
A High Rate Algal Pond Hosting a Dynamic Community of RNA Viruses

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Design and Sampling
2.2. RNA Extraction, cDNA Library Preparation, and Sequencing
2.3. Quality Control of Reads and Contig Assembly of Metagenomes
2.4. Taxonomic Assignment of Contigs and Annotation
2.5. Alpha and Beta Diversity by K-Mer Counts Produced from Metagenome Contigs
2.6. Phylogenetic Reconstruction of the RNA-Dependent RNA Polymerase (RdRp) Domain
2.7. qPCR Design for Viral Population Tracking
3. Results
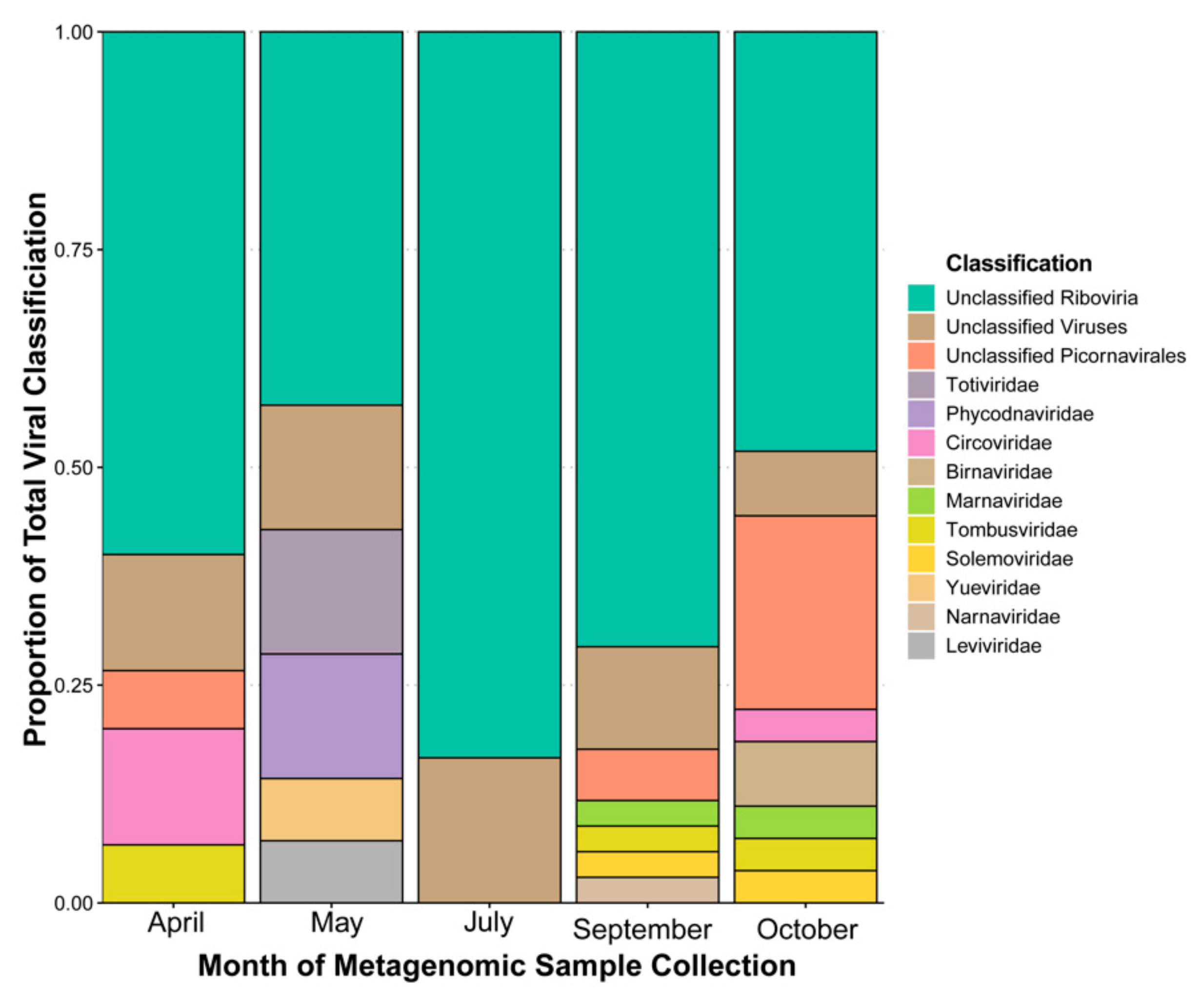
3.1. Taxonomic Classification by Database “Best Hit”, Sequencing Coverage by Contig, and Domain Prediction
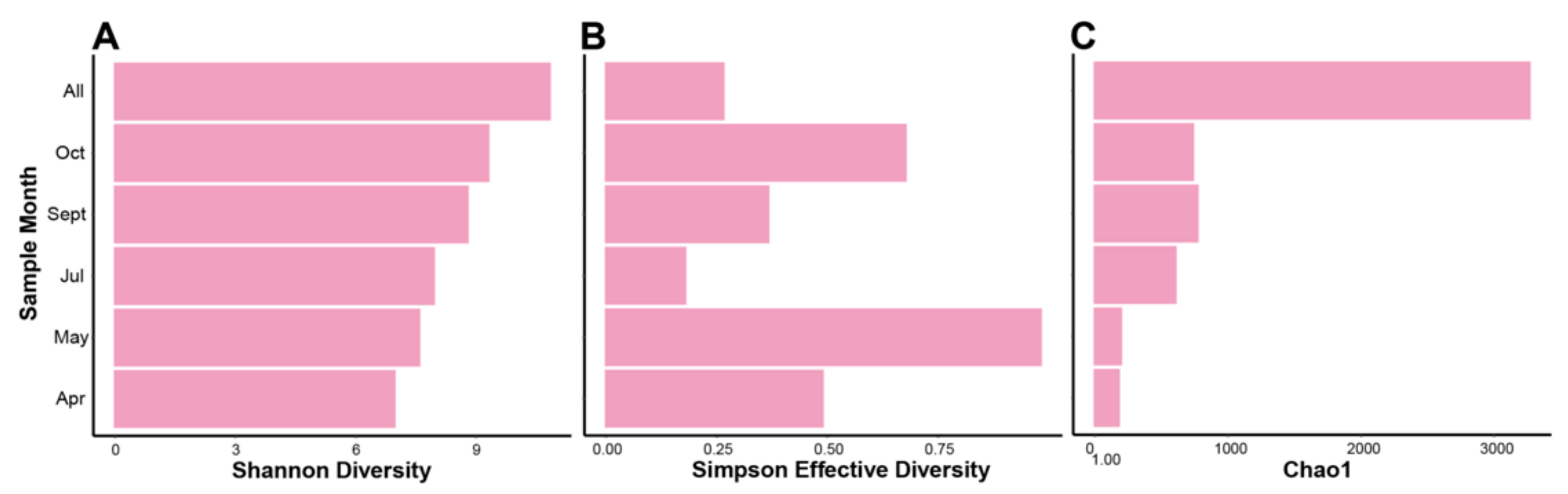
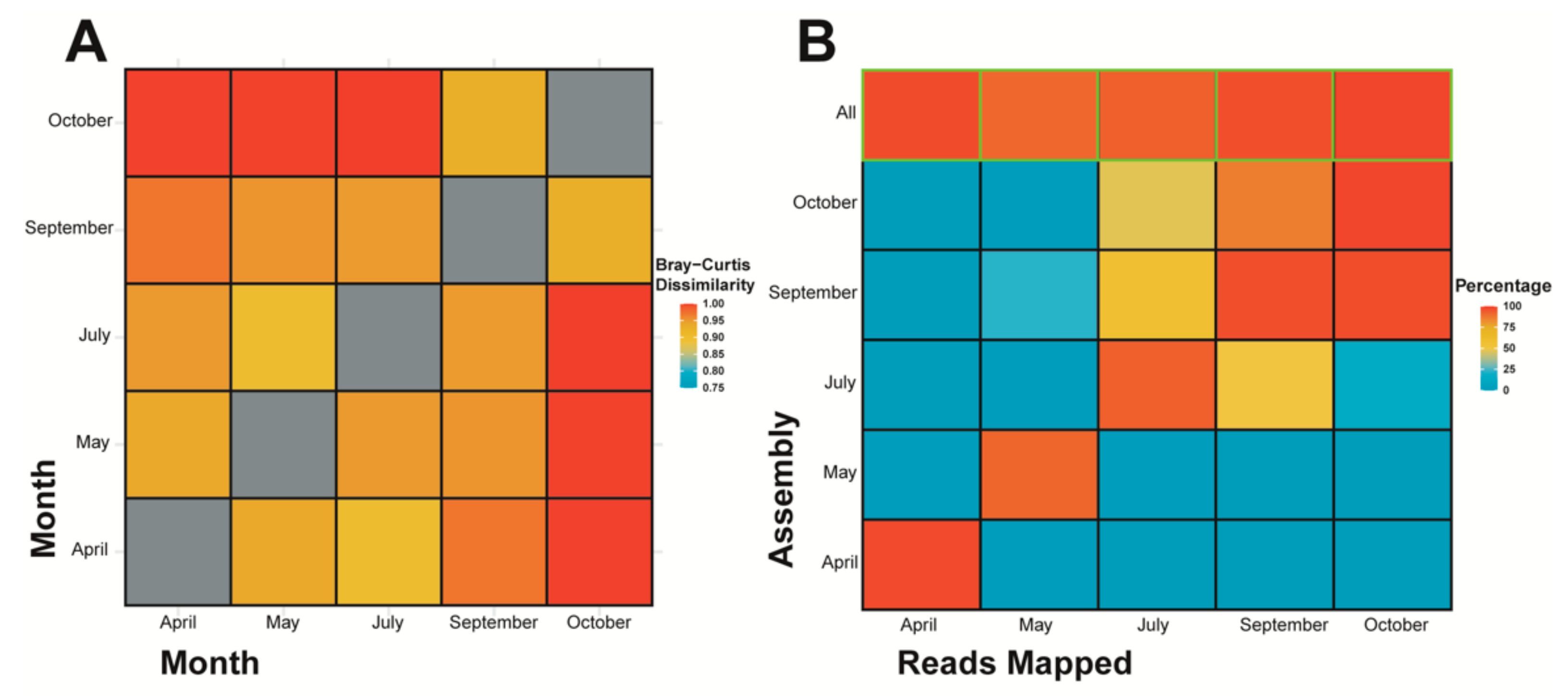
3.2. Alpha and Beta Diversity Indices, and Cross Sample Coverage
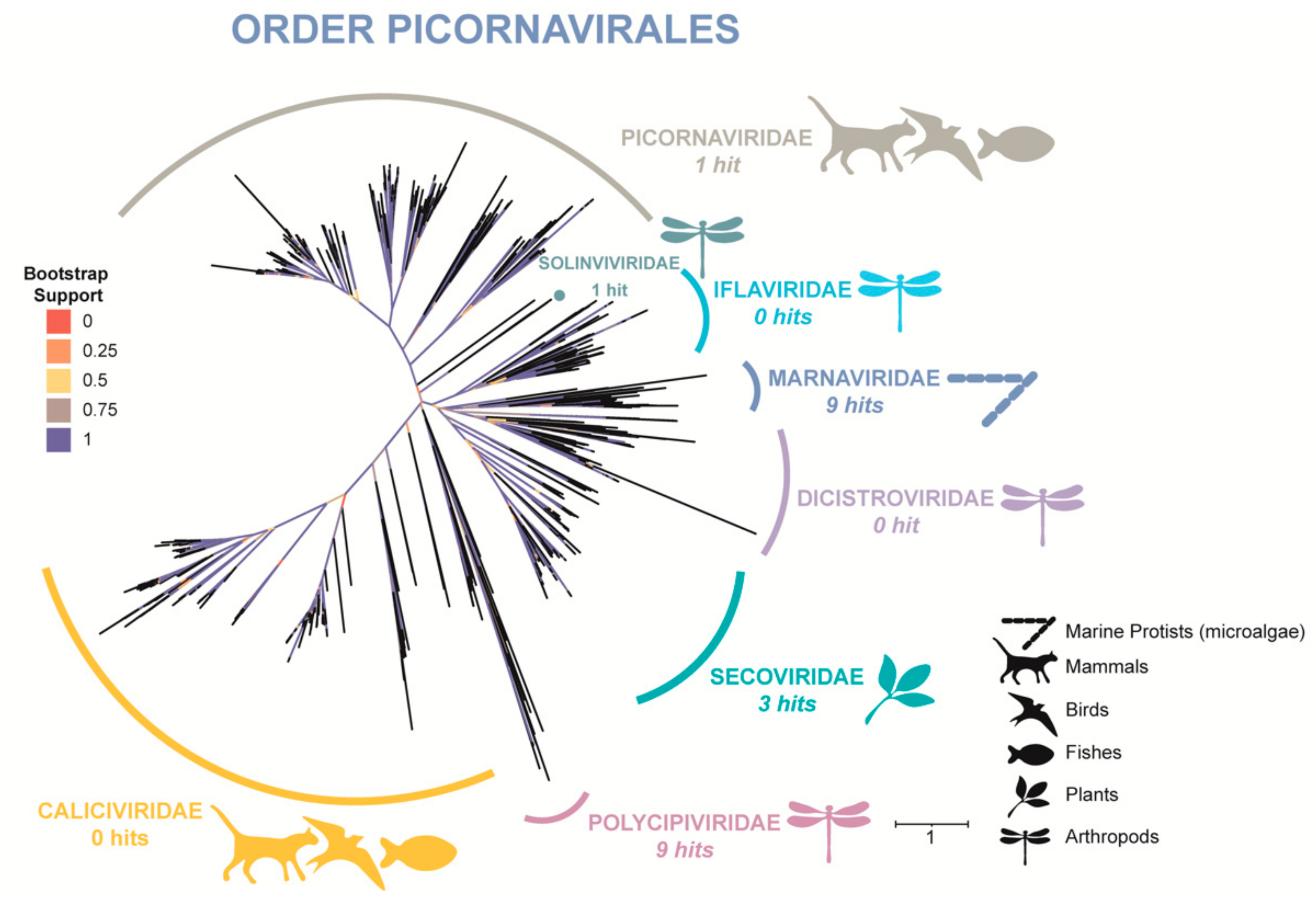
3.3. Phylogenetic Tree (RdRp) of Order Picornavirales
3.4. qPCR of Potential Marnaviridae and a Rotifera sp. Virus
4. Discussion
4.1. Taxonomic Classification and a Phylogenetic Study Provide Evidence of Microalgal Infecting RNA Viruses within the HRAP
4.2. Database-Independent Approaches to Quantifying Diversity Show Changes between and among Temporal Based Samples of the HRAP
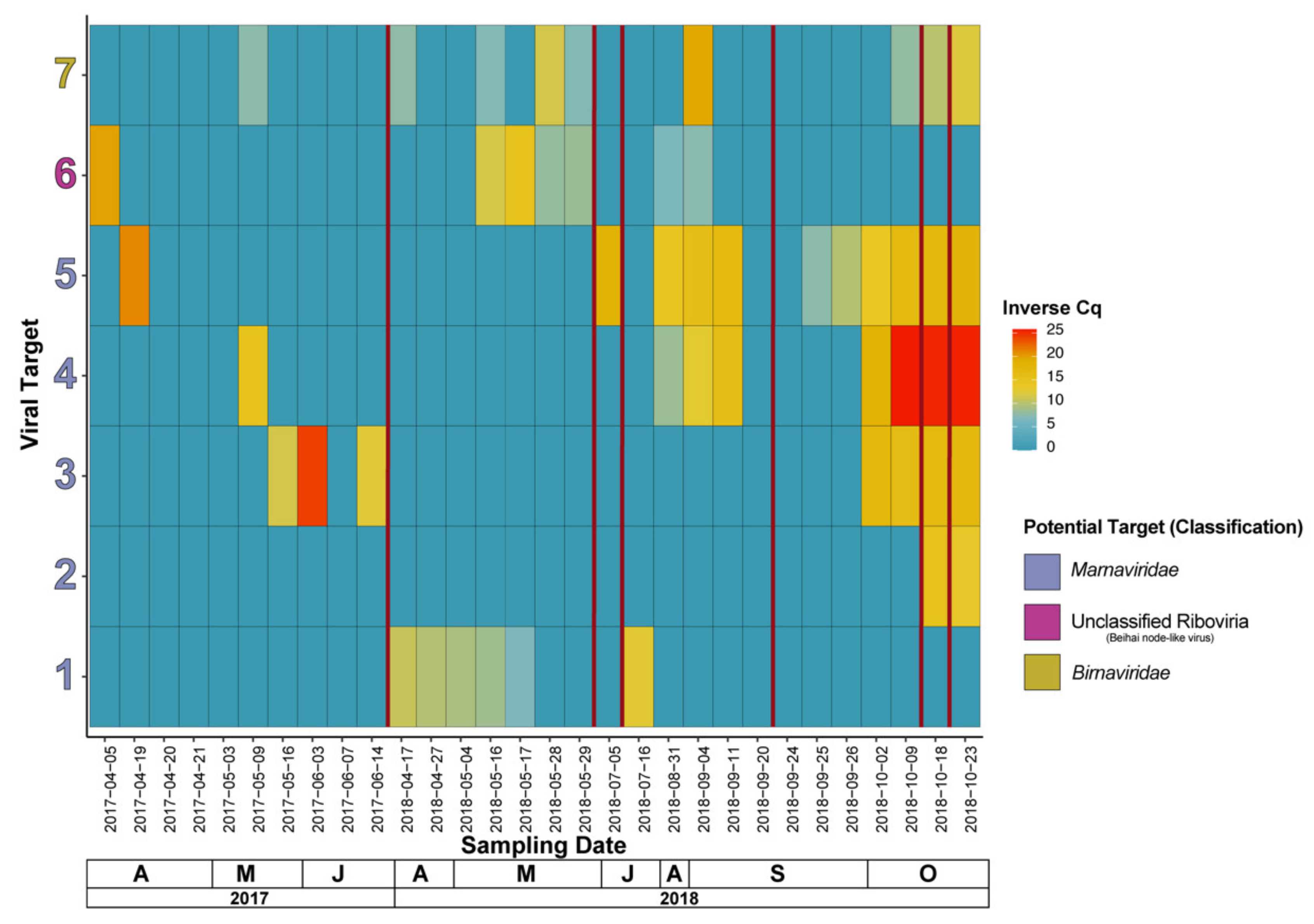
4.3. Viral Dynamics (RT-qPCR Based) Showcase a Variety of Different Dynamics Patterns among Putative Marnaviridae spp., and Other Viruses of Interest
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Starr, E.P.; Nuccio, E.E.; Pett-Ridge, J.; Banfield, J.F.; Firestone, M.K. Metatranscriptomic reconstruction reveals RNA viruses with the potential to shape carbon cycling in soil. Proc. Natl. Acad. Sci. USA 2019, 116, 25900–25908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, A.S.; Rise, M.L.; Culley, A.I.; Steward, G. RNA viruses in the sea. FEMS Microbiol. Rev. 2009, 33, 295–323. [Google Scholar] [CrossRef] [Green Version]
- Suttle, C.A. Viruses in the sea. Nat. Cell Biol. 2005, 437, 356–361. [Google Scholar] [CrossRef]
