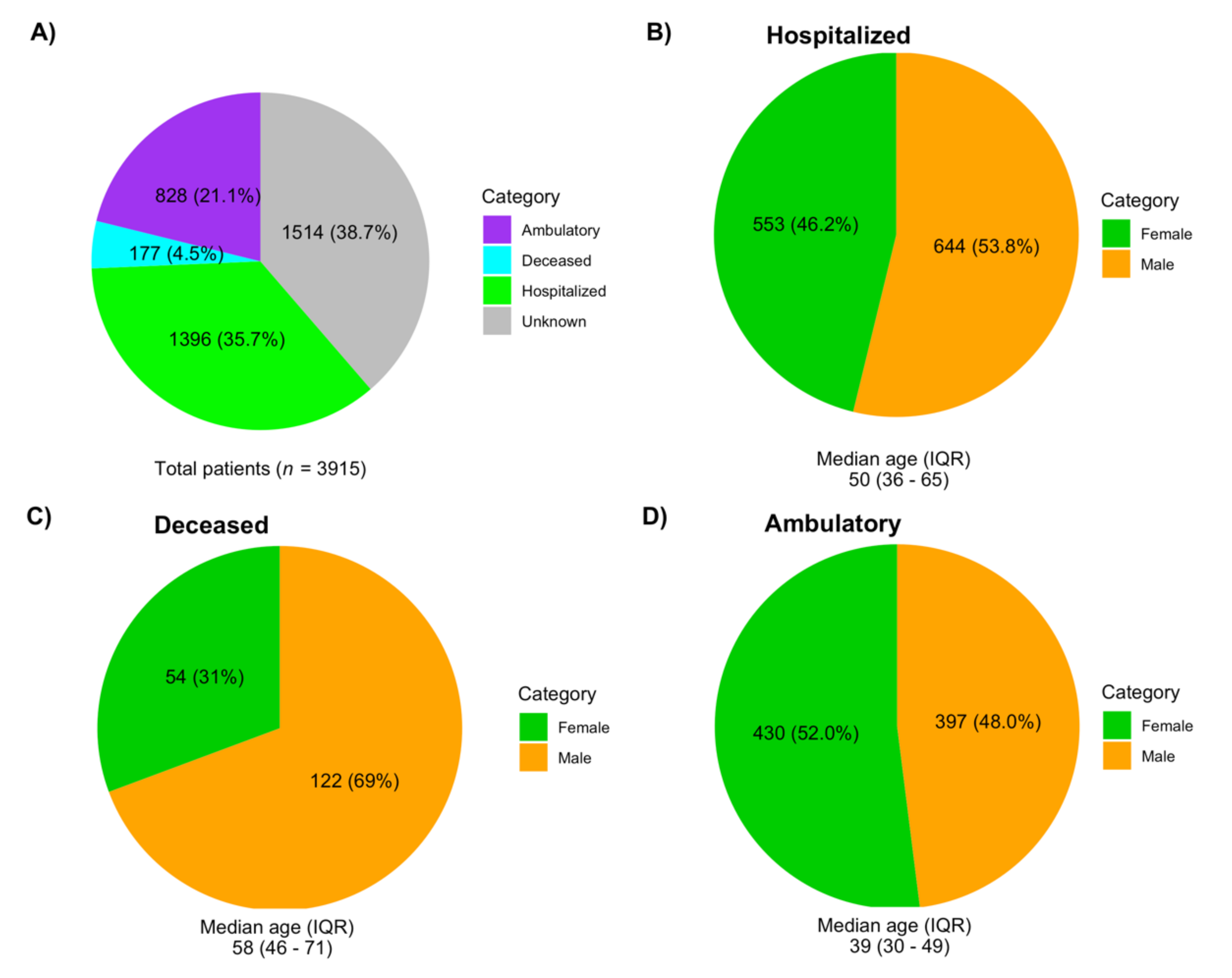
3.2. Demographic Data Associated with the Sequences Analyzed
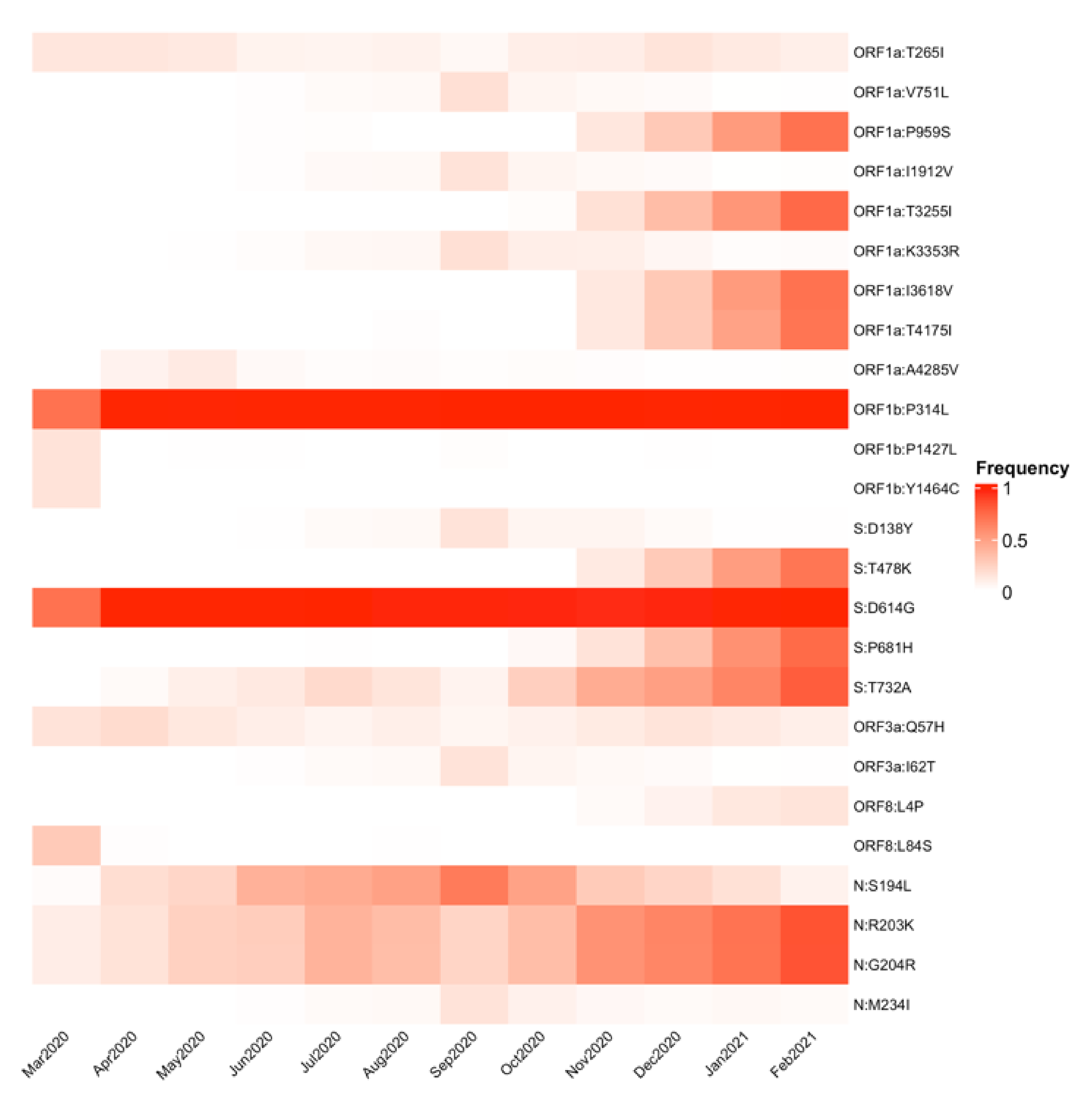
3.3. SARS-CoV-2 Genomic Substitutions
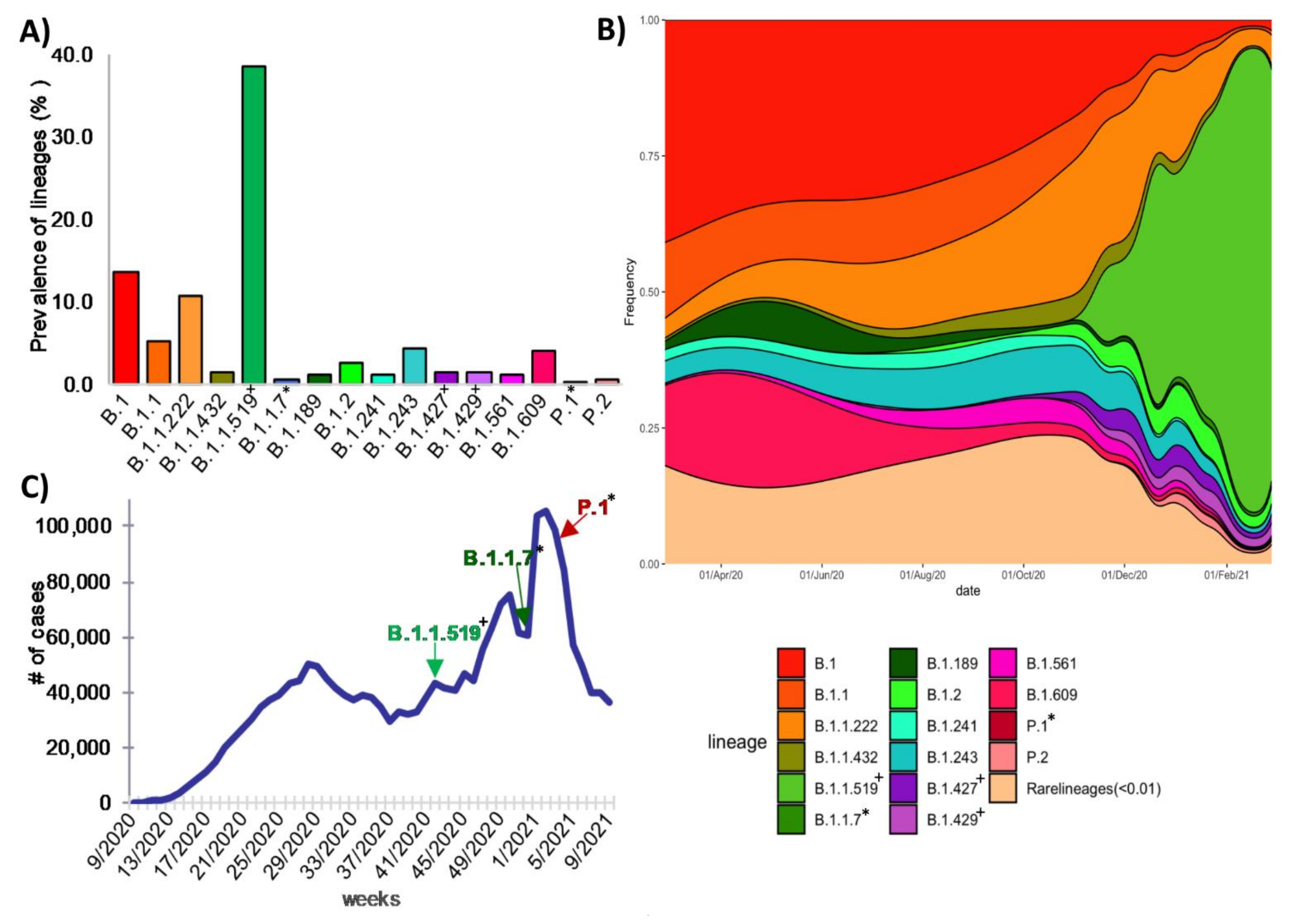
3.4. Global Genetic Diversity of SARS-CoV-2 in Mexico
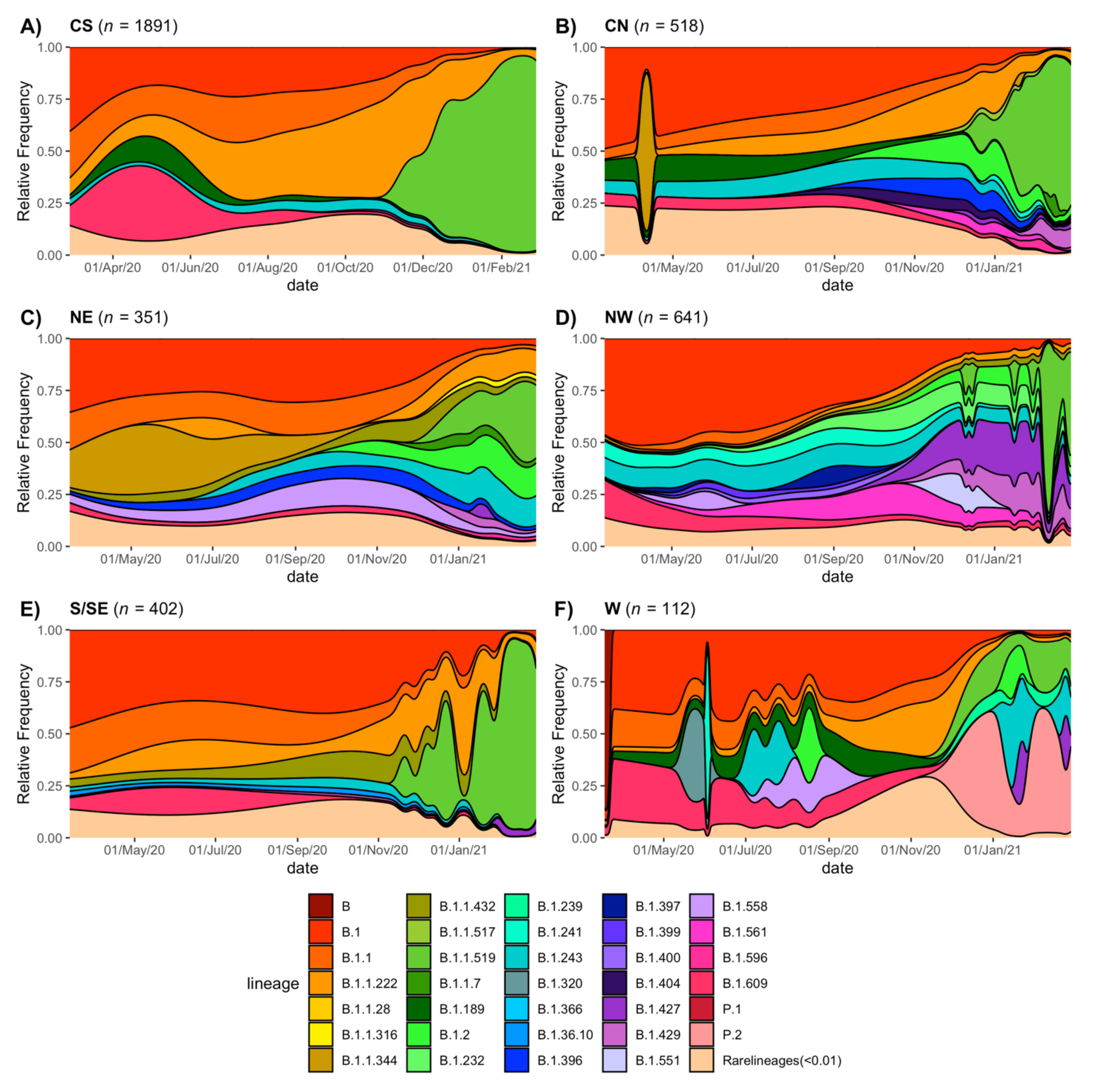
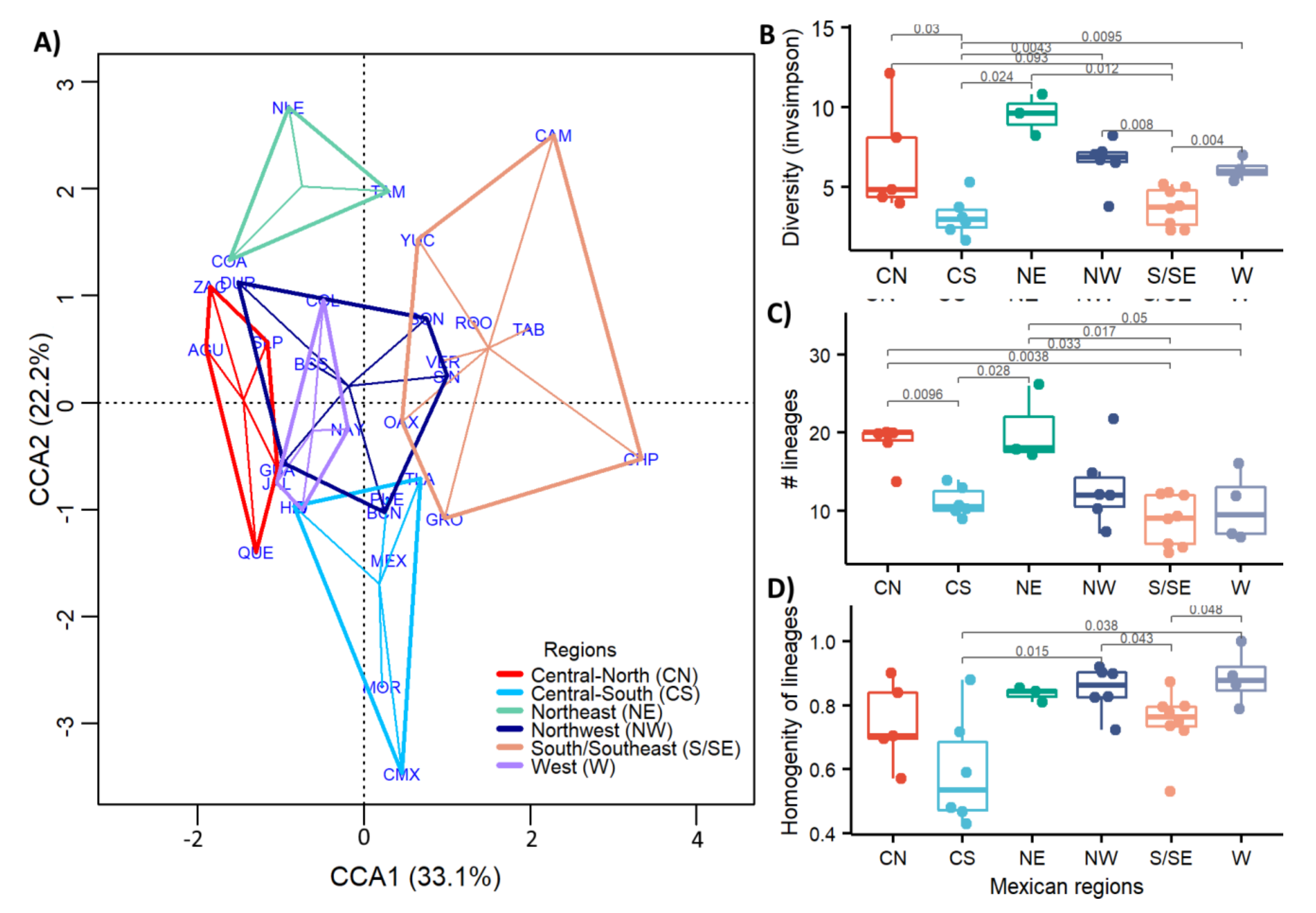
3.5. Regional Genetic Diversity
3.6. Lineages of National Interest
4. Discussion
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Garcés-Ayala, F.; Araiza-Rodríguez, A.; Mendieta-Condado, E.; Rodríguez-Maldonado, A.P.; Wong-Arámbula, C.; Landa-Flores, M.; del Mazo-López, J.C.; González-Villa, M.; Escobar-Escamilla, N.; Fragoso-Fonseca, D.E.; et al. Full Genome Sequence of the First SARS-CoV-2 Detected in Mexico. Arch. Virol. 2020, 165, 2095–2098. [Google Scholar] [CrossRef] [PubMed]
