
Epidemiology and Evolution of Emerging Porcine Circovirus-like Viruses in Pigs with Hemorrhagic Dysentery and Diarrhea Symptoms in Central China from 2018 to 2021

Abstract
:1. Introduction
2. Materials and Methods
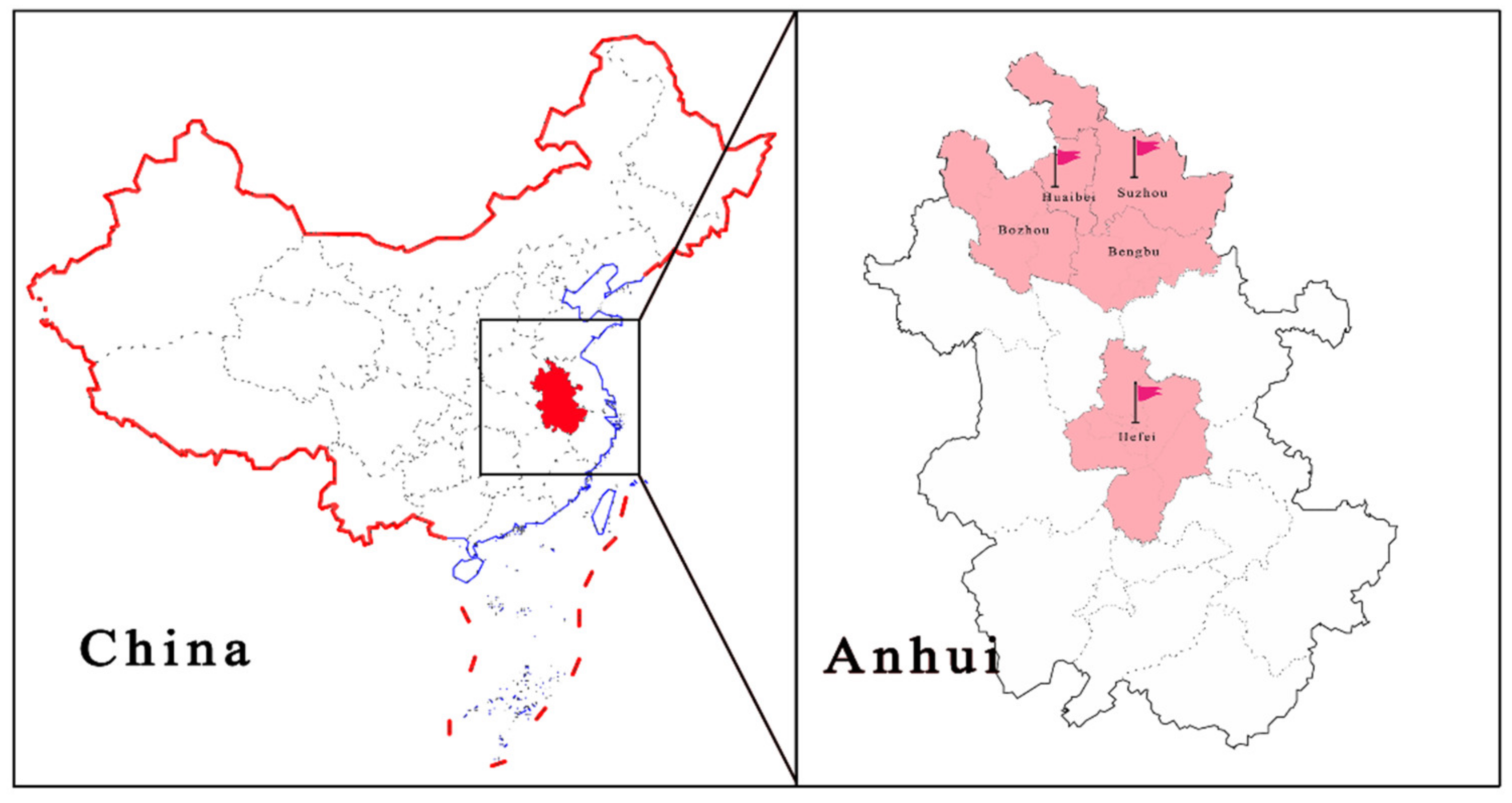
2.1. Sample Collection
2.2. Viral Nucleic Acid Extraction and PCR Detection
2.3. PCLV Sequencing of the Full-Length Genome
2.4. Sequence Alignment, Phylogenetic and Recombination Analysis
2.5. Selection Pressure, B-Cell Epitope Prediction and Codon Usage Bias Analyses
3. Results
3.1. Prevalence of PCLV
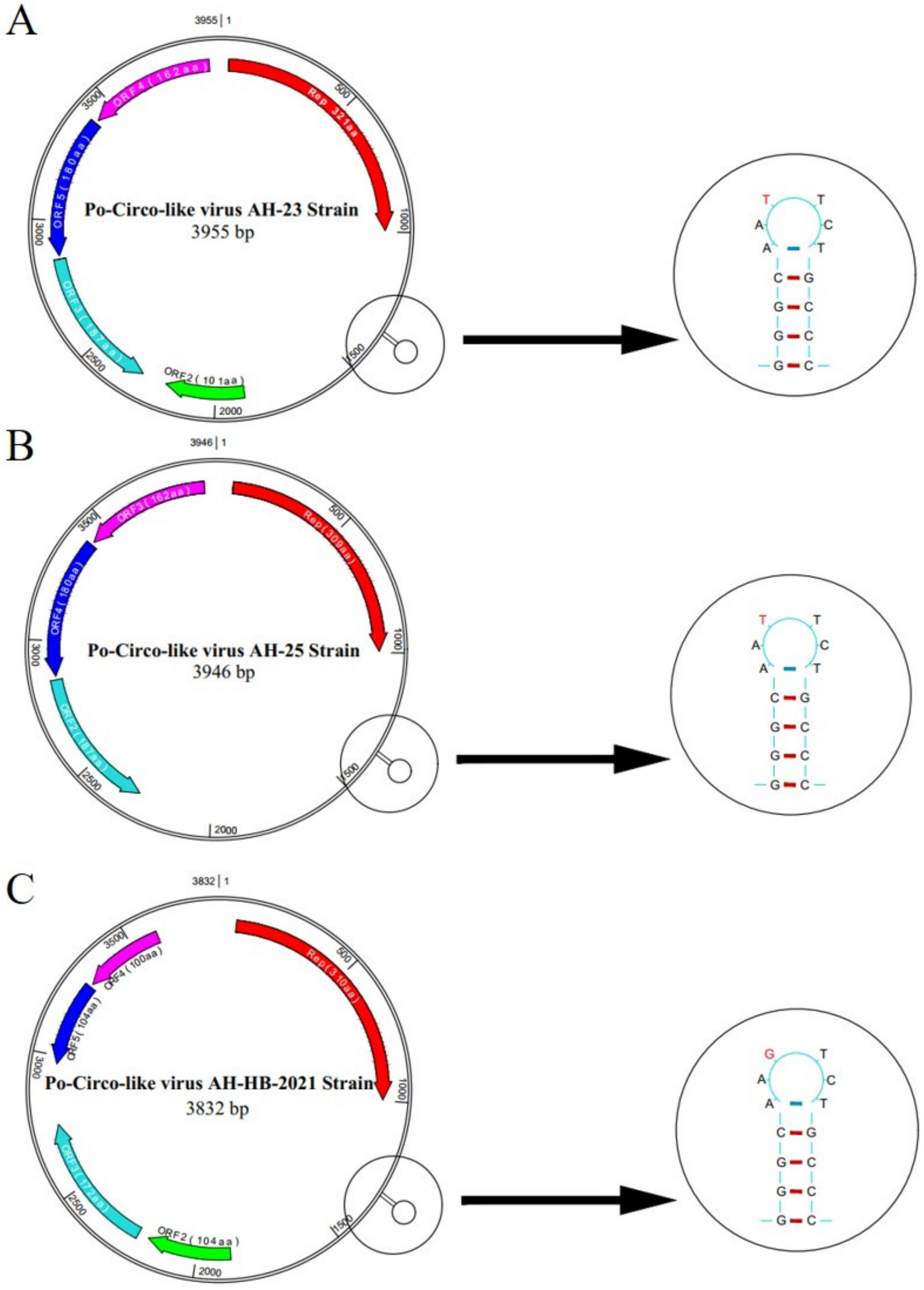
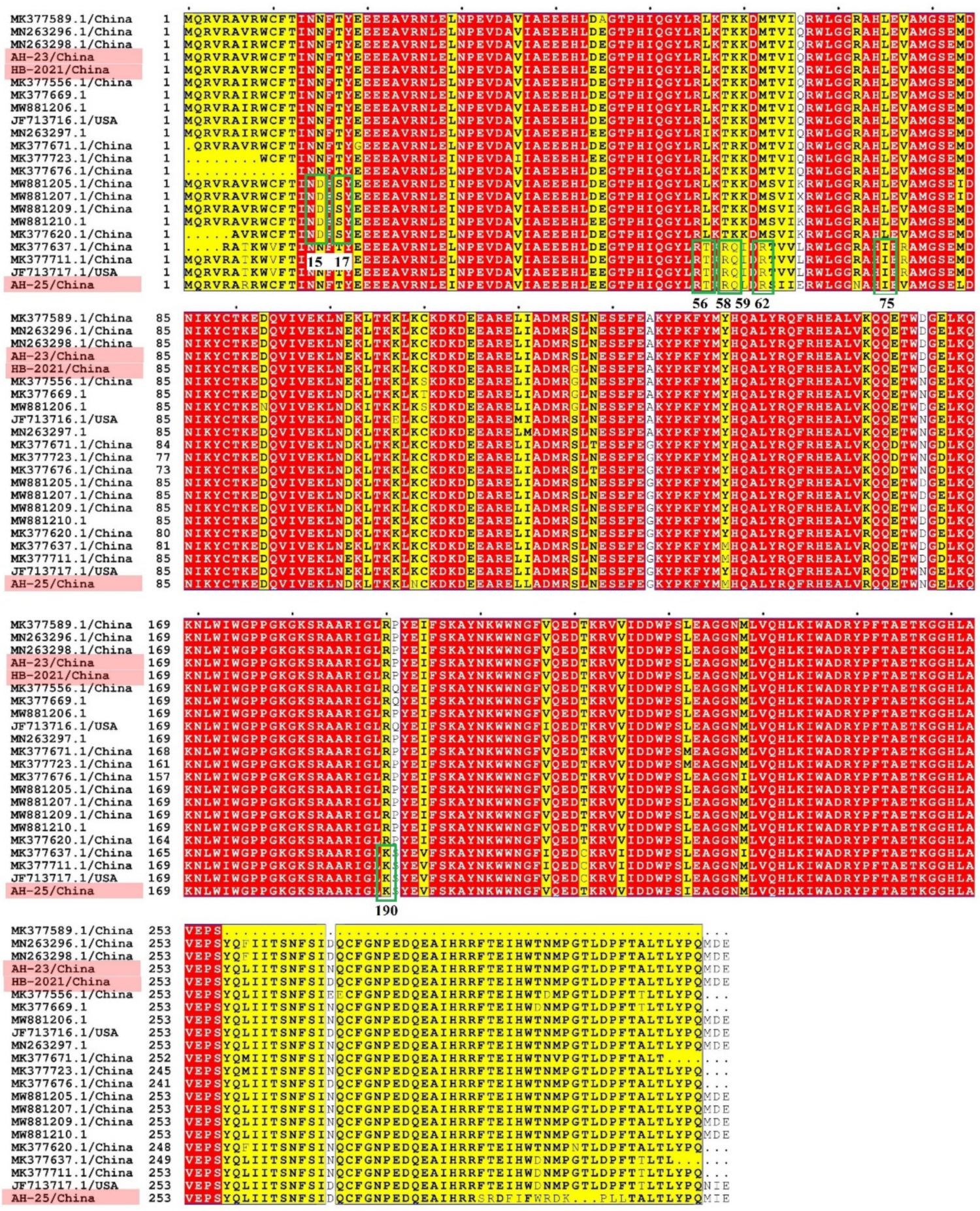
3.2. Full-Sequence Alignment of PCLV
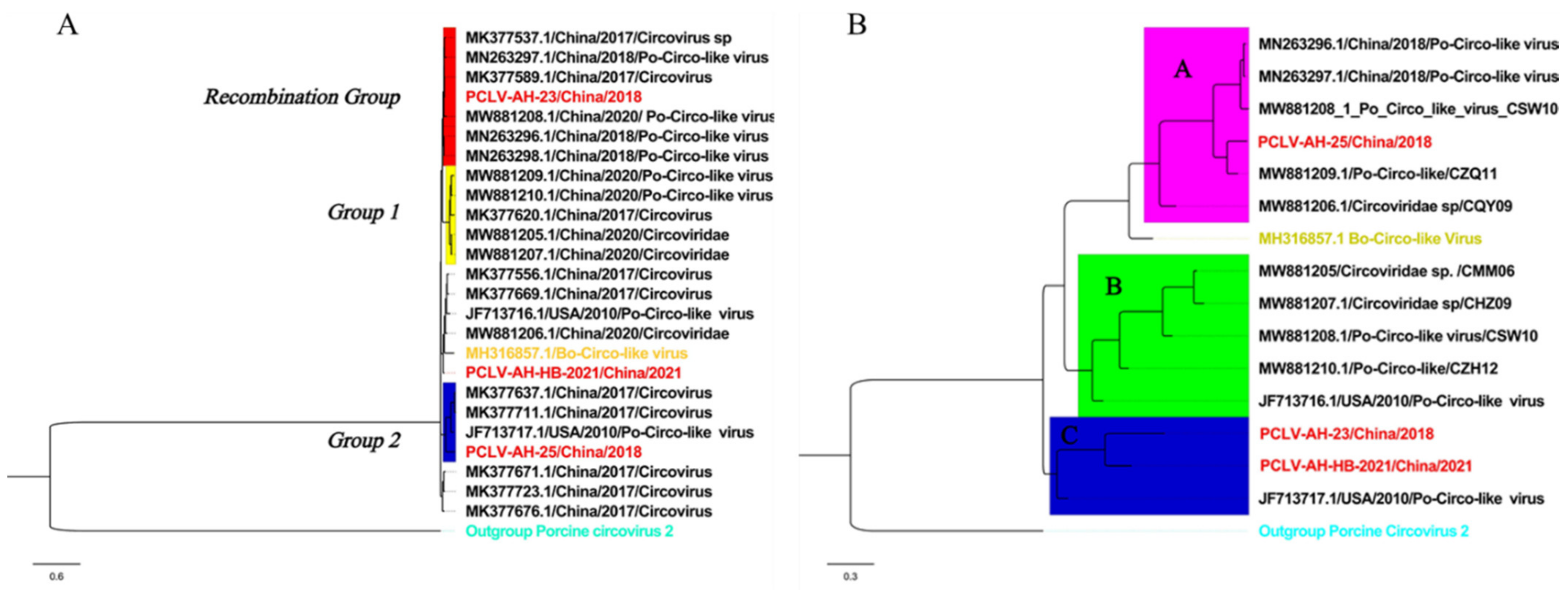
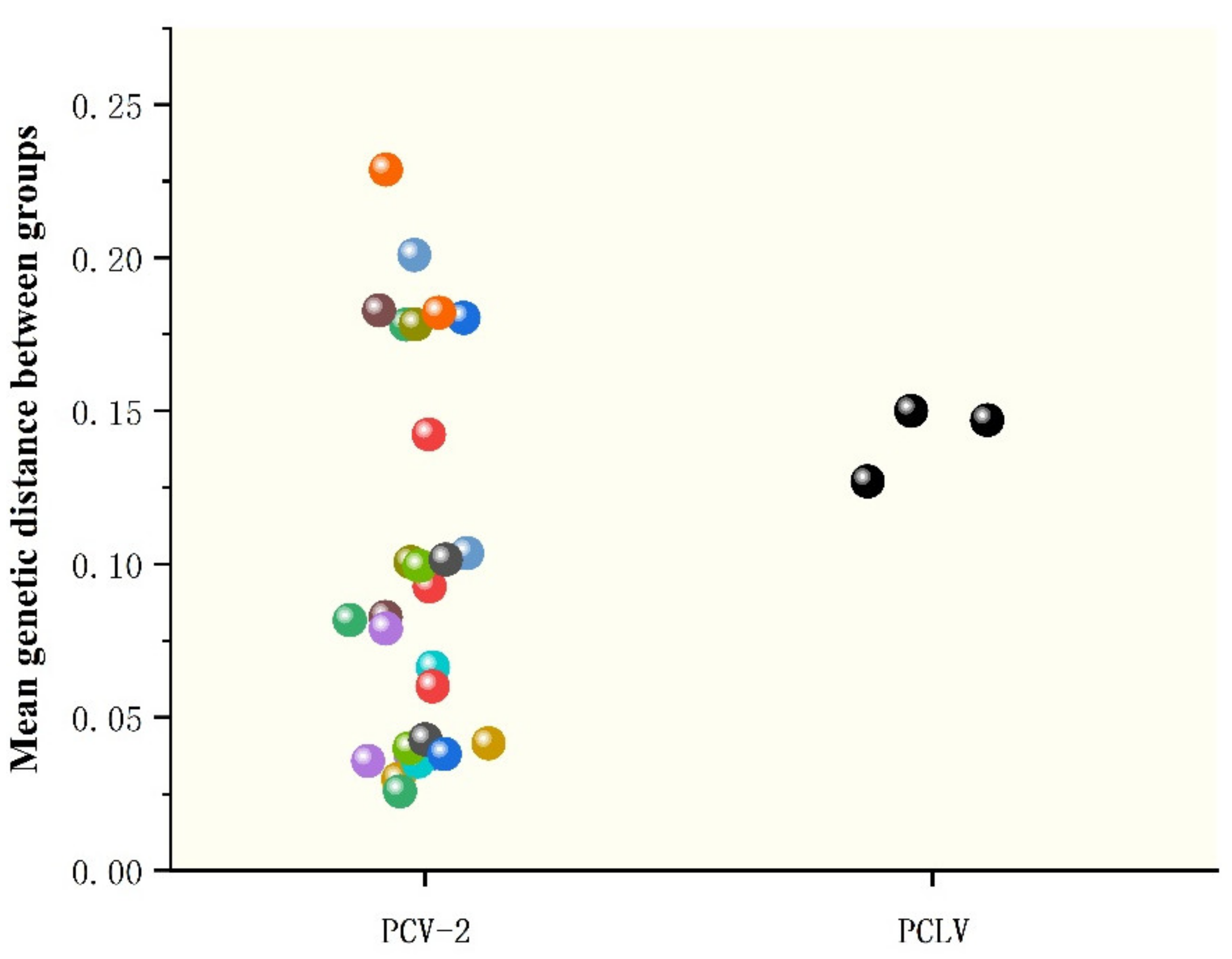
3.3. Phylogenetic Analysis and Genetic Divergence
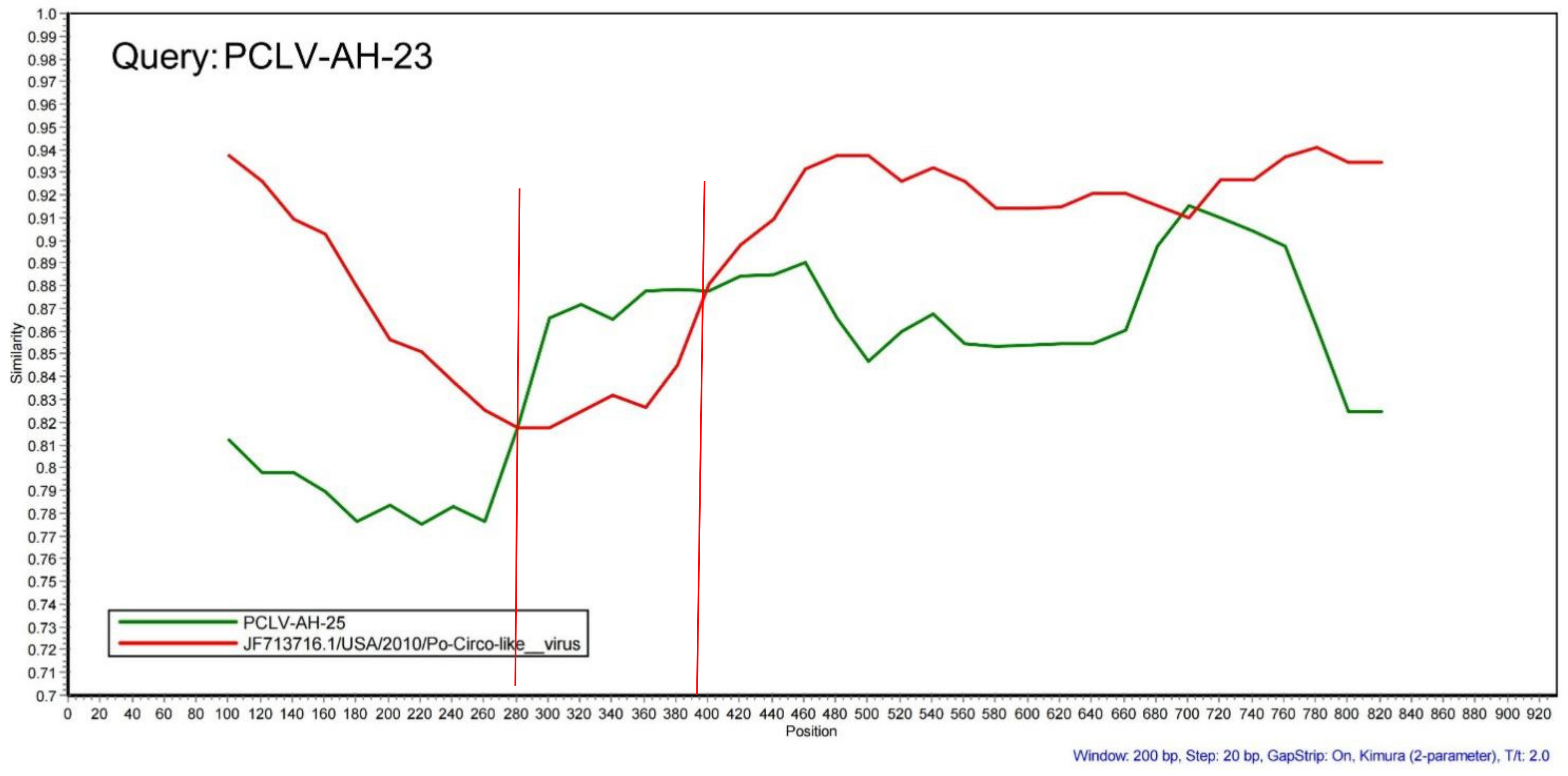
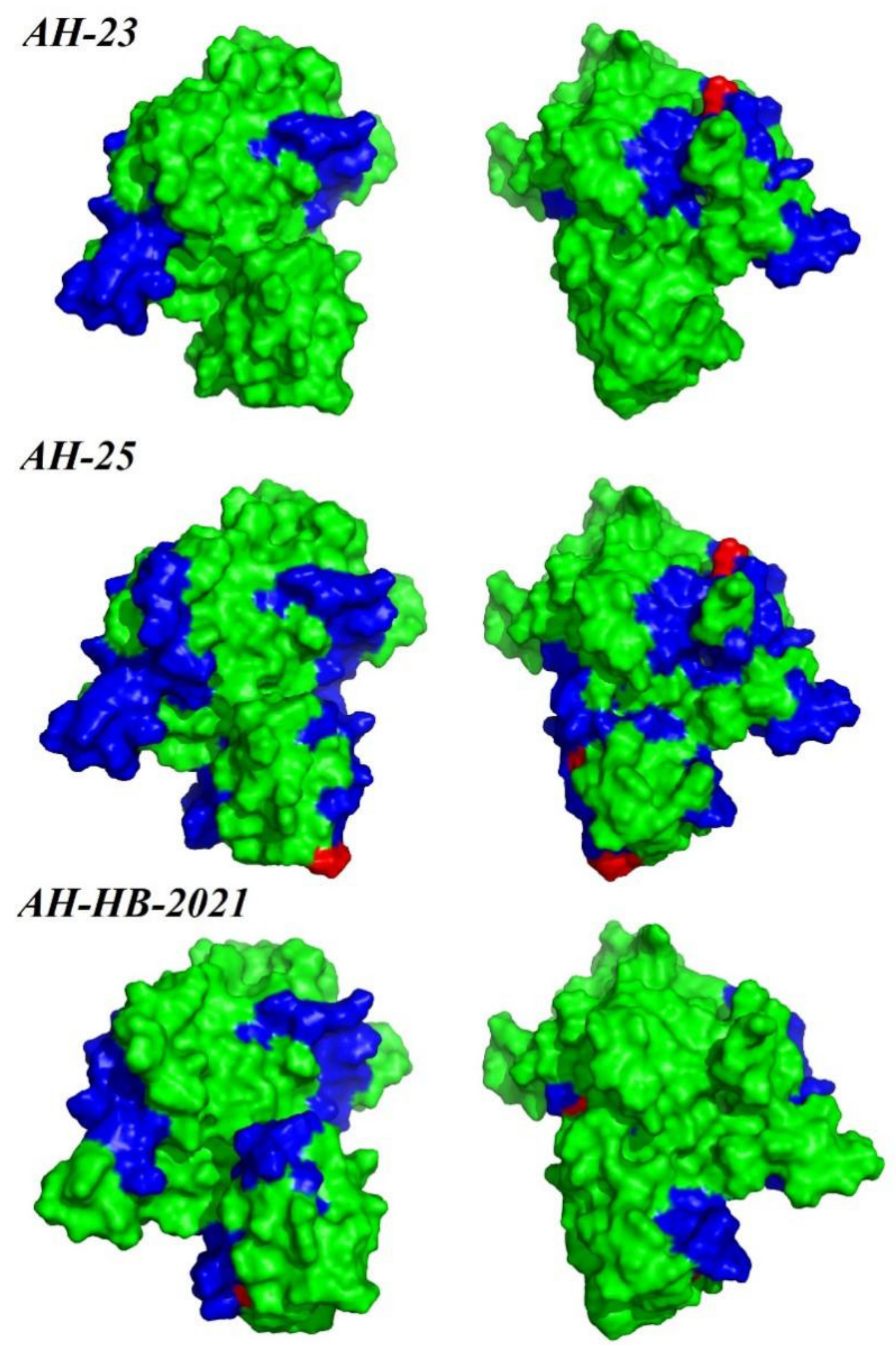
3.4. Recombination, Selection Pressure and B-Cell Antigenic Epitope Analysis
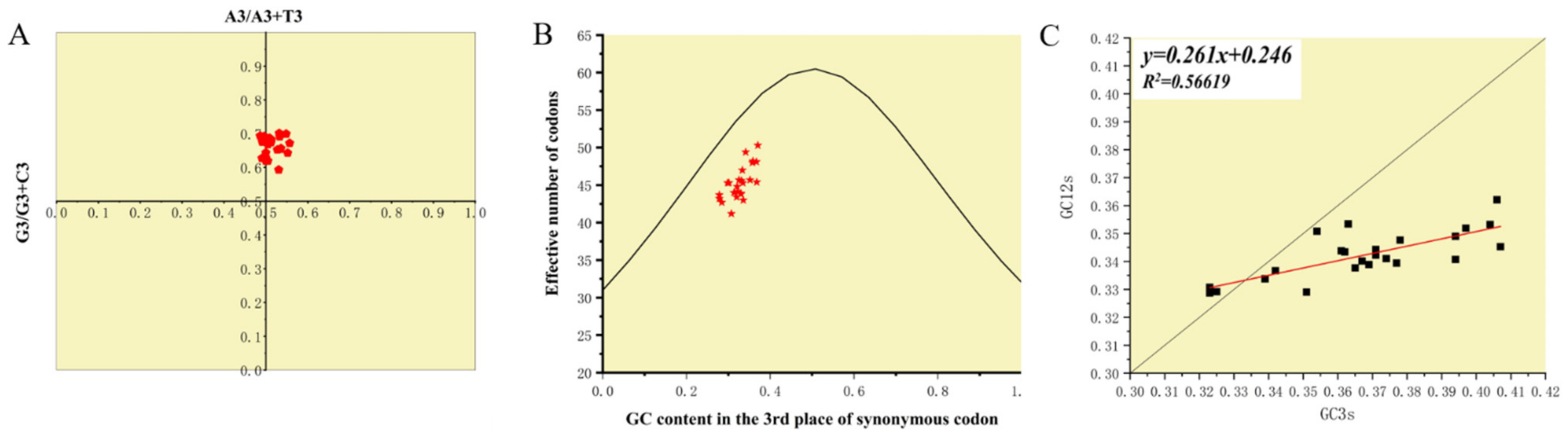
3.5. Codon Usage Bias Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kazlauskas, D.; Varsani, A.; Koonin, E.V.; Krupovic, M. Multiple origins of prokaryotic and eukaryotic single-stranded DNA viruses from bacterial and archaeal plasmids. Nat. Commun. 2019, 10, 3425. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Rosario, K.; Breitbart, M.; Duffy, S. Eukaryotic circular rep-encoding single-stranded DNA (CRESS DNA) viruses: Ubiquitous viruses with small genomes and a diverse host range. Adv. Virus Res. 2018, 103, 71–133. [Google Scholar] [CrossRef] [PubMed]