- Lu, G.; Ye, Z.-X.; He, Y.-J.; Zhang, Y.; Wang, X.; Huang, H.-J.; Zhuo, J.-C.; Sun, Z.-T.; Yan, F.; Chen, J.-P.; et al. Discovery of Two Novel Negeviruses in a Dungfly Collected from the Arctic. Viruses 2020, 12, 692. [Google Scholar] [CrossRef] [PubMed]
- Miranda, J.A.; Culley, A.I.; Schvarcz, C.; Steward, G.F. RNA viruses as major contributors to Antarctic virioplankton. Environ. Microbiol. 2016, 18, 3714–3727. [Google Scholar] [CrossRef]
- Djikeng, A.; Kuzmickas, R.; Anderson, N.G.; Spiro, D.J. Metagenomic Analysis of RNA Viruses in a Fresh Water Lake. PLoS ONE 2009, 4, e7264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skvortsov, T.; De Leeuwe, C.; Quinn, J.P.; McGrath, J.W.; Allen, C.; McELARNEY, Y.; Watson, C.; Arkhipova, K.; Lavigne, R.; Kulakov, L.A. Metagenomic Characterisation of the Viral Community of Lough Neagh, the Largest Freshwater Lake in Ireland. PLoS ONE 2016, 11, e0150361. [Google Scholar] [CrossRef] [Green Version]
- Steward, G.F.; I Culley, A.; A Mueller, J.; Wood-Charlson, E.M.; Belcaid, M.; Poisson, G. Are we missing half of the viruses in the ocean? ISME J. 2012, 7, 672–679. [Google Scholar] [CrossRef] [Green Version]
- Greninger, A. A decade of RNA virus metagenomics is (not) enough. Virus Res. 2018, 244, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Elena, S.F.; Bedhomme, S.; Carrasco, P.; Cuevas, J.; De La Iglesia, F.; Lafforgue, G.; Lalić, J.; Pròsper, À.; Tromas, N.; Zwart, M.P. The Evolutionary Genetics of Emerging Plant RNA Viruses. Mol. Plant-Microbe Interactions 2011, 24, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Lin, X.-D.; Chen, X.; Tian, J.-H.; Chen, L.-J.; Li, K.; Wang, W.; Eden, J.-S.; Shen, J.-J.; Liu, L.; et al. The evolutionary history of vertebrate RNA viruses. Nat. Cell Biol. 2018, 556, 197–202. [Google Scholar] [CrossRef]
- Zhang, Y.-Z.; Wu, W.-C.; Shi, M.; Holmes, E.C. The diversity, evolution and origins of vertebrate RNA viruses. Curr. Opin. Virol. 2018, 31, 9–16. [Google Scholar] [CrossRef]
- Ryabov, E.V. Invertebrate RNA virus diversity from a taxonomic point of view. J. Invertebr. Pathol. 2017, 147, 37–50. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the invertebrate RNA virosphere. Nat. Cell Biol. 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Hillman, B.I.; Cai, G. The Family Narnaviridae: Simplest of RNA Viruses. Adv. Virus Res. 2013, 86, 28. [Google Scholar]