- Johns Hopkins Coronavirus Resource Center Mortality Analyses -Johns Hopkins Coronavirus Resource Center. Available online: https://coronavirus.jhu.edu/data/mortality (accessed on 1 August 2021).
- Instituto Nacional de Salud Pública INSP. Tablero Interactivo Sobre COVID 19. Available online: https://www.insp.mx/informacion-institucional-covid-19.html (accessed on 1 August 2021).
- Mathieu, E.; Ritchie, H.; Ortiz-Ospina, E.; Roser, M.; Hasell, J.; Appel, C.; Giattino, C.; Rodés-Guirao, L. A Global Database of COVID-19 Vaccinations. Nat. Hum. Behav. 2021. [Google Scholar] [CrossRef]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75.e11. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G Variant Exhibits Efficient Replication Ex Vivo and Transmission in Vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef]
- Tian, F.; Tong, B.; Sun, L.; Shi, S.; Zheng, B.; Wang, Z.; Dong, X.; Zheng, P. N501Y Mutation of Spike Protein in SARS-CoV-2 Strengthens Its Binding to Receptor ACE2. eLife 2021, 10, e69091. [Google Scholar] [CrossRef]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated Transmissibility and Impact of SARS-CoV-2 Lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef]
- Lubinski, B.; Tang, T.; Daniel, S.; Jaimes, J.A.; Whittaker, G.R. Functional Evaluation of Proteolytic Activation for the SARS-CoV-2 Variant B.1.1.7: Role of the P681H