
G-Quadruplexes Formation at the Upstream Region of Replication Origin (OriL) of the Pseudorabies Virus: Implications for Antiviral Targets


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Oligonucleotides
2.2. Cells and Viruses
2.3. Circular Dichroism (CD) Spectroscopy
2.4. Gel Electrophoresis
2.5. Taqpolymerase Stop Assay
2.6. Fluorescence Resonance Energy Transfer (FRET) Melting Assay
2.7. Cell Viability Assays
2.8. Viral Titration
2.9. Western Blot Assay
2.10. Fluorescent Microscopy
2.11. Flow Cytometry
2.12. Time of Addition Assay (TOA)
2.13. Quantitative Polymerase Chain Reaction (q-PCR)
2.14. Statistical Analysis
3. Results
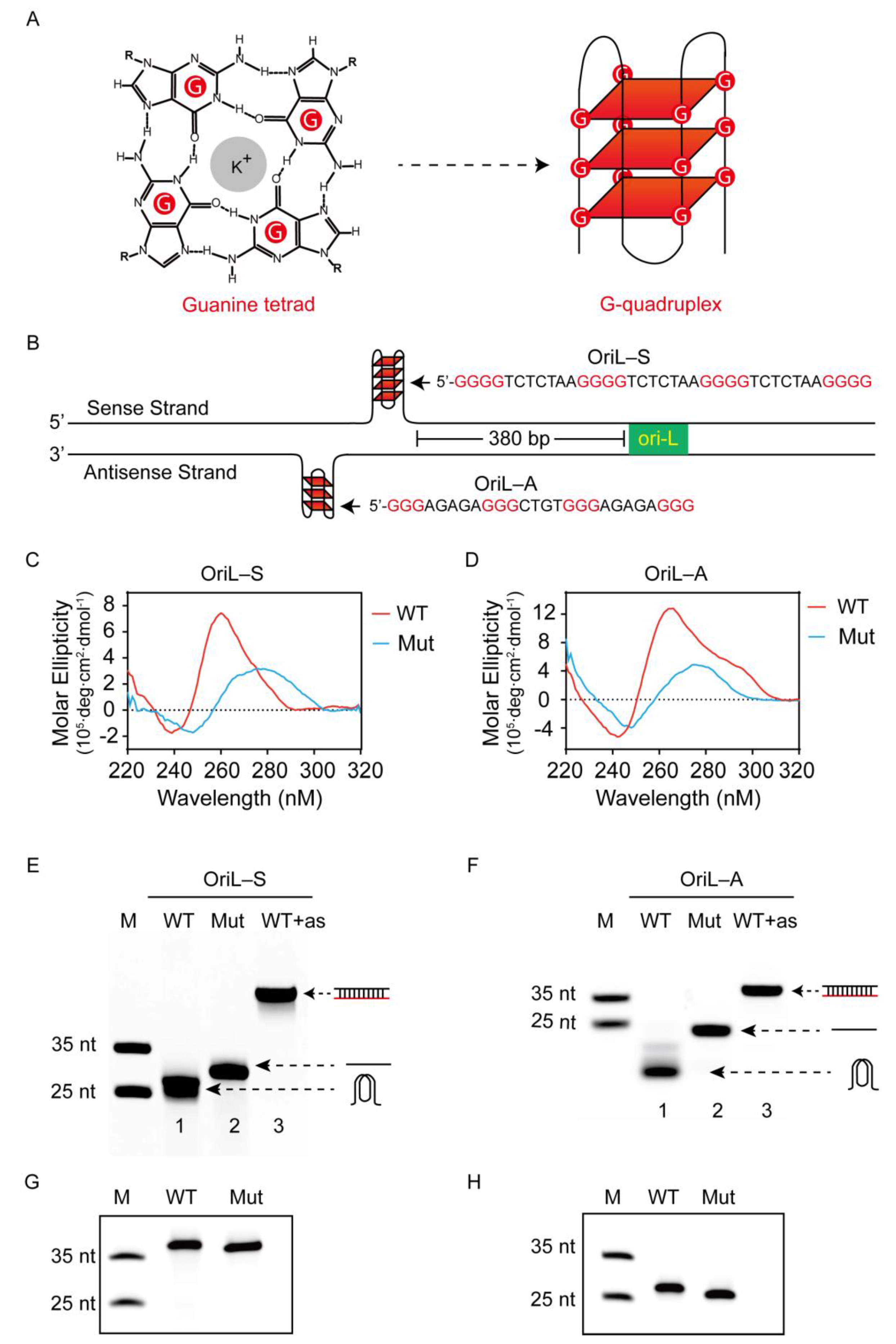
3.1. Two Conserved G-Rich Sequences at the Upstream Region of OriL Formed a Parallel G4 Structure In Vitro
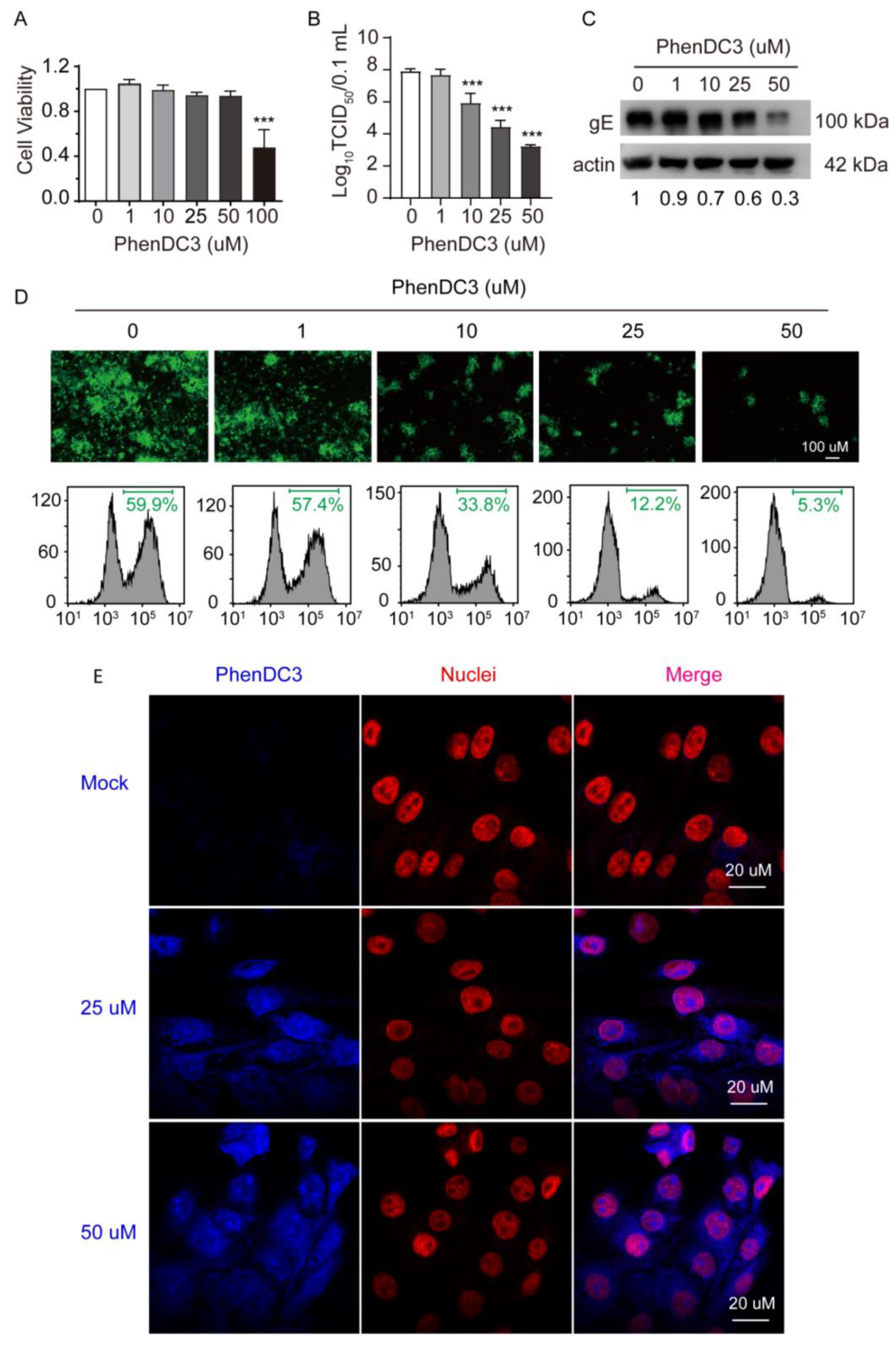
3.2. G4-Stabilizer PhenDC3 Binds and StabilizesG4 Structure of OriL-S and OriL-A
3.3. The Compound PhenDC3 Inhibits PRV Proliferation in PK-15 Cells
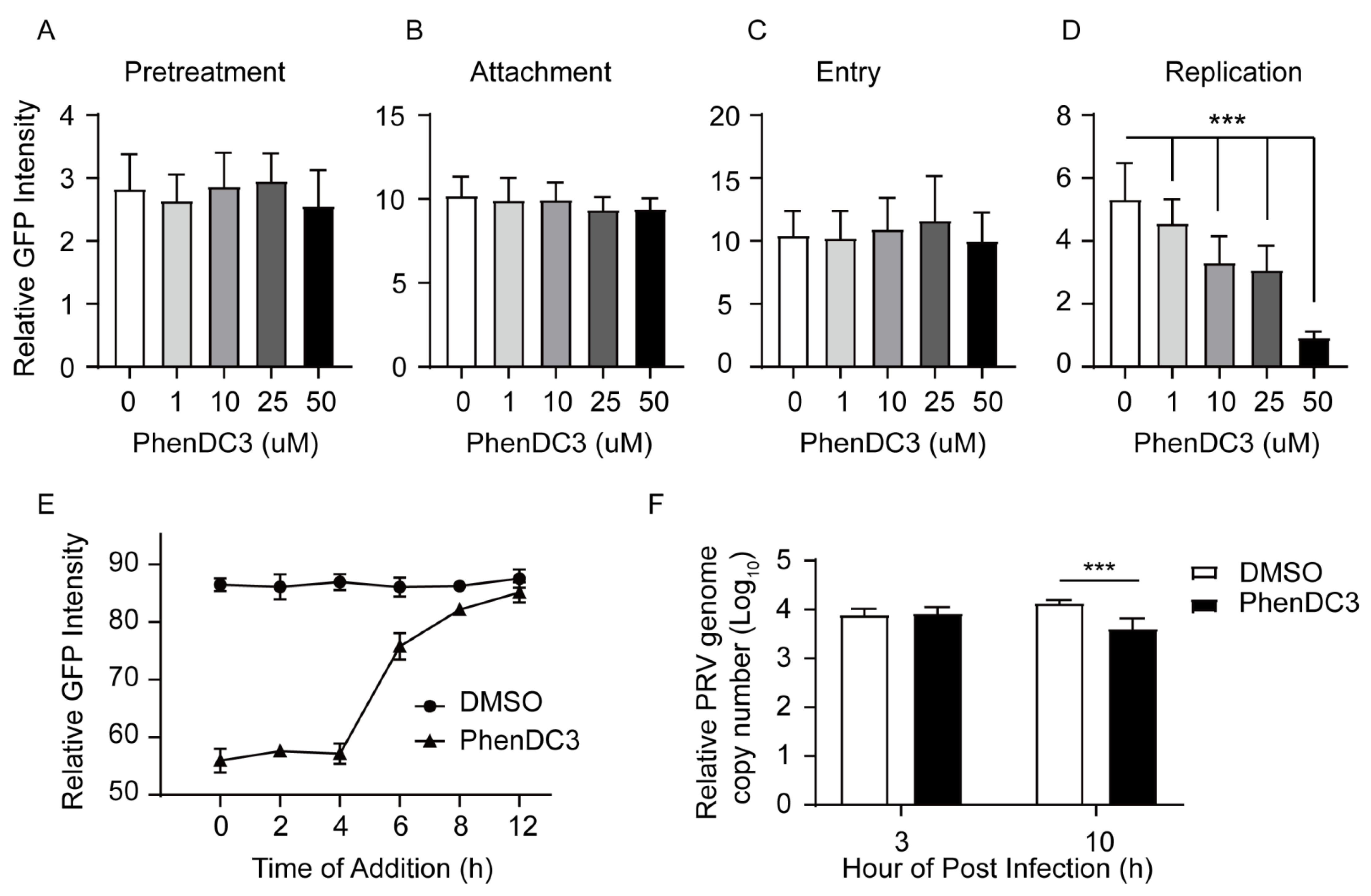
3.4. G4-Stabilizer PhenDC3 Impairs PRV DNA Replication
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mettenleiter, T.C. Aujeszky’s disease (pseudorabies) virus: The virus and molecular pathogenesis-state of the art, June 1999. Vet. Res. 2000, 31, 99–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, A.A., Jr.; Camargos, M.F.; de Oliveira, A.M.; Ciacci-Zanella, J.R.; Patrício, M.A.; Braga, A.C.; Cunha, E.S.; D’Ambros, R.; Heinemann, M.B.; Leite, R.C.; et al. Molecular epidemiology of Brazilian pseudorabies viral isolates. Vet. Microbiol. 2010, 141, 238–245. [Google Scholar] [CrossRef]