- Callanan, J.; Stockdale, S.R.; Shkoporov, A.; Draper, L.A.; Ross, R.P.; Hill, C. Expansion of known ssRNA phage genomes: From tens to over a thousand. Sci. Adv. 2020, 6, eaay5981. [Google Scholar] [CrossRef] [Green Version]
- Krupovic, M.; Cvirkaite-Krupovic, V.; Iranzo, J.; Prangishvili, D.; Koonin, E.V. Viruses of archaea: Structural, functional, environmental and evolutionary genomics. Virus Res. 2018, 244, 181–193. [Google Scholar] [CrossRef]
- Bolduc, B.; Wirth, J.F.; Mazurie, A.; Young, M.J. Viral assemblage composition in Yellowstone acidic hot springs assessed by network analysis. ISME J. 2015, 9, 2162–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadeghi, M.; Tomaru, Y.; Ahola, T. RNA Viruses in Aquatic Unicellular Eukaryotes. Viruses 2021, 13, 362. [Google Scholar] [CrossRef]
- Dolja, V.V.; Koonin, E.V. Metagenomics reshapes the concepts of RNA virus evolution by revealing extensive horizontal virus transfer. Virus Res. 2018, 244, 36–52. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, S.; Wang, D. Origins and challenges of viral dark matter. Virus Res. 2017, 239, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Wolf, Y.; Nagasaki, K.; Dolja, V.V. The Big Bang of picorna-like virus evolution antedates the radiation of eukaryotic supergroups. Nat. Rev. Genet. 2008, 6, 925–939. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Gall, O.; Christian, P.; Fauquet, C.M.; King, A.M.Q.; Knowles, N.J.; Nakashima, N.; Stanway, G.; Gorbalenya, A.E. Picornavirales, a proposed order of positive-sense single-stranded RNA viruses with a pseudo-T = 3 virion architecture. Arch. Virol. 2008, 153, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Tai, V.; Lawrence, J.E.; Lang, A.; Chan, A.M.; Culley, A.I.; Suttle, C.A. Characterization of HaRNAV, a single-stranded RNA virus causing lysis of Heterosigma akashiwo (raphidophyceae) 1. J. Phycol. 2003, 39, 343–352. [Google Scholar] [CrossRef]