Mutation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wilhelm, A.; Toptan, T.; Pallas, C.; Wolf, T.; Goetsch, U.; Gottschalk, R.; Vehreschild, M.J.G.T.; Ciesek, S.; Widera, M. Antibody-Mediated Neutralization of Authentic SARS-CoV-2 B.1.617 Variants Harboring L452R and T478K/E484Q. Viruses 2021, 13, 1693. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-González, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, Infectivity, and Neutralization of a Spike L452R SARS-CoV-2 Variant. Cell 2021, 184, 3426–3437.e8. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, C.K.V.; Franco, M.M.; Gräf, T.; Barcia, C.A.d.L.; Mendonça, R.N.d.Á.; de Sousa, K.A.F.; Neiva, L.M.C.; Fosenca, V.; Mendes, A.V.A.; de Aguiar, R.S.; et al. Genomic Evidence of SARS-CoV-2 Reinfection Involving E484K Spike Mutation, Brazil. Emerg. Infect. Dis. 2021, 27, 1522. [Google Scholar] [CrossRef] [PubMed]
- Taboada, B.; Vazquez-Perez, J.A.; Muñoz-Medina, J.E.; Ramos-Cervantes, P.; Escalera-Zamudio, M.; Boukadida, C.; Sanchez-Flores, A.; Isa, P.; Mendieta-Condado, E.; Martínez-Orozco, J.A.; et al. Genomic Analysis of Early SARS-CoV-2 Variants Introduced in Mexico. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Instituto de Diagnóstico y Referencia Epidemiológicos Lineamientos Para La Toma, Manejo y Envió de Muestras Para El Diagnóstico a La Red Nacional de Laboratorios de Salud Pública; 2020. Available online: https://www.gob.mx/cms/uploads/attachment/file/558702/Lineamientos_TMEM_2020_180620.pdf (accessed on 20 August 2021).