- An, T.Q.; Peng, J.M.; Tian, Z.J.; Zhao, H.Y.; Li, N.; Liu, Y.M.; Chen, J.Z.; Leng, C.L.; Sun, Y.; Chang, D.; et al. Pseudorabies virus variant in Bartha-K61-vaccinated pigs, China, 2012. Emerg. Infect. Dis. 2013, 19, 1749–1755. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.; Liu, F.; Zheng, H.; Liang, C.; Zhou, Y.J.; Jiang, Y.F.; Shan, T.L.; Gao, F.; Li, G.X.; Tong, G.Z. Emergence of a Pseudorabies virus variant with increased virulence to piglets. Vet. Microbiol. 2015, 181, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Li, N.; Cong, X.; Wang, C.H.; Du, M.; Li, L.; Zhao, B.; Yuan, J.; Liu, D.D.; Li, S.; et al. Pathogenicity and genomic characterization of a pseudorabies virus variant isolated from Bartha-K61-vaccinated swine population in China. Vet. Microbiol. 2014, 174, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Ai, J.W.; Weng, S.S.; Cheng, Q.; Cui, P.; Li, Y.J.; Wu, H.L.; Zhu, Y.M.; Xu, B.; Zhang, W.H. Human endophthalmitis caused by pseudorabies virus infection, China, 2017. Emerg. Infect. Dis. 2018, 24, 1087–1090. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Han, H.; Wang, H.; Cui, Y.; Liu, H.; Ding, S. A case of human viral encephalitis caused by pseudorabies virus infection in China. Front. Neurol. 2019, 10, 534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Guan, H.; Li, C.; Li, Y.; Wang, S.; Zhao, X.; Zhao, Y.; Liu, Y. Characteristics of human encephalitis caused by pseudorabies virus: A case series study. Int. J. Infect. Dis. 2019, 87, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Wang, X.; Xie, C.; Ding, S.; Yang, H.; Guo, S.; Li, J.; Qin, L.; Ban, F.; Wang, D.; et al. A novel human acute encephalitis caused by pseudorabies virus variant strain. Clin. Infect. Dis. 2020, ciaa987. [Google Scholar] [CrossRef]
- Biffi, G.; Tannahill, D.; McCafferty, J.; Balasubramanian, S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013, 5, 182–186. [Google Scholar] [CrossRef]
- Chambers, V.S.; Marsico, G.; Boutell, J.M.; Di Antonio, M.; Smith, G.P.; Balasubramanian, S. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat. Biotechnol. 2015, 33, 877–881. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 15. [Google Scholar] [CrossRef] [Green Version]