- Saunders, K.; Bedford, I.D.; Yahara, T.; Stanley, J. The earliest recorded plant virus disease. Nature 2003, 422, 831. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Varsani, A.; Kazlauskas, D.; Breitbart, M.; Delwart, E.; Rosario, K.; Yutin, N.; Wolf, Y.I.; Harrach, B.; Zerbini, F.M.; et al. Cressdnaviricota: A virus phylum unifying seven families of rep-encoding viruses with single-stranded, circular DNA genomes. J. Virol. 2020, 94, 12. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, H.; Ling, Y.; Yang, S.-X.; Wang, X.-C.; Zhou, R.; Xiao, Y.-Q.; Chen, X.; Yang, J.; Fu, W.-G.; et al. Viral metagenomics revealed diverse CRESS-DNA virus genomes in faeces of forest musk deer. Virol. J. 2020, 17, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosario, K.; Breitbart, M.; Harrach, B.; Segalés, J.; Delwart, E.; Biagini, P.; Varsani, A. Revisiting the taxonomy of the family Circoviridae: Establishment of the genus Cyclovirus and removal of the genus Gyrovirus. Arch. Virol. 2017, 162, 1447–1463. [Google Scholar] [CrossRef] [Green Version]
- Varsani, A.; Krupovic, M. Correction to: Smacoviridae: A new family of animal-associated single-stranded DNA viruses. Arch. Virol. 2018, 163, 3213–3214. [Google Scholar] [CrossRef] [Green Version]
- Varsani, A.; Krupovic, M. Smacoviridae: A new family of animal-associated single-stranded DNA viruses. Arch. Virol. 2018, 163, 2005–2015. [Google Scholar] [CrossRef]
- Li, L.; Giannitti, F.; Ullmann, L.S.U.; Deng, X.; Pesavento, P.A.; Delwart, E.; Pusterla, N.; Keyes, C.; Low, J.; Aleman, M. Exploring the virome of diseased horses. J. Gen. Virol. 2015, 96, 2721–2733. [Google Scholar] [CrossRef] [Green Version]
- Rybicki, E. A phylogenetic and evolutionary justification for three genera of Geminiviridae. Arch. Virol. 1994, 139, 49–77. [Google Scholar] [CrossRef]
- Guo, Z.; He, Q.; Tang, C.; Zhang, B.; Yue, H. Identification and genomic characterization of a novel CRESS DNA virus from a calf with severe hemorrhagic enteritis in China. Virus Res. 2018, 255, 141–146. [Google Scholar] [CrossRef]