- Nagasaki, K.; Tomaru, Y.; Katanozaka, N.; Shirai, Y.; Nishida, K.; Itakura, S.; Yamaguchi, M. Isolation and Characterization of a Novel Single-Stranded RNA Virus Infecting the Bloom-Forming Diatom Rhizosolenia setigera. Appl. Environ. Microbiol. 2004, 70, 704–711. [Google Scholar] [CrossRef] [Green Version]
- Shirai, Y.; Tomaru, Y.; Takao, Y.; Suzuki, H.; Nagumo, T.; Nagasaki, K. Isolation and Characterization of a Single-Stranded RNA Virus Infecting the Marine Planktonic Diatom Chaetoceros tenuissimus Meunier. Appl. Environ. Microbiol. 2008, 74, 4022–4027. [Google Scholar] [CrossRef] [Green Version]
- Tomaru, Y.; Takao, Y.; Suzuki, H.; Nagumo, T.; Nagasaki, K. Isolation and Characterization of a Single-Stranded RNA Virus Infecting the Bloom-Forming Diatom Chaetoceros socialis. Appl. Environ. Microbiol. 2009, 75, 2375–2381. [Google Scholar] [CrossRef] [Green Version]
- Tomaru, Y.; Nagasaki, K. Diatom Viruses. In The Diatom World; Seckbach, J., Kociolek, P., Eds.; Cellular Origin, Life in Extreme Habitats and Astrobiology; Springer Netherlands: Dordrecht, The Netherlans, 2011; pp. 211–225. ISBN 978-94-007-1327-7. [Google Scholar]
- Alexandersen, S.; Knowles, N.J.; Belsham, G.J.; Dekker, A.; Nfon, C.; Zhang, Z.; Koenen, F. Picornaviruses. In Diseases of Swine; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2019; pp. 641–684. ISBN 978-1-119-35092-7. [Google Scholar]
- Morocho-Jácome, A.L.; Ruscinc, N.; Martinez, R.M.; de Carvalho, J.C.M.; de Almeida, T.S.; Rosado, C.; Costa, J.G.; Velasco, M.V.R.; Baby, A.R. (Bio)Technological aspects of microalgae pigments for cosmetics. Appl. Microbiol. Biotechnol. 2020, 104, 9513–9522. [Google Scholar] [CrossRef]
- Barkia, I.; Saari, N.; Manning, S.R. Microalgae for High-Value Products Towards Human Health and Nutrition. Mar. Drugs 2019, 17, 304. [Google Scholar] [CrossRef] [Green Version]
- Barolo, L.; Abbriano, R.M.; Commault, A.S.; George, J.; Kahlke, T.; Fabris, M.; Padula, M.P.; Lopez, A.; Ralph, P.J.; Pernice, M. Perspectives for Glyco-Engineering of Recombinant Biopharmaceuticals from Microalgae. Cells 2020, 9, 633. [Google Scholar] [CrossRef] [Green Version]