- Hansel-Hertsch, R.; Antonio, M.D.; Balasubramanian, S. DNA G-quadruplexes in the human genome: Detection, functions and therapeutic potential. Nat. Rev. Mol. Cell Biol. 2017, 18, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, E.; Richter, S.N. Viral G-quadruplexes: New frontiers in virus pathogenesis and antiviral therapy. Ann. Rep. Med. Chem. 2020, 46, 3270–3283. [Google Scholar]
- Abiri, A.; Lavigne, M.; Rezaei, M.; Nikzad, S.; Zare, P.; Mergny, J.L.; Rahimi, H.R. Unlocking G-Quadruplexes as Antiviral targets. Pharmacol. Rev. 2021, 73, 897–923. [Google Scholar] [CrossRef]
- Metifiot, M.; Amrane, S.; Litvak, S.; Andreola, M.L. G-quadruplexes in viruses: Function and potential therapeutic applications. Nucleic Acids Res. 2014, 42, 12352–12366. [Google Scholar] [CrossRef] [Green Version]
- Perrone, R.; Nadai, M.; Frasson, I.; Poe, J.A.; Butovskaya, E.; Smithgall, T.E.; Palumbo, M.; Palu, G.; Richter, S.N. A dynamic G-quadruplex region regulates the HIV-1 long terminal repeat promoter. J. Med. Chem. 2013, 56, 6521–6530. [Google Scholar] [CrossRef] [Green Version]
- Murat, P.; Zhong, J.; Lekieffre, L.; Cowieson, N.P.; Clancy, J.L.; Preiss, T.; Balasubramanian, S.; Khanna, R.; Tellam, J. G-quadruplexes regulate Epstein-Barr virus-encoded nuclear antigen 1 mRNA translation. Nat. Chem. Biol. 2014, 10, 358–364. [Google Scholar] [CrossRef]
- Artusi, S.; Nadai, M.; Perrone, R.; Biasolo, M.A.; Palu, G.; Flamand, L.; Calistri, A.; Richter, S.N. The Herpes Simplex Virus-1 genome contains multiple clusters of repeated G-quadruplex: Implications for the antiviral activity of a G-quadruplex ligand. Antivir. Res. 2015, 118, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.R.; Min, Y.Q.; Wang, J.Q.; Liu, C.X.; Fu, B.S.; Wu, F.; Wu, L.Y.; Qiao, Z.X.; Song, Y.Y.; Xu, G.H.; et al. A highly conserved G-rich consensus sequence in hepatitis