- Shan, T.; Li, L.; Simmonds, P.; Wang, C.; Moeser, A.; Delwart, E. The fecal virome of pigs on a high-density farm. J. Virol. 2011, 85, 11697–11708. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, X.; Xu, G.; Wang, Z.; Shen, H.; Lian, K.; Lin, Y.; Zheng, J.; Liang, P.; Zhang, L.; et al. Emergence of porcine circovirus-like viruses associated with porcine diarrheal disease in China. Transbound. Emerg. Dis. 2021. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Wang, W.; Cao, L.; Zheng, M.; Zhuang, X.; Zhang, H.; Yu, N.; Tian, M.; Lu, H.; Jin, N. Genetic characterization of three porcine circovirus-like viruses in pigs with diarrhoea in China. Transbound. Emerg. Dis. 2020, 68, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zhou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2019, 20, 348–355. [Google Scholar] [CrossRef]
- Minh, B.Q.; Nguyen, M.A.T.; Von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pond, S.; Frost, S.D.W. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [Green Version]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.; Wertheim, J.O.; Weaver, S.; Murrell, B.; Scheffler, K.; Pond, S.L.K. Less is more: An adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol. Biol. Evol. 2015, 32, 1342–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, B.; Zhang, L.; Liang, S.; Zhang, C. SVMTriP: A method to predict antigenic epitopes using support vector machine to integrate tri-peptide similarity and propensity. PLoS ONE 2012, 7, e45152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sueoka, N. Intrastrand parity rules of DNA base composition and usage biases of synonymous codons. J. Mol. Evol. 1995, 40, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Sueoka, N. Translation-coupled violation of parity rule 2 in human genes is not the cause of heterogeneity of the DNA G+C content of third codon position. Gene 1999, 238, 53–58. [Google Scholar] [CrossRef]
