
Stress Induced Transcription Factors Transactivate the Herpes Simplex Virus 1 Infected Cell Protein 27 (ICP27) Transcriptional Enhancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Virus Infection
2.3. Primers
2.4. Plasmids
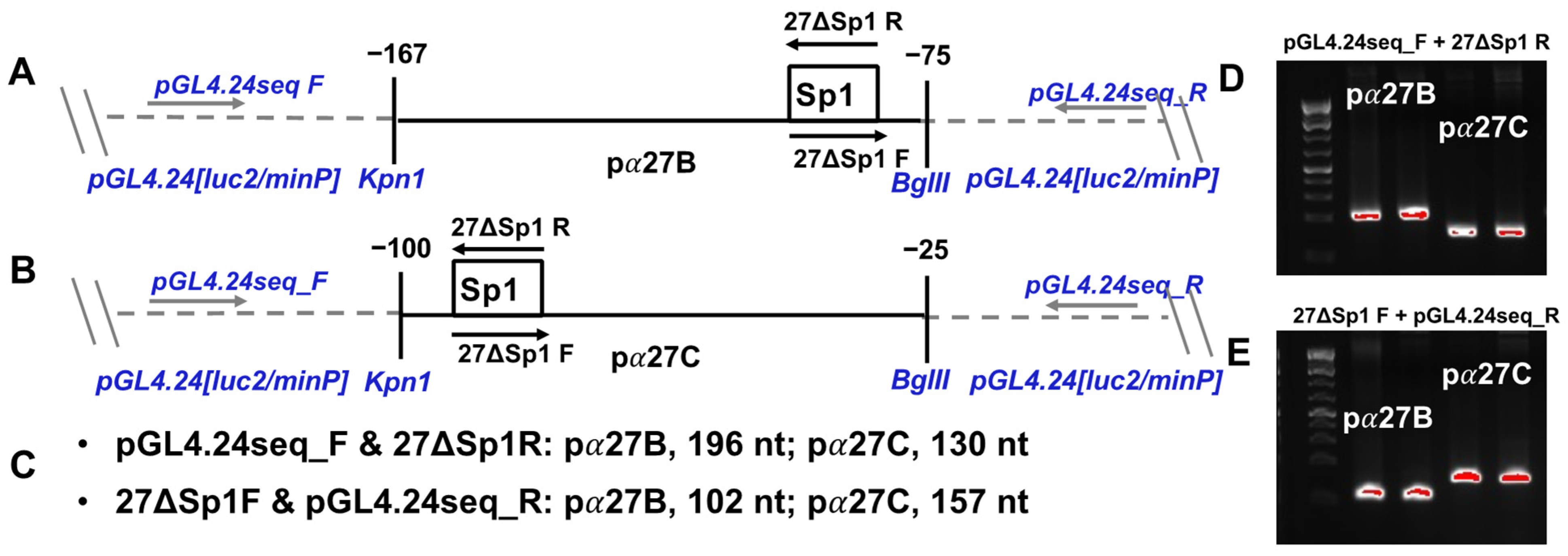
2.5. PCR-Directed Mutagenesis
2.6. Dual Luciferase Assay
2.7. Chromatin Immunoprecipitation
3. Results
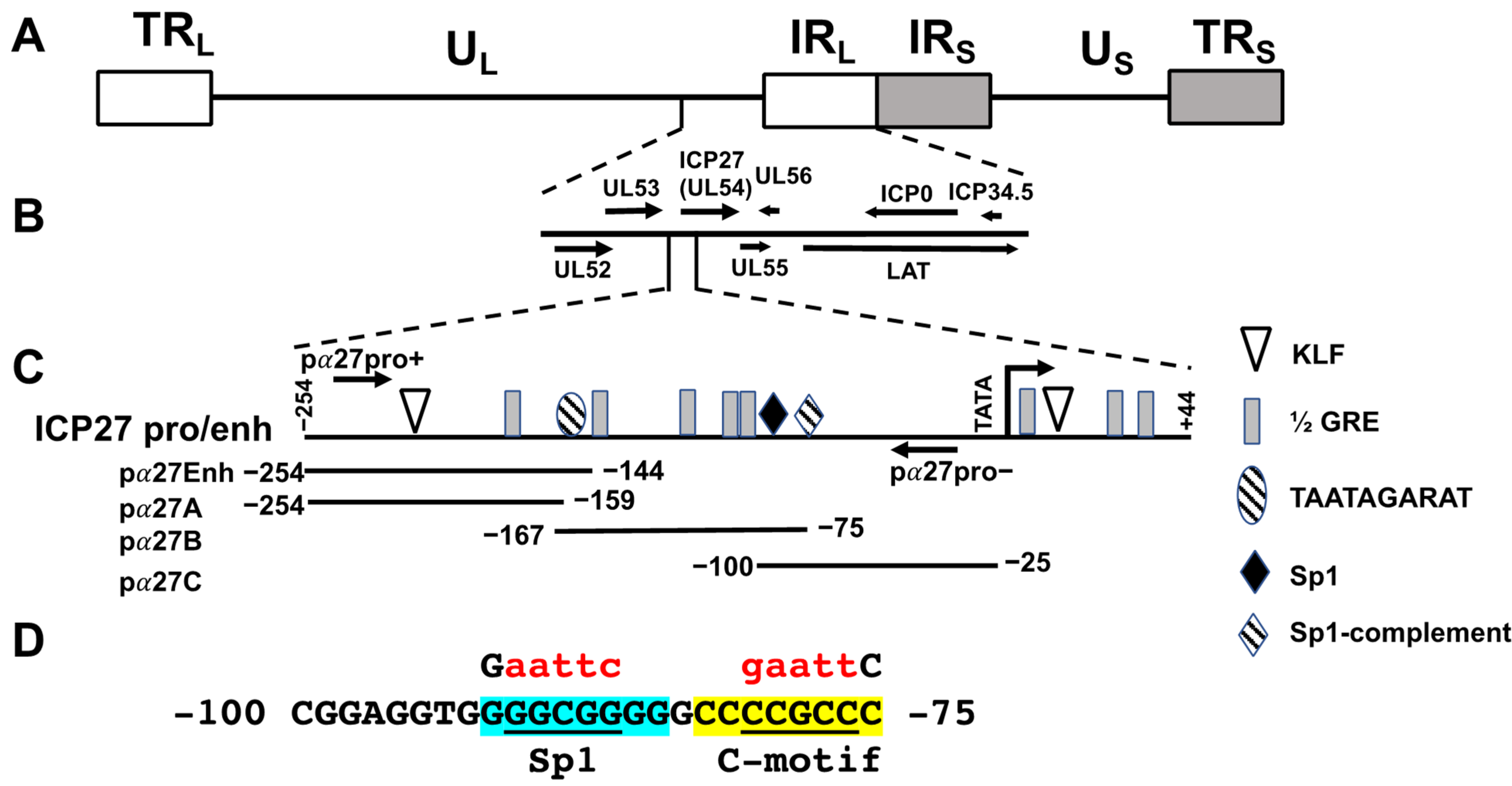
3.1. Identification of Enhancer Sequences in the ICP27 Promoter
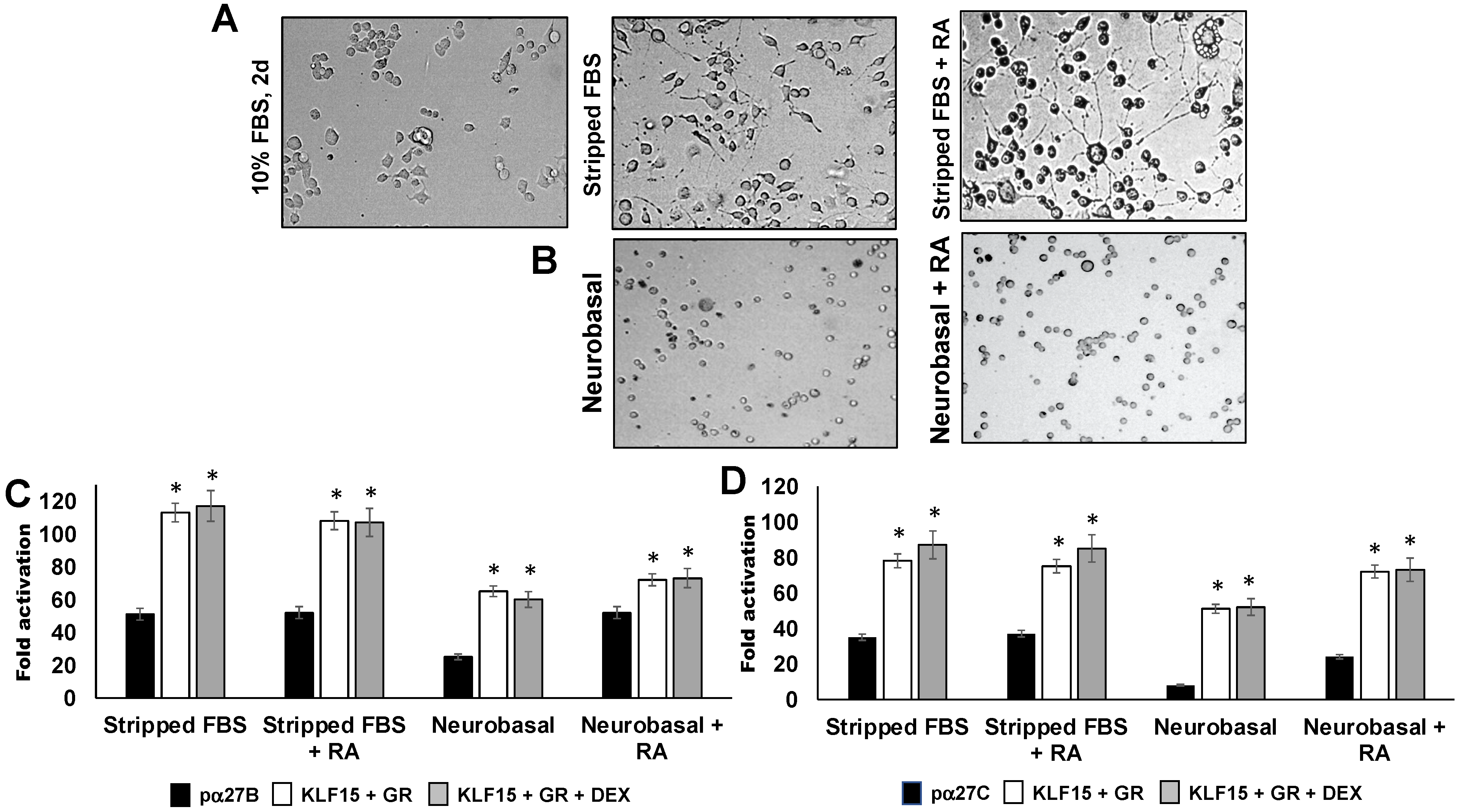
3.2. Influence of Neurite Formation in Neuro-2A Cells on ICP27 Enhancer Activity
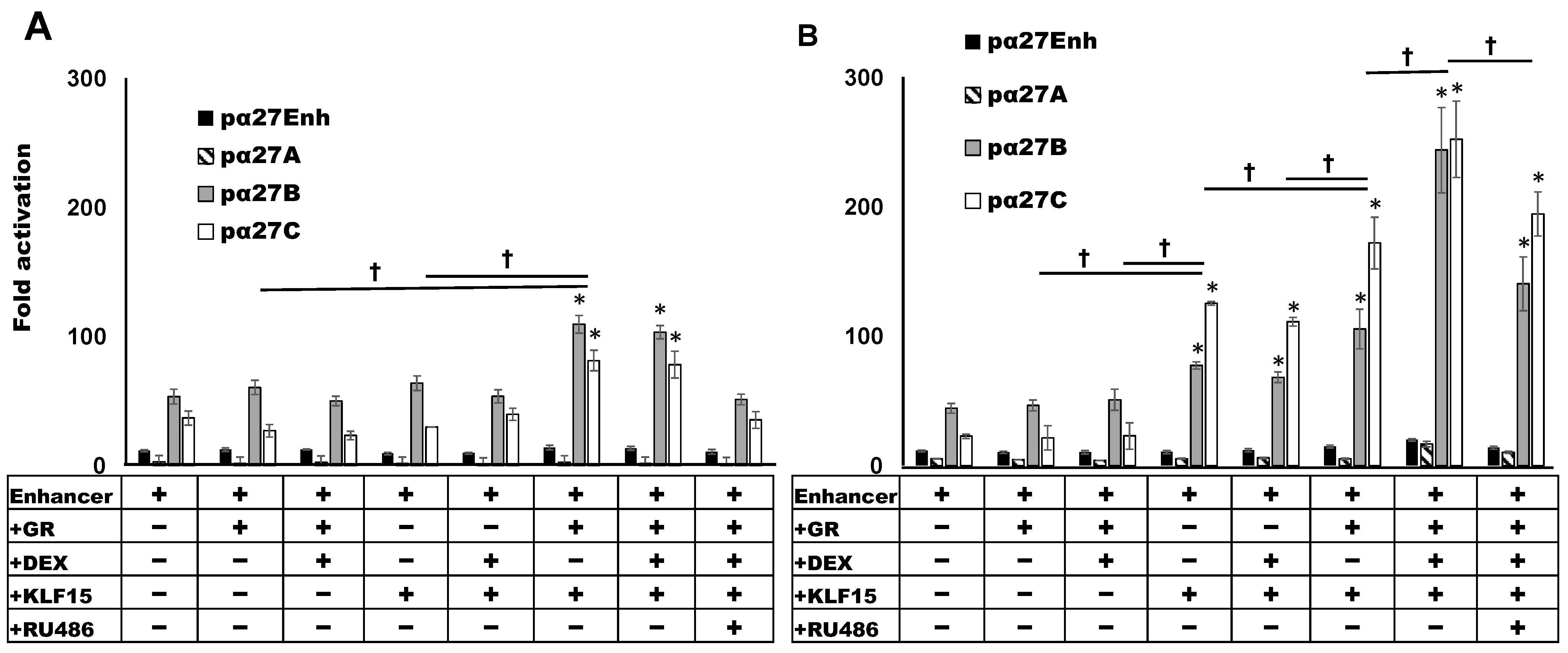
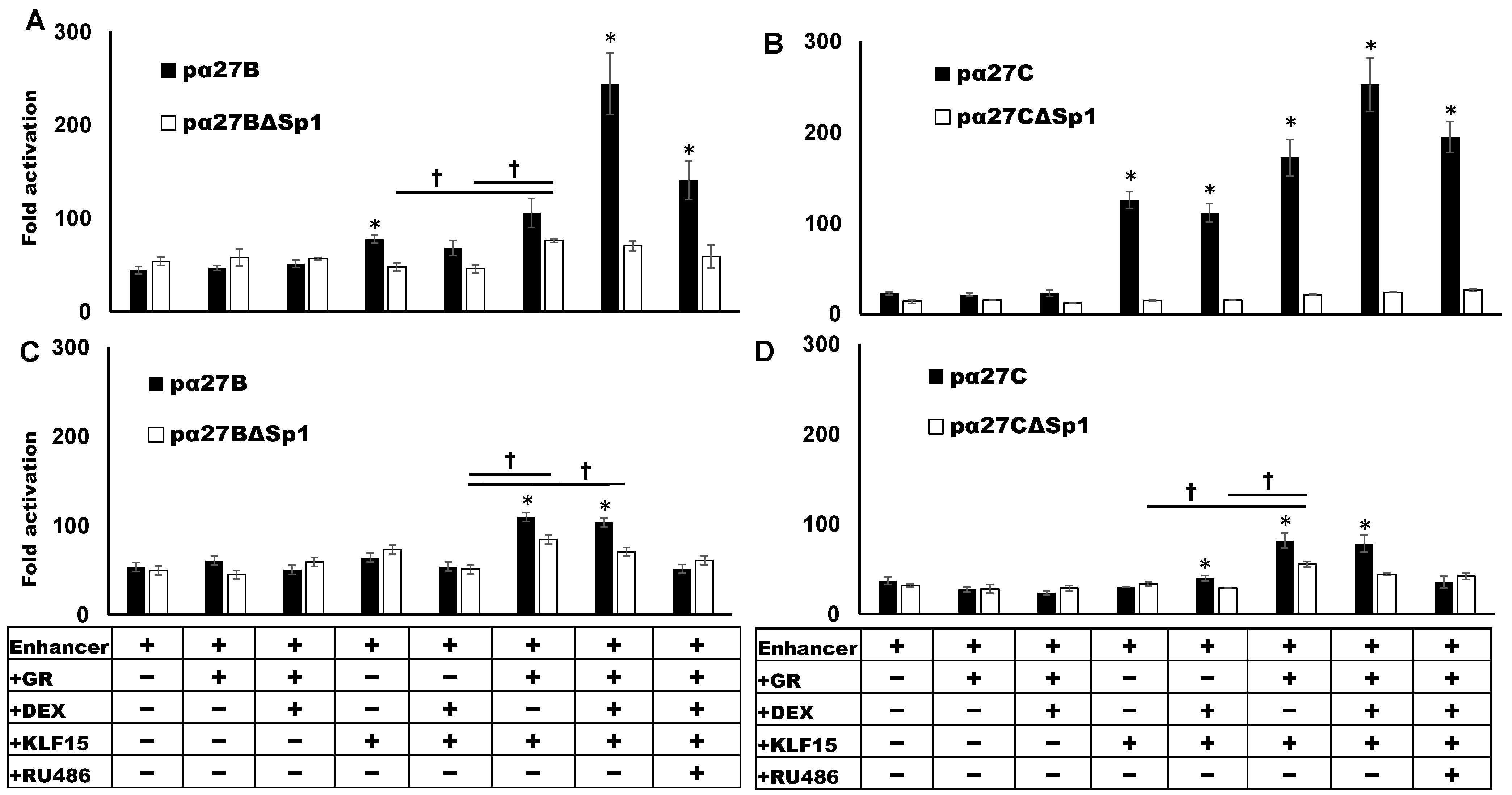
3.3. Sp1 Binding Site Mediates GR- and KLF15-Dependant Transactivation
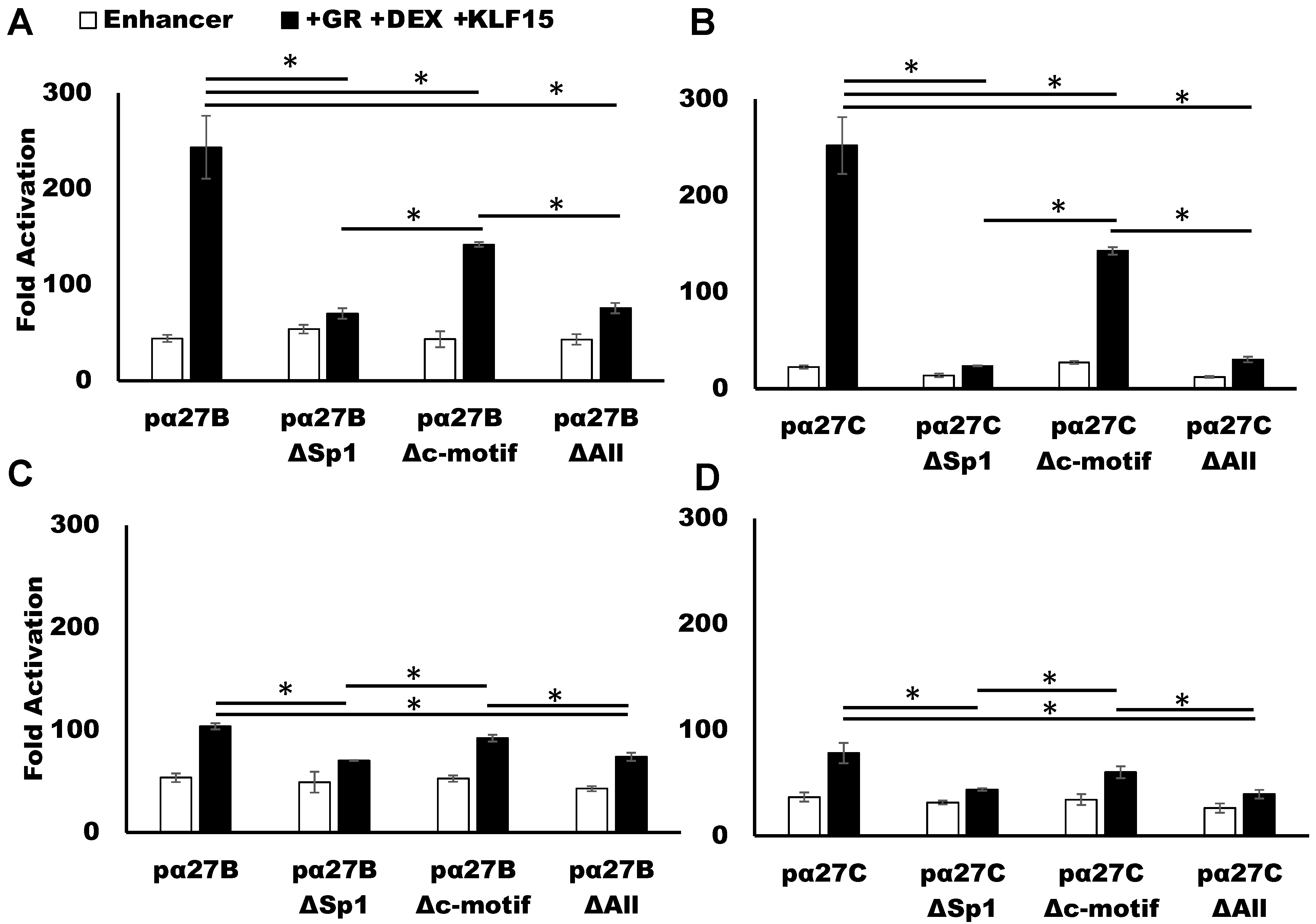