C virus core gene represents a new anti-hepatitis C target. Sci. Adv. 2016, 2, e1501535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.R.; Zhang, Q.Y.; Wang, J.Q.; Ge, X.Y.; Song, Y.Y.; Wang, Y.F.; Li, X.D.; Fu, B.S.; Xu, G.H.; Shu, B.; et al. Chemical targeting of a G-quadruplex RNA in the Ebola virus L gene. Cell Chem. Biol. 2016, 23, 1113–1122. [Google Scholar] [CrossRef] [Green Version]
- Fleming, A.M.; Ding, Y.; Alenko, A.; Burrows, C.J. Zika Virus Genomic RNA possesses conserved G-quadruplexes characteristic of the flaviviridae family. ACS Infect. Dis. 2016, 2, 674–681. [Google Scholar] [CrossRef]
- Bezzi, G.; Piga, E.J.; Binolfi, A.; Armas, P. CNBP Binds and unfolds in vitro G-quadruplexes formed in the SARS-CoV-2 positive and negative genome strands. Int. J. Mol. Sci. 2021, 22, 2614. [Google Scholar] [CrossRef]
- Belmonte-Reche, E.; Serrano-Chacón, I.; Gonzalez, C.; Gallo, J.; Bañobre-López, M. Potential G-quadruplexes and i-Motifs in the SARS-CoV-2. PLoS ONE 2021, 16, e0250654. [Google Scholar] [CrossRef]
- Zhao, C.; Qin, G.; Niu, J.; Wang, Z.; Wang, C.; Ren, J.; Qu, X. Targeting RNA G-Quadruplex in SARS-CoV-2: A Promising Therapeutic Target for COVID-19? Angew. Chem. Int. Ed. Engl. 2021, 60, 432–438. [Google Scholar] [CrossRef]
- Kong, J.N.; Zhang, C.; Zhu, Y.C.; Zhong, K.; Wang, J.; Chu, B.B.; Yang, G.Y. Identification and characterization of G-quadruplex formation within the EP0 promoter of pseudorabies virus. Sci. Rep. 2018, 8, 14029. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, S.; Jiang, H.; Deng, H.; Dong, C.; Shen, W.; Chen, H.; Gao, C.; Xiao, S.; Liu, Z.F.; et al. G(2)-quadruplex in the 3′UTR of IE180 regulates Pseudorabies virus replication by enhancing gene expression. RNA Biol. 2020, 17, 816–827. [Google Scholar] [CrossRef]
- Paramasivan, S.; Rujan, I.; Bolton, P.H. Circular dichroism of quadruplex DNAs: Applications to structure, cation effects and ligand binding. Methods 2007, 43, 324–331. [Google Scholar] [CrossRef]