- Tian, H.-F.; Hu, Q.-M.; Xiao, H.-B.; Zeng, L.-B.; Meng, Y.; Li, Z. Genetic and codon usage bias analyses of major capsid protein gene in Ranavirus. Infect. Genet. Evol. 2020, 84, 104379. [Google Scholar] [CrossRef]
- Wang, L.Y.; Xing, H.X.; Yuan, Y.C.; Wang, X.L.; Saeed, M.; Tao, J.C.; Feng, W.; Zhang, G.H.; Song, X.L.; Sun, X.Z. Genome-wide analysis of codon usage bias in four sequenced cotton species. PLoS ONE 2018, 13, e0194372. [Google Scholar]
- Sun, J.; Zhao, W.; Wang, R.; Zhang, W.; Li, G.; Lu, M.; Shao, Y.; Yang, Y.; Wang, N.; Gao, Q.; et al. Analysis of the codon usage pattern of HA and NA genes of H7N9 influenza a virus. Int. J. Mol. Sci. 2020, 21, 7129. [Google Scholar] [CrossRef]
- Sibila, M.; Rocco, C.; Franzo, G.; Huerta, E.; Domingo, M.; Núñez, J.I.; Segalés, J. Genotyping of porcine circovirus 2 (PCV-2) in vaccinated pigs suffering from PCV-2-systemic disease between 2009 and 2020 in Spain. Pathogens 2021, 10, 1016. [Google Scholar] [CrossRef]
- Ghosh, S.; Chakraborty, S. Phylogenomics analysis of SARS-CoV2 Genomes reveals distinct selection pressure on different viral strains. Biomed Res. Int. 2020, 2020, 5746461. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, X.; Li, W.; Cui, Y.Q.; Zhang, D.; Xu, F.Z.; Jiang, S.D.; Zhou, T.H. Phylogenetic analysis and evolution of feline bocavirus in Anhui Province, eastern China. Comp. Immunol. Microb. 2021, 77, 101676. [Google Scholar] [CrossRef]