3.4. C-Motif Was Not as Important as Sp1 Binding Site for GR and KLF15-Mediated Transactivation
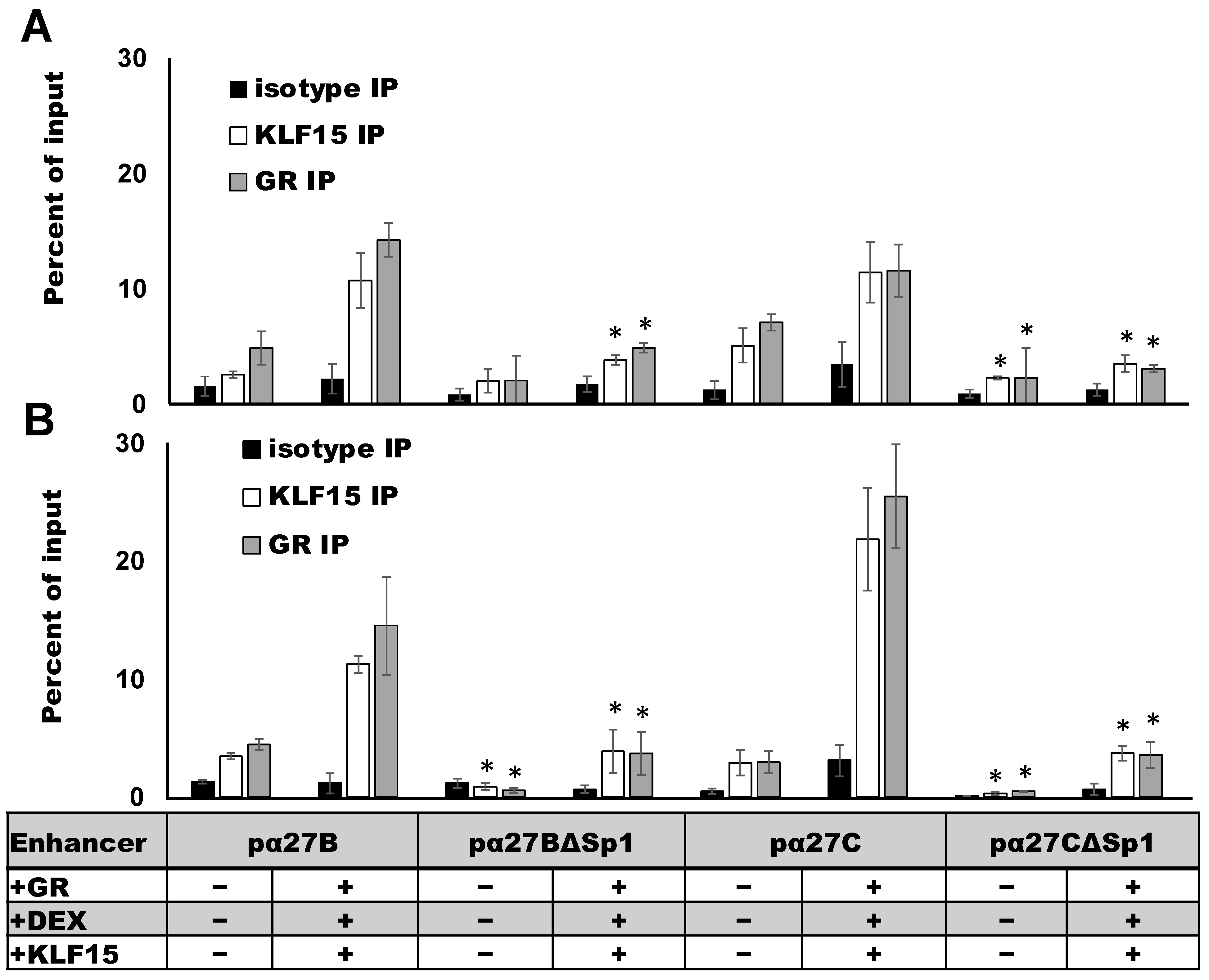
3.5. ICP27 Enhancer Occupancy by GR and KLF15 Requires an Intact Sp1 Binding Site
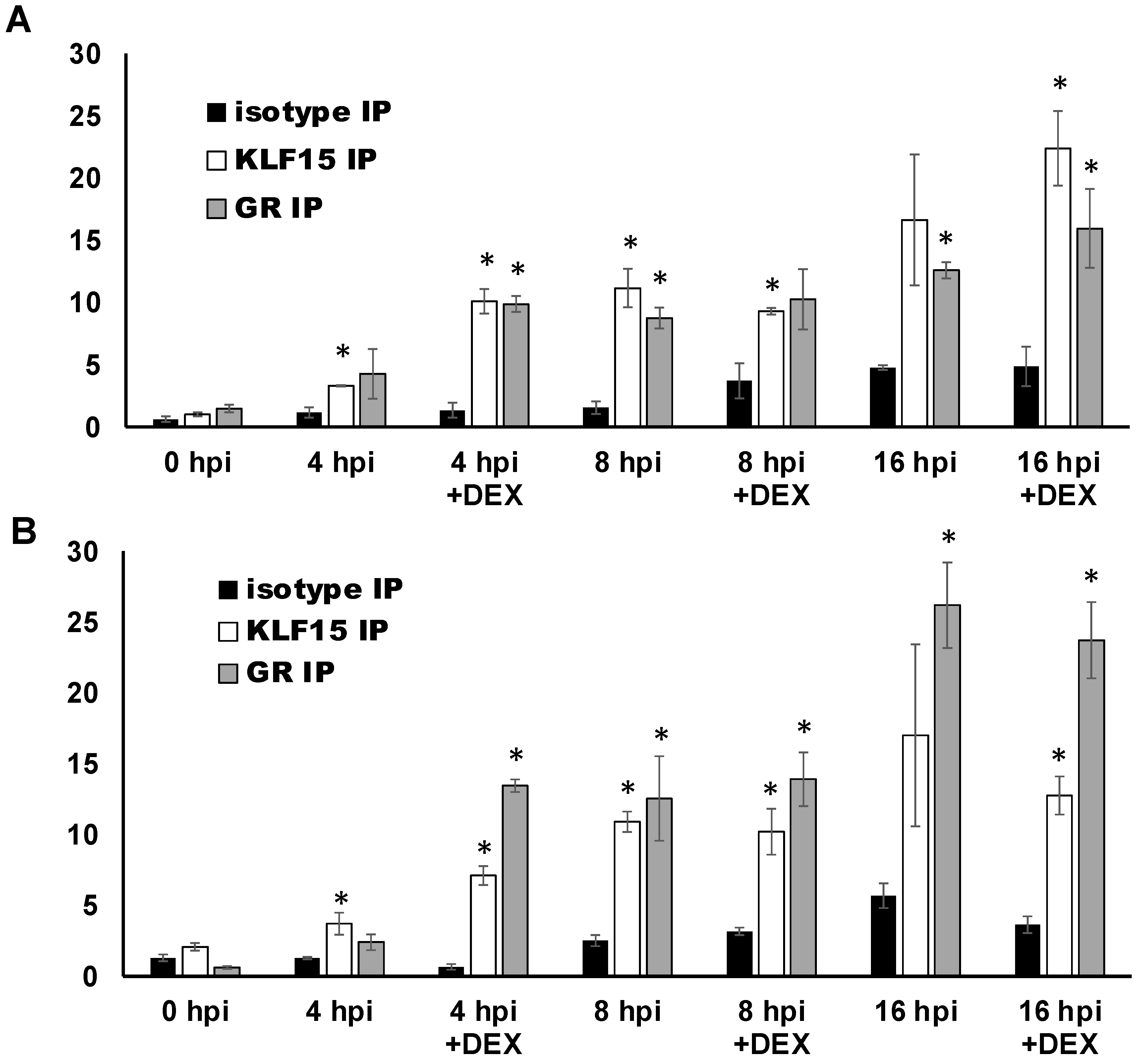
3.6. ICP27 Promoter/Enhancer Sequences Are Occupied by GR and KLF15 during Productive Infection
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liesegang, T.J. Classification of herpes simplex virus keratitis and anterior uveitis. Cornea 1999, 18, 127–143. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Kameyama, T.; Nagaya, S.; Hashizume, Y.; Yoshida, M. Relapsing herpes simplex encephalitis: Pathological confirmation of viral reactivation. J. Neurol. Neurosurg. Psychiatry 2002, 74, 