- Qari, H.; Rehan, M.; Nizami, A.-S. Key Issues in Microalgae Biofuels: A Short Review. Energy Procedia 2017, 142, 898–903. [Google Scholar] [CrossRef]
- Kamyab, H.; Chelliapan, S.; Kumar, A.; Rezania, S.; Talaiekhozani, A.; Khademi, T.; Rupani, P.F.; Sharma, S. Microalgal Biotechnology Application Towards Environmental Sustainability. In Application of Microalgae in Wastewater Treatment; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2019; pp. 445–465. [Google Scholar]
- Chew, K.W.; Yap, J.Y.; Show, P.L.; Suan, N.H.; Juan, J.C.; Ling, T.C.; Lee, D.-J.; Chang, J.-S. Microalgae biorefinery: High value products perspectives. Bioresour. Technol. 2017, 229, 53–62. [Google Scholar] [CrossRef]
- Vuppaladadiyam, A.K.; Prinsen, P.; Raheem, A.; Luque, R.; Zhao, M. Sustainability Analysis of Microalgae Production Systems: A Review on Resource with Unexploited High-Value Reserves. Environ. Sci. Technol. 2018, 52, 14031–14049. [Google Scholar] [CrossRef] [PubMed]
- McGlathery, K.J.; Sundbäck, K.; Anderson, I.C. The Importance of Primary Producers for Benthic Nitrogen and Phosphorus Cycling. In Estuarine Nutrient Cycling: The Influence of Primary Producers: The Fate of Nutrients and Biomass; Nielsen, S.L., Banta, G.T., Pedersen, M.F., Eds.; Aquatic Ecology Book Series; Springer: Dordrecht, The Netherlands, 2004; pp. 231–261. ISBN 978-1-4020-3021-5. [Google Scholar]
- Singh, U.B.; Ahluwalia, A.S. Microalgae: A promising tool for carbon sequestration. Mitig. Adapt. Strat. Glob. Chang. 2012, 18, 73–95. [Google Scholar] [CrossRef]
- Banerjee, I.; Dutta, S.; Pohrmen, C.B.; Verma, R.; Singh, D. Microalgae-Based Carbon Sequestration to Mitigate Climate Change and Application of Nanomaterials in Algal Biorefinery. Octa J. Biosci. 2020, 8, 129–136. [Google Scholar]
- Wang, H.; Zhang, W.; Chen, L.; Wang, J.; Liu, T. The contamination and control of biological pollutants in mass cultivation of microalgae. Bioresour. Technol. 2013, 128, 745–750. [Google Scholar] [CrossRef]
- Suttle, C.A. Marine viruses-major players in the global ecosystem. Nat. Rev. Genet. 2007, 5, 801–812. [Google Scholar] [CrossRef]