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 Novel Coronavirus (2019-NCoV) by Real-Time RT-PCR. Eurosurveillance 2020, 25, 2000045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An Amplicon-Based Sequencing Framework for Accurately Measuring Intrahost Virus Diversity Using PrimalSeq and IVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, Y.; McCauley, J. GISAID: Global Initiative on Sharing All Influenza Data—from Vision to Reality. Euro Surveill 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team R. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; ggplot2; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Haeseler, A.; von Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- To, T.-H.; Jung, M.; Lycett, S.; Gascuel, O. Fast Dating Using Least-Squares Criteria and Algorithms. Syst. Biol. 2016, 65, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. Ggtree: An r Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Wang, L.-G.; Lam, T.T.-Y.; Xu, S.; Dai, Z.; Zhou, L.; Feng, T.; Guo, P.; Dunn, C.W.; Jones, B.R.; Bradley, T.; et al. Treeio: An R Package for Phylogenetic Tree Input and Output with Richly Annotated and Associated Data. Mol. Biol. Evol. 2020, 37, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H.; et al. Package Vegan: Community Ecol. Package Version 2.5.7, 2014, R Package. Available online: https://cran.ism.ac.jp/web/packages/vegan/vegan.pdf (accessed on 20 August 2021).
- Anderson, M.J. A New Method for Non-Parametric Multivariate Analysis of Variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- Mexico | MapChart. Available online: https://mapchart.net/mexico.html (accessed on 14 May 2021).
- Velazquez-Salinas, L.; Zarate, S.; Eberl, S.; Gladue, D.P.; Novella, I.; Borca, M.V. Positive Selection of ORF1ab, ORF3a, and ORF8 Genes Drives the Early Evolutionary Trends of SARS-CoV-2 During the 2020 COVID-19 Pandemic. Front. Microbiol. 2020, 11, 2592. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Maldonado, A.P.; Vázquez-Pérez, J.A.; Cedro-Tanda, A.; Taboada, B.; Boukadida, C.; Wong-Arámbula, C.; Nuñez-García, T.E.; Cruz-Ortiz, N.; Barrera-Badillo, G.; Hernández-Rivas, L.; et al. Emergence and Spread of the Potential Variant of Interest (VOI) B.1.1.519 of SARS-CoV-2 Predominantly Present in Mexico. Arch. Virol. 2021, 166, 3173–3177. [Google Scholar] [CrossRef]
- Wu, H.; Xing, N.; Meng, K.; Fu, B.; Xue, W.; Dong, P.; Xiao, Y.; Liu, G.; Luo, H.; Zhu, W.; et al. Nucleocapsid Mutation R203K/G204R Increases the Infectivity, Fitness and Virulence of SARS-CoV-2. bioRxiv 2021. [Google Scholar] [CrossRef]
- Liu, C.; Ginn, H.M.; Dejnirattisai, W.; Supasa, P.; Wang, B.; Tuekprakhon, A.; Nutalai, R.; Zhou, D.; Mentzer, A.J.; Zhao, Y.; et al. Reduced Neutralization of SARS-CoV-2 B.1.617 by Vaccine and Convalescent Serum. Cell 2021, 184, 4220–4236.e13. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Ju, X.; Gong, M.; Lan, J.; Yu, Y.; Long, Q.; Zhang, Y.; Zhong, J.; Zhong, G.; Wang, X.; et al. Characterization of SARS-CoV-2 Variants B.1.617.1 (Kappa), B.1.617.2 (Delta) and B.1.618 on Cell Entry, Host Range, and Sensitivity to Convalescent Plasma and ACE2 Decoy Receptor. bioRxiv 2021. [Google Scholar] [CrossRef]
- Jin, J.-M.; Bai, P.; He, W.; Wu, F.; Liu, X.-F.; Han, D.-M.; Liu, S.; Yang, J.-K. Gender Differences in Patients With COVID-19: Focus on Severity and Mortality. Front. Public Health 2020, 8, 152. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, M.; Ribeiro Dos Santos, G.; Wang, L.; Cummings, D.A.T.; Azman, A.S.; Paireau, J.; Fontanet, A.; Cauchemez, S.; Salje, H. Age-Specific Mortality and Immunity Patterns of SARS-CoV-2. Nature 2021, 590, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Kammar-García, A.; Vidal-Mayo, J.d.J.; Vera-Zertuche, J.M.; Lazcano-Hernández, M.; Vera-López, O.; Segura-Badilla, O.; Aguilar-Alonso, P.; Navarro-Cruz, A.R. Impact of Comorbidities in Mexican SARS-CoV-2-Positive Patients: A Retrospective Analysis in a National Cohort. Rev. Investig. Clin. 2020, 72, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Secretaria de Comunicacione y Transportes Estadística Mensual Del Sector de Comunicaciones y TRansportes. Available online: http://www.sct.gob.mx/fileadmin/DireccionesGrales/DGP/estadistica/Indicador-Mensual/INDI-2020/CI-ENERO_2020.pdf (accessed on 20 July 2021).