262–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimeld, C.; Efstathiou, S.; Hill, T. Tracking the Spread of a lacZ-Tagged Herpes Simplex Virus Type 1 between the Eye and the Nervous System of the Mouse: Comparison of Primary and Recurrent Infection. J. Virol. 2001, 75, 5252–5262. [Google Scholar] [CrossRef] [Green Version]
- Perng, G.-C.; Jones, C. Towards an understanding of the Herpes Simplex Virus Type 1 latency-reactivation cycle. Interdiscip. Perspect. Infect. Dis. 2010, 2010, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, C. Herpes Simplex Virus Type 1 and Bovine Herpesvirus 1 Latency. Clin. Microbiol. Rev. 2003, 16, 79. [Google Scholar] [CrossRef] [Green Version]
- Phelan, D.; Barrozo, E.R.; Bloom, D.C. HSV1 latent transcription and non-coding RNA: A critical retrospective. J. Neuroimmunol. 2017, 308, 65–101. [Google Scholar] [CrossRef]
- Wechsler, S.L.; Nesburn, A.B.; Watson, R.; Slanina, S.; Ghiasi, H. Fine mapping of the major latency-related RNA of herpes simplex virus type 1 in humans. J. Gen. Virol. 1988, 69 Pt 12, 3101–3106. [Google Scholar] [CrossRef]
- Perng, G.C.; Dunkel, E.C.; Geary, P.A.; Slanina, S.M.; Ghiasi, H.; Kaiwar, R.; Nesburn, A.B.; Wechsler, S.L. The latency-associated transcript gene of herpes simplex virus type 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J. Virol. 1994, 68, 8045–8055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perng, G.C.; Ghiasi, H.; Slanina, S.M.; Nesburn, A.B.; Wechsler, S.L. The spontaneous reactivation function of the herpes simplex virus type 1 LAT gene resides completely within the first 1.5 kilobases of the 8.3-kilobase primary transcript. J. Virol. 1996, 70, 976–984. [Google Scholar] [CrossRef] [Green Version]