- Galès, A.; Triplet, S.; Geoffroy, T.; Roques, C.; Carré, C.; Le Floc’H, E.; Lanfranchi, M.; Simier, M.; D’Orbcastel, E.R.; Przybyla, C.; et al. Control of the pH for marine microalgae polycultures: A key point for CO2 fixation improvement in intensive cultures. J. CO2 Util. 2020, 38, 187–193. [Google Scholar] [CrossRef]
- Froussard, P. A random-PCR method (rPCR) to construct whole cDNA library from low amounts of RNA. Nucleic Acids Res. 1992, 20, 2900. [Google Scholar] [CrossRef] [Green Version]
- Monteil-Bouchard, S.; Temmam, S.; Desnues, C. Protocol for Generating Infectious RNA Viromes from Complex Biological Samples. In Advanced Structural Safety Studies; Springer International Publishing: Berlin/Heidelberg, Germany, 2018; pp. 25–36. [Google Scholar]
- Brown, J.W.; Pirrung, M.; McCue, L.A. FQC Dashboard: Integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 2017, 33, 3137–3139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antipov, D.; Raiko, M.; Lapidus, A.; A Pevzner, P. Metaviral SPAdes: Assembly of viruses from metagenomic data. Bioinformatics 2020, 36, 4126–4129. [Google Scholar] [CrossRef] [PubMed]
- Mikheenko, A.; Saveliev, V.; Gurevich, A. MetaQUAST: Evaluation of metagenome assemblies. Bioinformatics 2016, 32, 1088–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Steinegger, M.; Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef] [Green Version]
- nbsp; NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- White, R.A., III; Panyala, A.; Glass, K.; Colby, S.; Glaesemann, K.R.; Jansson, C.; Jansson, J.K. MerCat: A Versatile k-Mer Counter and Diversity Estimator for Database-Independent Property Analysis Obtained from Metagenomic and/or Metatranscriptomic Sequencing Data. PeerJ Preprints 2017. [Google Scholar] [CrossRef]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef] [Green Version]
- Simpson, E. Measurement of diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Benoit, G. Simka: Fast Kmer-Based Method for Estimating the Similarity between Numerous Metagenomic Datasets. Available online: https://hal.inria.fr/hal-01231795. (accessed on 25 October 2021).