- Nguyen, V.; Do, H.; Huynh, T.; Park, Y.; Park, B.; Chung, H. Molecular-based detection, genetic characterization and phylogenetic analysis of porcine circovirus 4 from Korean domestic swine farms. Transbound. Emerg. Dis. 2021. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, S.; Xin, C.; Li, C.; Shi, J.; Peng, Z.; Liu, C.; Han, H.; Yang, Y.; Tian, Y.; et al. Mutation of the sixth amino acid of the Rep protein has no effect on porcine circovirus 2b but enhances porcine circovirus 2d replication in vitro. Arch. Virol. 2021, 166, 3189–3192. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Cai, X.; Zhan, Y.; Yuan, X.; Liu, T.; Tan, L.; Li, Y.; Zhang, L.; Yang, L.; Liu, W.; et al. Truncated rep protein of porcine circovirus 2 (PCV2) caused by a naturally occurring mutation reduced virus replication in PK15 cells. BMC Vet. Res. 2019, 15, 248. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Peng, Z.; Fu, F.; Xu, S.; Xu, S.; Cong, X.; Yuan, X.; Yu, J.; Wu, J.; Sun, W.; et al. Mutant Rep protein of the porcine circovirus type 2 N-glycosylation: 23-25aa, 256-258aa mutation reduced virus replication but 286-288aa mutation enhanced virus replication in PK-15 cells. Vet. Microbiol. 2015, 177, 370–372. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Liu, T.; Du, Y.; Cui, X.; Yu, Q.; Wang, Z.; Zhang, J.; Li, Y.; Zhu, Q. A novel genotype VII Newcastle disease virus vaccine candidate generated by mutation in the L and F genes confers improved protection in chickens. Veter.-Microbiol. 2018, 216, 99–106. [Google Scholar] [CrossRef]
- Pang, H.; Li, L.; Liu, H.; Pan, Z. Proline to threonine mutation at position 162 of NS5B of classical swine fever virus vaccine C strain promoted genome replication and infectious virus production by facilitating initiation of RNA synthesis. Viruses 2021, 13, 1523. [Google Scholar] [CrossRef]
- Wang, L.; Han, F.; Duan, H.; Ji, F.; Yan, X.; Fan, Y.; Wang, K. Hepatitis B virus pre-existing drug resistant mutation is related to the genotype and disease progression. J. Infect. Dev. Ctries. 2017, 11, 727–732. [Google Scholar] [CrossRef] [Green Version]