- Callegaro, S.; Perrone, R.; Scalabrin, M.; Doria, F.; Palu, G.; Richter, S.N. A core extended naphtalenediimide G-quadruplex ligand potently inhibits herpes simplex virus 1 replication. Sci. Rep. 2017, 7, 2341. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Williamson, J.R.; Raghuraman, M.K.; Cech, T.R. Monovalent cation-induced structure of telomeric DNA: The G-quartet model. Cell 1989, 59, 871–880. [Google Scholar] [CrossRef]
- Murchie, A.I.; Lilley, D.M. Tetraplex folding of telomere sequences and the inclusion of adenine bases. EMBO J. 1994, 13, 993–1001. [Google Scholar] [CrossRef]
- Balagurumoorthy, P.; Brahmachari, S.K. Structure and stability of human telomeric sequence. J. Biol. Chem. 1994, 269, 21858–21869. [Google Scholar] [CrossRef]
- Cocchi, F.; Fusco, D.; Menotti, L.; Gianni, T.; Eisenberg, R.J.; Cohen, G.H.; Campadelli-Fiume, G. The soluble ectodomain of herpes simplex virus gD contains a membrane-proximal pro-fusion domain and suffices to mediate virus entry. Proc. Natl. Acad. Sci. USA 2004, 101, 7445–7450. [Google Scholar] [CrossRef] [Green Version]
- Krummenacher, C.; Supekar, V.M.; Whitbeck, J.C.; Lazear, E.; Connolly, S.A.; Eisenberg, R.J.; Cohen, G.H.; Wiley, D.C.; Carfí, A. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J. 2005, 24, 4144–4153. [Google Scholar] [CrossRef]
- Li, A.; Lu, G.; Qi, J.; Wu, L.; Tian, K.; Luo, T.; Shi, Y.; Yan, J.; Gao, G.F. Structural basis of nectin-1 recognition by pseudorabies virus glycoprotein D. PLoS Pathog. 2017, 13, e1006314. [Google Scholar] [CrossRef] [Green Version]
- Daelemans, D.; Pauwels, R.; De Clercq, E.; Pannecouque, C. A time-of-drug addition approach to target identification of antiviral compounds. Nat. Protoc. 2011, 6, 925–933. [Google Scholar] [CrossRef]