- Perng, G.C.; Slanina, S.M.; Ghiasi, H.; Nesburn, A.B.; Wechsler, S.L. A 371-nucleotide region between the herpes simplex virus type 1 (HSV-1) LAT promoter and the 2-kilobase LAT is not essential for efficient spontaneous reactivation of latent HSV-1. J. Virol. 1996, 70, 2014–2018. [Google Scholar] [CrossRef] [Green Version]
- Perng, G.C.; Slanina, S.M.; Yukht, A.; Ghiasi, H.; Nesburn, A.B.; Wechsler, S.L. The latency-associated transcript gene enhances establishment of herpes simplex virus type 1 latency in rabbits. J. Virol. 2000, 74, 1885–1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaser, R.; Kiecolt-Glaser, J.K.; Speicher, C.E.; Holliday, J.E. Stress, loneliness, and changes in herpesvirus latency. J. Behav. Med. 1985, 8, 249–260. [Google Scholar] [CrossRef]
- Padgett, D.A.; Sheridan, J.F.; Dorne, J.; Berntson, G.G.; Candelora, J.; Glaser, R. Social stress and the reactivation of latent herpes simplex virus type 1. Proc. Natl. Acad. Sci. USA 1998, 95, 7231–7235. [Google Scholar] [CrossRef] [Green Version]
- Noisakran, S.; Halford, W.P.; Veress, L.; Carr, D.J. Role of the hypothalamic pituitary adrenal axis and IL-6 in stress-induced reactivation of latent herpes simplex virus type 1. J. Immunol. 1998, 160, 5441–5447. [Google Scholar]
- Glaser, R.; Kiecolt-Glaser, J.K. Stress-induced immune dysfunction: Implications for health. Nat. Rev. Immunol. 2005, 5, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, L.; Meadows, J.; Catalan, J.; Barton, S. Are reported stress and