- Alteri, C.; Cento, V.; Piralla, A.; Costabile, V.; Tallarita, M.; Colagrossi, L.; Renica, S.; Giardina, F.; Novazzi, F.; Gaiarsa, S.; et al. Genomic Epidemiology of SARS-CoV-2 Reveals Multiple Lineages and Early Spread of SARS-CoV-2 Infections in Lombardy, Italy. Nat. Commun. 2021, 12, 434. [Google Scholar] [CrossRef] [PubMed]
- Franco, D.; Gonzalez, C.; Abrego, L.E.; Carrera, J.-P.; Diaz, Y.; Caicedo, Y.; Moreno, A.; Chavarria, O.; Gondola, J.; Castillo, M.; et al. Early Transmission Dynamics, Spread, and Genomic Characterization of SARS-CoV-2 in Panama. Emerg. Infect. Dis. 2021, 27, 612–615. [Google Scholar] [CrossRef] [PubMed]
- du Plessis, L.; McCrone, J.T.; Zarebski, A.E.; Hill, V.; Ruis, C.; Gutierrez, B.; Raghwani, J.; Ashworth, J.; Colquhoun, R.; Connor, T.R.; et al. Establishment and Lineage Dynamics of the SARS-CoV-2 Epidemic in the UK. Science 2021, 371, 708–712. [Google Scholar] [CrossRef]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Vaughan, T.G.; Crawford, K.H.D.; Althaus, C.L.; Reichmuth, M.L.; Bowen, J.E.; Walls, A.C.; Corti, D.; et al. Spread of a SARS-CoV-2 Variant through Europe in the Summer of 2020. Nature 2021, 595, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Wells, C.R.; Sah, P.; Moghadas, S.M.; Pandey, A.; Shoukat, A.; Wang, Y.; Wang, Z.; Meyers, L.A.; Singer, B.H.; Galvani, A.P. Impact of International Travel and Border Control Measures on the Global Spread of the Novel 2019 Coronavirus Outbreak. Proc. Natl. Acad. Sci. USA 2020, 117, 7504–7509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pond, S. Natural Selection Analysis of Global SARS-CoV-2/COVID-19. Available online: https://observablehq.com/@spond/revised-sars-cov-2-analytics-page (accessed on 20 July 2021).
- Wang, R.; Chen, J.; Gao, K.; Wei, G.W. Vaccine-Escape and Fast-Growing Mutations in the United Kingdom, the United States, Singapore, Spain, India, and Other COVID-19-Devastated Countries. Genomics 2021, 113, 2158–2170. [Google Scholar] [CrossRef]
- Zahradník, J.; Marciano, S.; Shemesh, M.; Zoler, E.; Chiaravalli, J.; Meyer, B.; Rudich, Y.; Dym, O.; Elad, N.; Schreiber, G. SARS-CoV-2 RBD in Vitro Evolution Follows Contagious Mutation Spread, yet Generates an Able Infection Inhibitor. bioRxiv 2021. [Google Scholar] [CrossRef]
- Liu, Z.; VanBlargan, L.A.; Bloyet, L.-M.; Rothlauf, P.W.; Chen, R.E.; Stumpf, S.; Zhao, H.; Errico, J.M.; Theel, E.S.; Liebeskind, M.J.; et al. Identification of SARS-CoV-2 Spike Mutations That Attenuate Monoclonal and Serum Antibody Neutralization. Cell Host Microbe 2021, 29, 477–488.e4. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VOC | VUM | ||||
|---|---|---|---|---|---|
| Variants | Alpha (B.1.1.7) | Gamma (P.1) | B.1.427 + B.1.429 | B.1.1.519 | P.2 |
| Identified for the first time | UK | Japan | USA—California | Multiple | Brazil |
| First date identified in Mexico | 31 December 2020 | 28 January 2021 | 20 July 2020 | 11 November 2021 | 4 January 2021 |
| Number of samples | 20 | 3 | 119 | 1512 | 23 |
| Number of states where they were identified | 6 | 3 | 19 | 30 | 2 |