- Pomeranz, L.E.; Reynolds, A.E.; Hengartner, C.J. Molecular biology of pseudorabies virus: Impact on neurovirology and veterinary medicine. Microbiol. Mol. Biol. Rev. MMBR 2005, 69, 462–500. [Google Scholar] [CrossRef] [Green Version]
- Frasson, I.; Nadai, M.; Richter, S.N. Conserved G-quadruplexes regulate the immediate early promoters of human alphaherpesviruses. Molecules 2019, 24, 2375. [Google Scholar] [CrossRef] [Green Version]
- Artusi, S.; Perrone, R.; Lago, S.; Raffa, P.; Di Iorio, E.; Palu, G.; Richter, S.N. Visualization of DNA G-quadruplexes in herpes simplex virus 1-infected cells. Nucleic Acids Res. 2016, 44, 10343–10353. [Google Scholar] [CrossRef]
- VyThi Le, T.; Han, S.; Chae, J.; Park, H.J. G-quadruplex binding ligands: From naturally occurring to rationally designed molecules. Curr. Pharm. Des. 2012, 18, 1948–1972. [Google Scholar] [CrossRef] [PubMed]
- De Cian, A.; Delemos, E.; Mergny, J.L.; Teulade-Fichou, M.P.; Monchaud, D. Highly efficient G-quadruplex recognition by bisquinolinium compounds. J. Am. Chem. Soc. 2007, 129, 1856–1857. [Google Scholar] [CrossRef]
- Monchaud, D.; Allain, C.; Bertrand, H.; Smargiasso, N.; Rosu, F.; Gabelica, V.; De Cian, A.; Mergny, J.L.; Teulade-Fichou, M.P. Ligands playing musical chairs with G-quadruplex DNA: A rapid and simple displacement assay for identifying selective G-quadruplex binders. Biochimie 2008, 90, 1207–1223. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.; Guedin, A.; Mergny, J.L.; Salles, B.; Riou, J.F.; Teulade-Fichou, M.P.; Calsou, P. A G-quadruplex structure within the 5′-UTR of TRF2 mRNA represses translation in human cells. Nucleic Acids Res. 2010, 38, 7187–7198. [Google Scholar] [CrossRef] [Green Version]
- Lopes, J.; Piazza, A.; Bermejo, R.; Kriegsman, B.; Colosio, A.; Teulade-Fichou, M.P.; Foiani, M.; Nicolas, A. G-quadruplex-induced instability during leading-strand replication. EMBO J. 2011, 30, 4033–4046. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.J.; Heddi, B.; Hamon, F.; Teulade-Fichou, M.P.; Phan, A.T. Solution structure of a G-quadruplex bound to the bisquinolinium compound Phen-DC(3). Angew. Chem. Int. Ed. Engl. 2014, 53, 999–1002. [Google Scholar] [CrossRef]