- Kans, J. Entrez Direct: E-Utilities on the Unix Command Line; National Center for Biotechnology Information (US). 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK179288/ (accessed on 25 October 2021).
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; López, R.; Finn, R.D. HMMER web server: 2018 update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bateman, A. The Pfam protein families database. Nucleic Acids Res. 2004, 32, 138D–141. [Google Scholar] [CrossRef]
- Rueckert, R.R.; Wimmer, E. Systematic nomenclature of picornavirus proteins. J. Virol. 1984, 50, 957–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlok, M.; Lang, A.S.; A Suttle, C. Application of a sequence-based taxonomic classification method to uncultured and unclassified marine single-stranded RNA viruses in the order Picornavirales. Virus Evol. 2019, 5. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Le, S.Q.; Gascuel, O. An Improved General Amino Acid Replacement Matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [Green Version]
- Marshall, O.J. PerlPrimer: Cross-platform, graphical primer design for standard, bisulphite and real-time PCR. Bioinformatics 2004, 20, 2471–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Ghabrial, S.A.; Nibert, M.L. Victorivirus, a new genus of fungal viruses in the family Totiviridae. Arch. Virol. 2009, 154, 373–379. [Google Scholar] [CrossRef]
- Stuart, G.; Moffett, K.; Bozarth, R.F. A whole genome perspective on the phylogeny of the plant virus family Tombusviridae. Arch. Virol. 2004, 149, 1595–1610. [Google Scholar] [CrossRef] [PubMed]
- Delmas, B.; Attoui, H.; Ghosh, S.; Malik, Y.S.; Mundt, E.; Vakharia, V.N. ICTV Report Consortium ICTV virus taxonomy profile: Birnaviridae. J. Gen. Virol. 2019, 100, 5–6. [Google Scholar] [CrossRef]
- Bollback, J.P.; Huelsenbeck, J.P. Phylogeny, Genome Evolution, and Host Specificity of Single-Stranded RNA Bacteriophage (Family Leviviridae). J. Mol. Evol. 2001, 52, 117–128. [Google Scholar] [CrossRef]
- Charon, J.; Murray, S.; Holmes, E.C. Revealing RNA virus diversity and evolution in unicellular algae transcriptomes. Virus Evol. 2021, 7. [Google Scholar] [CrossRef]
- Wilson, W.H.; Van Etten, J.L.; Allen, M.J. The Phycodnaviridae: The Story of How Tiny Giants Rule the World. Curr. Top. Microbiol. Immunol. 2009, 328, 1–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, L.Z.; McCrow, J.P.; Ininbergs, K.; Dupont, C.L.; Badger, J.H.; Hoffman, J.M.; Ekman, M.; Allen, A.E.; Bergman, B.; Venter, J.C. The Baltic Sea Virome: Diversity and Transcriptional Activity of DNA and RNA Viruses. mSystems 2017, 2, e00125-16. [Google Scholar] [CrossRef] [Green Version]
- Breitbart, M.; Delwart, E.; Rosario, K.; Segalés, J.; Varsani, A. ICTV Report Consortium ICTV Virus Taxonomy Profile: Circoviridae. J. Gen. Virol. 2017, 98, 1997–1998. [Google Scholar] [CrossRef]
- Daly, A.J.; Baetens, J.M.; De Baets, B. Ecological Diversity: Measuring the Unmeasurable. Mathematics 2018, 6, 119. [Google Scholar] [CrossRef] [Green Version]
- Gibrat, J.-F.; Mariadassou, M.; Boudinot, P.; Delmas, B. Analyses of the radiation of birnaviruses from diverse host phyla and of their evolutionary affinities with other double-stranded RNA and positive strand RNA viruses using robust structure-based multiple sequence alignments and advanced phylogenetic methods. BMC Evol. Biol. 2013, 13, 154. [Google Scholar] [CrossRef] [Green Version]
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Virus taxonomy in the age of metagenomics. Nat. Rev. Genet. 2017, 15, 161–168. [Google Scholar] [CrossRef]
- Aiewsakun, P.; Simmonds, P. The genomic underpinnings of eukaryotic virus taxonomy: Creating a sequence-based framework for family-level virus classification. Microbiome 2018, 6, 1–24. [Google Scholar] [CrossRef]
- Roux, S.; Enault, F.; Bronner, G.; Vaulot, D.; Forterre, P.; Krupovic, M. Chimeric viruses blur the borders between the major groups of eukaryotic single-stranded DNA viruses. Nat. Commun. 2013, 4, 2700. [Google Scholar] [CrossRef] [PubMed]
- Parras-Moltó, M.; Rodríguez-Galet, A.; Suárez-Rodríguez, P.; López-Bueno, A. Evaluation of bias induced by viral enrichment and random amplification protocols in metagenomic surveys of saliva DNA viruses. Microbiome 2018, 6, 1–18. [Google Scholar] [CrossRef]
- Brinkman, N.E.; Villegas, E.N.; Garland, J.L.; Keely, S.P. Reducing inherent biases introduced during DNA viral metagenome analyses of municipal wastewater. PLoS ONE 2018, 13, e0195350. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Cataluña, A.; Cuevas-Ferrando, E.; Randazzo, W.; Sánchez, G. Bias of library preparation for virome characterization in untreated and treated wastewaters. Sci. Total. Environ. 2021, 767, 144589. [Google Scholar] [CrossRef] [PubMed]