- Franzo, G.; Delwart, E.; Fux, R.; Hause, B.; Su, S.; Zhou, J.Y.; Segales, J. Genotyping porcine circovirus 3 (PCV-3) nowadays: Does it make sense? Viruses 2020, 12, 265. [Google Scholar] [CrossRef] [Green Version]
- Franzo, G.; Segalés, J. Porcine circovirus 2 (PCV-2) genotype update and proposal of a new genotyping methodology. PLoS ONE 2018, 13, e0208585. [Google Scholar] [CrossRef] [Green Version]
- Saporiti, V.; Cruz, T.F.; Correa-Fiz, F.; Núñez, J.I.; Sibila, M.; Segalés, J. Similar frequency of Porcine circovirus 3 (PCV-3) detection in serum samples of pigs affected by digestive or respiratory disorders and age-matched clinically healthy pigs. Transbound. Emerg. Dis. 2019, 67, 199–205. [Google Scholar] [CrossRef]
- Ji, J.; Chen, Q.; Sui, C.; Yu, Z.; Xu, X.; Yao, L.; Kan, Y.; Bi, Y.; Xie, Q. Novel genotype definition and genome characteristics of duck circovirus in central and Eastern China. Transbound. Emerg. Dis. 2020, 67, 2993–3004. [Google Scholar] [CrossRef]
- Wu, X.; Wang, X.; Shi, T.; Luo, L.; Qiao, D.; Wang, Z.; Han, C.; Du, Q.; Tong, D.; Huang, Y. Porcine circovirus type 2 rep enhances IL-10 production in macrophages via activation of p38-MAPK pathway. Viruses 2019, 11, 1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalifeh, A.; Kraberger, S.; Dziewulska, D.; Varsani, A.; Stenzel, T. A pilot study investigating the dynamics of pigeon circovirus recombination in domesticated pigeons housed in a single loft. Viruses 2021, 13, 964. [Google Scholar] [CrossRef]
- Piewbang, C.; Jo, W.K.; Puff, C.; Van Der Vries, E.; Kesdangsakonwut, S.; Rungsipipat, A.; Kruppa, J.; Jung, K.; Baumgärtner, W.; Techangamsuwan, S.; et al. Novel canine circovirus strains from Thailand: Evidence for genetic recombination. Sci. Rep. 2018, 8, 7524. [Google Scholar] [CrossRef]