- Harris, L.M.; Merrick, C.J. G-quadruplexes in pathogens: A common route to virulence control? PLoS Pathog. 2015, 11, e1004562. [Google Scholar] [CrossRef]
- Bryan, T.M. Mechanisms of DNA replication and repair: Insights from the study of G-quadruplexes. Molecules 2019, 24, 3439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madireddy, A.; Purushothaman, P.; Loosbroock, C.P.; Robertson, E.S.; Schildkraut, C.L.; Verma, S.C. G-quadruplex-interacting compounds alter latent DNA replication and episomal persistence of KSHV. Nucleic Acids Res. 2016, 44, 3675–3694. [Google Scholar] [CrossRef] [Green Version]
- Prorok, P.; Artufel, M.; Aze, A.; Coulombe, P.; Peiffer, I.; Lacroix, L.; Guedin, A.; Mergny, J.L.; Damaschke, J.; Schepers, A.; et al. Involvement of G-quadruplex regions in mammalian replication origin activity. Nat. Commun. 2019, 10, 3274. [Google Scholar] [CrossRef] [Green Version]
- Prioleau, M.N. G-Quadruplexes and DNA Replication Origins. Adv. Exp. Med. Biol. 2017, 1042, 273–286. [Google Scholar]
- Besnard, E.; Babled, A.; Lapasset, L.; Milhavet, O.; Parrinello, H.; Dantec, C.; Marin, J.M.; Lemaitre, J.M. Unraveling cell type-specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat. Struct. Mol. Biol. 2012, 19, 837–844. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay | Name | Sequence (5′ to 3′) * |
|---|---|---|
| CD | OriL-S-WT | GGGGTCTCTAAGGGGTCTCTAAGGGGTCTCTAAGGGG |
| OriL-A-WT | GGGAGAGAGGGCTGTGGGAGAGAGGG | |
| OriL-S-Mut | AGGATCTCTAAGAAGTCTCTAAGAAATCTCTAGGAAG | |
| OriL-A-Mut | GAGAGAGAGAGCTGTGAGAGAGAGGA | |
| PAGE | OriL-S-WT | FAM-GGGGTCTCTAAGGGGTCTCTAAGGGGTCTCTAAGGGG |
| OriL-A-WT | FAM-GGGAGAGAGGGCTGTGGGAGAGAGGG | |