- Kustin, T.; Stern, A. Biased Mutation and Selection in RNA Viruses. Mol. Biol. Evol. 2021, 38, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.P.; Ogura, Y.; Nakamura, K.; Nishida, R.; Gotoh, Y.; Hayashi, M.; Hisatsune, J.; Sugai, M.; Takehiko, I.; Hayashi, T. Comparison of the sequencing bias of currently available library preparation kits for Illumina sequencing of bacterial genomes and metagenomes. DNA Res. 2019, 26, 391–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, A.; Garhyan, J.; Gibas, C. The impact of RNA secondary structure on read start locations on the Illumina sequencing platform. PLoS ONE 2017, 12, e0173023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaouri, N.; Jumat, M.R.; Cheema, T.; Hong, P.-Y. Metagenomics-based evaluation of groundwater microbial profiles in response to treated wastewater discharge. Environ. Res. 2020, 180, 108835. [Google Scholar] [CrossRef]
- Culley, A.I.; Lang, A.S.; Suttle, C.A. Metagenomic Analysis of Coastal RNA Virus Communities. Science 2006, 312, 1795–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlok, M.; Lang, A.S.; Suttle, C.A. Marine RNA Virus Quasispecies Are Distributed throughout the Oceans. mSphere 2019, 4, e00157-19. [Google Scholar] [CrossRef] [Green Version]
- López-Bueno, A.; Rastrojo, A.; Peiró, R.; Arenas, M.; Alcamí, A. Ecological connectivity shapes quasispecies structure of RNA viruses in an Antarctic lake. Mol. Ecol. 2015, 24, 4812–4825. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Correa, I.; Truchado, D.A.; Gomez, A.D.; Doménech, A.; Pérez-Tris, J.; Schmidt-Chanasit, J.; Cadar, D.; Benítez, L. A novel group of avian astroviruses from Neotropical passerine birds broaden the diversity and host range of Astroviridae. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Salomon, P.S.; Imai, I. Pathogens of Harmful Microalgae. In Ecology of Harmful Algae; Ecological Studies; Granéli, E., Turner, J.T., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 189, pp. 271–282. ISBN 978-3-540-32209-2. [Google Scholar]
- Negreiros, N.F.; Dos Santos-Wisniewski, M.J.; Dos Santos, R.M.; Rocha, O. The influence of environmental factors on the seasonal dynamics and composition of Rotifera in the Sapucaí River arm of Furnas Reservoir, MG, Brazil. Biota Neotropica 2010, 10, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Pagarete, A.; Chow, C.-E.T.; Johannessen, T.; Fuhrman, J.; Thingstad, T.F.; Sandaa, R.A. Strong Seasonality and Interannual Recurrence in Marine Myovirus Communities. Appl. Environ. Microbiol. 2013, 79, 6253–6259. [Google Scholar] [CrossRef] [Green Version]
- Mojica, K.D.A.; Brussaard, C.P.D. Factors affecting virus dynamics and microbial host-virus interactions in marine environments. FEMS Microbiol. Ecol. 2014, 89, 495–515. [Google Scholar] [CrossRef] [Green Version]
- Duffy, S. Why are RNA virus mutation rates so damn high? PLoS Biol. 2018, 16, e3000003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewson, I.; Bistolas, K.S.I.; Button, J.B.; Jackson, E.W. Occurrence and seasonal dynamics of RNA viral genotypes in three contrasting temperate lakes. PLoS ONE 2018, 13, e0194419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abou-Shanab, R.; Singh, M.; Rivera-Cruz, A.; Power, G.; Bagby-Moon, T.; Das, K. Effect of Brachionus rubens on the growth characteristics of various species of microalgae. Electron. J. Biotechnol. 2016, 22, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Delpy, F.; Pagano, M.; Blanchot, J.; Carlotti, F.; Thibault-Botha, D. Man-induced hydrological changes, metazooplankton communities and invasive species in the Berre Lagoon (Mediterranean Sea, France). Mar. Pollut. Bull. 2012, 64, 1921–1932. [Google Scholar] [CrossRef] [PubMed]
- Conde-Porcuna, J.M. Relative importance of competition with Daphnia (Cladocera) and nutrient limitation on Anuraeopsis (Rotifera) population dynamics in a laboratory study. Freshw. Biol. 2000, 44, 423–430. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chase, E.E.; Monteil-Bouchard, S.; Gobet, A.; Andrianjakarivony, F.H.; Desnues, C.; Blanc, G. A High Rate Algal Pond Hosting a Dynamic Community of RNA Viruses. Viruses 2021, 13, 2163. https://doi.org/10.3390/v13112163
Chase EE, Monteil-Bouchard S, Gobet A, Andrianjakarivony FH, Desnues C, Blanc G. A High Rate Algal Pond Hosting a Dynamic Community of RNA Viruses. Viruses. 2021; 13(11):2163. https://doi.org/10.3390/v13112163
Chicago/Turabian StyleChase, Emily E., Sonia Monteil-Bouchard, Angélique Gobet, Felana H. Andrianjakarivony, Christelle Desnues, and Guillaume Blanc. 2021. "A High Rate Algal Pond Hosting a Dynamic Community of RNA Viruses" Viruses 13, no. 11: 2163. https://doi.org/10.3390/v13112163
APA StyleChase, E. E., Monteil-Bouchard, S., Gobet, A., Andrianjakarivony, F. H., Desnues, C., & Blanc, G. (2021). A High Rate Algal Pond Hosting a Dynamic Community of RNA Viruses. Viruses, 13(11), 2163. https://doi.org/10.3390/v13112163