- Rajkhowa, T.K.; Lalnunthanga, P.; Rao, P.L.; Subbiah, M.; Lalrohlua, B. Emergence of porcine circovirus 2g (PCV2g) and evidence for recombination between genotypes 2g, 2b and 2d among field isolates from non-vaccinated pigs in Mizoram, India. Infect. Genet. Evol. 2021, 90, 104775. [Google Scholar] [CrossRef]
- Stenzel, T.; Dziewulska, D.; Muhire, B.M.; Hartnady, P.; Kraberger, S.; Martin, D.P.; Varsani, A. Recombinant goose circoviruses circulating in domesticated and wild geese in Poland. Viruses 2018, 10, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; He, W.; Zhu, H.; Bi, Y.; Wang, R.; Xing, G.; Zhang, C.; Zhou, J.; Yuen, K.-Y.; Gao, G.F.; et al. Origin, genetic diversity, and evolutionary dynamics of novel porcine circovirus 3. Adv. Sci. 2018, 5, 1800275. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Yin, S.; Shang, Y.; Liu, B.; Yuan, L.; Khan, M.U.Z.; Liu, X.; Cai, J. Phylogenetic and genetic variation analyses of porcine circovirus type 2 isolated from China. Transbound. Emerg. Dis. 2017, 65, e383–e392. [Google Scholar] [CrossRef]
- Yao, X.; Fan, Q.; Yao, B.; Lu, P.; Rahman, S.U.; Chen, D.; Tao, S. Codon usage bias analysis of bluetongue virus causing livestock infection. Front. Microbiol. 2020, 11, 655. [Google Scholar] [CrossRef]
- Wang, L.; Li, Y.; Guo, Z.; Yi, Y.; Zhang, H.; Shangguan, H.; Huang, C.; Ge, J. Genetic changes and evolutionary analysis of canine circovirus. Arch. Virol. 2021, 166, 2235–2247. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Gao, K.; Pi, M.; Li, H.; Zhong, W.; Li, B.; Ning, Z. Phylogenetic and codon usage analysis for replicase and capsid genes of porcine circovirus 3. Veter.-Res. Commun. 2021, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Jia, R.Y.; Zhang, Z.L.; Lu, Y.Y.; Wang, M.S.; Zhu, D.K.; Chen, S.; Liu, M.F.; Yin, Z.Q.; Cheng, A.C. Analysis of syn-onymous codon usage pattern in duck circovirus. Gene 2015, 557, 138–145. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Positive Selection Sites | Threshold |