| OriL-S-Mut | FAM-AGGATCTCTAAGAAGTCTCTAAGAAATCTCTAGGAAG | |
| OriL-A-Mut | FAM- GAGAGAGAGAGCTGTGAGAGAGAGGA | |
| Marker-25nt | FAM-TTTTTTTTTTTTTTTTTTTTTTTTT | |
| Marker-35nt | FAM-TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT | |
| FRET Melting | OriL-S-WT | FAM-GGGGTCTCTAAGGGGTCTCTAAGGGGTCTCTAAGGGG-TAMAR |
| OriL-A-WT | FAM-GGGAGAGAGGGCTGTGGGAGAGAGGG-TAMAR | |
| Taq polymerase stop assay | OriL-S-WT | TTTTTGGGGTCTCTAAGGGGTCTCTAAGGGGTCTCTAAGGGGTTTTTCGCACTGAGCGAAGATACGGAGCCACGCCA |
| OriL-A-WT | TTTTTGGGAGAGAGGGCTGTGGGAGAGAGGGTTTTTCGCACTGAGCGAAGATACGGAGCCACGCCA | |
| OriL-S-Mut | TTTTTAGGATCTCTAAGAAGTCTCTAAGAAATCTCTAGGAAGTTTTTCGCACTGAGCGAAGATACGGAGCCACGCCA | |
| OriL-A-Mut | TTTTTGAGAGAGAGAGCTGTGAGAGAGAGGATTTTTCGCACTGAGCGAAGATACGGAGCCACGCCA | |
| Primer | FAM-TGGCGTGGCTCCGTATCTTCGCTCAG | |
| qPCR | gH primer F | CTCGCCCTCGTCAGCAA |
| gH primer R | GCTGCTCCTCCATGTCCTT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Y.; Liu, W.; Zhang, C. G-Quadruplexes Formation at the Upstream Region of Replication Origin (OriL) of the Pseudorabies Virus: Implications for Antiviral Targets. Viruses 2021, 13, 2219. https://doi.org/10.3390/v13112219
Zhu Y, Liu W, Zhang C. G-Quadruplexes Formation at the Upstream Region of Replication Origin (OriL) of the Pseudorabies Virus: Implications for Antiviral Targets. Viruses. 2021; 13(11):2219. https://doi.org/10.3390/v13112219
Chicago/Turabian StyleZhu, Yance, Wenhao Liu, and Chao Zhang. 2021. "G-Quadruplexes Formation at the Upstream Region of Replication Origin (OriL) of the Pseudorabies Virus: Implications for Antiviral Targets" Viruses 13, no. 11: 2219. https://doi.org/10.3390/v13112219
APA StyleZhu, Y., Liu, W., & Zhang, C. (2021). G-Quadruplexes Formation at the Upstream Region of Replication Origin (OriL) of the Pseudorabies Virus: Implications for Antiviral Targets. Viruses, 13(11), 2219. https://doi.org/10.3390/v13112219