| Mutation | Presence in B.1.1.222 | Presence in B.1.1.519 | Presence in VOCs and VOIs | Phenotypic Effect (If Known) |
|---|---|---|---|---|
| ORF1a:P959S | X | ✓ | No | No |
| ORF1a:T3255I | X | ✓ | Delta sublineages and Lambda | No |
| ORF1a:I3618V | X | ✓ | No | No |
| ORF1a:T4175I | X | ✓ | No | No |
| ORF1b:P314L | ✓ | ✓ | Present in all B.1 derived lineages | No |
| S:T478K | X | ✓ | Delta | Increased receptor binding, cell entry, and transmissibility [33,34] |
| S:D614G | ✓ | ✓ | Present in all B.1 derived lineages | Increase transmissibility [5,6] |
| S:P681H | X | ✓ | Alpha, Mu | Increased spike cleavage [1,9] |
| S:T732A | ✓ | ✓ | No | |
| N:R203K | ✓ | ✓ | Alpha, Gamma, Lambda | Increased infectivity and disease severity [32] |
| N:G204R | ✓ | ✓ | Alpha, Gamma, Lambda | Increased infectivity and disease severity [32] |
| Month | B.1.1.222 | B.1.1.519 | ||
|---|---|---|---|---|
| Mexico | USA | Mexico | USA | |
| 2020-04 | 2.10 | 0.02 | 0.00 | 0.00 |
| 2020-05 | 6.60 | 0.01 | 0.00 | 0.00 |
| 2020-06 | 8.80 | 0.01 | 0.00 | 0.00 |
| 2020-07 | 13.90 | 0.07 | 0.00 | 0.00 |
| 2020-08 | 7.30 | 0.41 | 0.00 | 0.00 |
| 2020-09 | 1.30 | 0.67 | 0.00 | 0.00 |
| 2020-10 | 5.60 | 0.90 | 0.00 | 0.00 |
| 2020-11 | 12.10 | 0.97 | 6.10 | 0.02 |
| 2020-12 | 4.70 | 1.00 | 11.5 | 0.15 |
| 2021-01 | 6.70 | 0.65 | 36.50 | 1.11 |
| 2021-02 | 6.00 | 0.43 | 62.30 | 2.69 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taboada, B.; Zárate, S.; Iša, P.; Boukadida, C.; Vazquez-Perez, J.A.; Muñoz-Medina, J.E.; Ramírez-González, J.E.; Comas-García, A.; Grajales-Muñiz, C.; Rincón-Rubio, A.; et al. Genetic Analysis of SARS-CoV-2 Variants in Mexico during the First Year of the COVID-19 Pandemic. Viruses 2021, 13, 2161. https://doi.org/10.3390/v13112161
Taboada B, Zárate S, Iša P, Boukadida C, Vazquez-Perez JA, Muñoz-Medina JE, Ramírez-González JE, Comas-García A, Grajales-Muñiz C, Rincón-Rubio A, et al. Genetic Analysis of SARS-CoV-2 Variants in Mexico during the First Year of the COVID-19 Pandemic. Viruses. 2021; 13(11):2161. https://doi.org/10.3390/v13112161
Chicago/Turabian StyleTaboada, Blanca, Selene Zárate, Pavel Iša, Celia Boukadida, Joel Armando Vazquez-Perez, José Esteban Muñoz-Medina, José Ernesto Ramírez-González, Andreu Comas-García, Concepción Grajales-Muñiz, Alma Rincón-Rubio, and et al. 2021. "Genetic Analysis of SARS-CoV-2 Variants in Mexico during the First Year of the COVID-19 Pandemic" Viruses 13, no. 11: 2161. https://doi.org/10.3390/v13112161
APA StyleTaboada, B., Zárate, S., Iša, P., Boukadida, C., Vazquez-Perez, J. A., Muñoz-Medina, J. E., Ramírez-González, J. E., Comas-García, A., Grajales-Muñiz, C., Rincón-Rubio, A., Matías-Florentino, M., Sanchez-Flores, A., Mendieta-Condado, E., Verleyen, J., Barrera-Badillo, G., Hernández-Rivas, L., Mejía-Nepomuceno, F., Martínez-Orozco, J. A., Becerril-Vargas, E., ... Arias, C. F. (2021). Genetic Analysis of SARS-CoV-2 Variants in Mexico during the First Year of the COVID-19 Pandemic. Viruses, 13(11), 2161. https://doi.org/10.3390/v13112161