|---|---|---|
| FUBAR | 29, 142 | p < 0.1 |
| FEL | 29 | |
| MEME | 47, 81, 142, 211, 290, 8, 309, 72, 29 | |
| SLAC | / | Posterior probability of 0.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, K.; Zhang, M.; Liu, Q.; Cao, Y.; Zhang, W.; Liang, Y.; Song, X.; Ji, K.; Shao, Y.; Qi, K.; et al. Epidemiology and Evolution of Emerging Porcine Circovirus-like Viruses in Pigs with Hemorrhagic Dysentery and Diarrhea Symptoms in Central China from 2018 to 2021. Viruses 2021, 13, 2282. https://doi.org/10.3390/v13112282
Yang K, Zhang M, Liu Q, Cao Y, Zhang W, Liang Y, Song X, Ji K, Shao Y, Qi K, et al. Epidemiology and Evolution of Emerging Porcine Circovirus-like Viruses in Pigs with Hemorrhagic Dysentery and Diarrhea Symptoms in Central China from 2018 to 2021. Viruses. 2021; 13(11):2282. https://doi.org/10.3390/v13112282
Chicago/Turabian StyleYang, Kankan, Menghuan Zhang, Qi Liu, Yingli Cao, Wuyin Zhang, Yueqiao Liang, Xiangjun Song, Kaiyuan Ji, Ying Shao, Kezong Qi, and et al. 2021. "Epidemiology and Evolution of Emerging Porcine Circovirus-like Viruses in Pigs with Hemorrhagic Dysentery and Diarrhea Symptoms in Central China from 2018 to 2021" Viruses 13, no. 11: 2282. https://doi.org/10.3390/v13112282
APA StyleYang, K., Zhang, M., Liu, Q., Cao, Y., Zhang, W., Liang, Y., Song, X., Ji, K., Shao, Y., Qi, K., & Tu, J. (2021). Epidemiology and Evolution of Emerging Porcine Circovirus-like Viruses in Pigs with Hemorrhagic Dysentery and Diarrhea Symptoms in Central China from 2018 to 2021. Viruses, 13(11), 2282. https://doi.org/10.3390/v13112282