coping style associated with frequent recurrence of genital herpes? Genitourin. Med. 1997, 73, 263–266. [Google Scholar] [CrossRef]
- Oakley, R.H.; Cidlowski, J.A. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 2013, 132, 1033–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funder, J.W. Glucocorticoids and mineralocorticoid receptors: Biology and clinical relevance. Annu. Rev. Med. 1997, 48, 231–240. [Google Scholar] [CrossRef] [Green Version]
- Workman, A.; Eudy, J.; Smith, L.; Frizzo da Silva, L.; Sinani, D.; Bricker, H.; Cook, E.; Doster, A.; Jones, C. Cellular Transcription Factors Induced in Trigeminal Ganglia during Dexamethasone-Induced Reactivation from Latency Stimulate Bovine Herpesvirus 1 Productive Infection and Certain Viral Promoters. J. Virol. 2012, 86, 2459–2473. [Google Scholar] [CrossRef] [Green Version]
- Sinani, D.; Cordes, E.; Workman, A.; Thunuguntia, P.; Jones, C. Stress-induced cellular transcription factors expressed in trigeminal ganglionic neurons stimulate the herpes simplex virus 1 ICP0 promoter. J. Virol. 2013, 87, 13042–13047. [Google Scholar] [CrossRef] [Green Version]
- Black, A.R.; Black, J.D.; Azizkhan-Clifford, J. Sp1 and kruppel-like factor family of transcription factors in cell growth regulation and cancer. J. Cell. Physiol. 2001, 188, 143–160. [Google Scholar] [CrossRef]
- Kaczynski, J.; Cook, T.; Urrutia, R. Sp1-and Krüppel-like transcription factors. Genome Biol. 2003, 4, 206. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.A.; Tjian, R. Sp1 binds to promoter sequences and activates herpes simplex virus ‘immediate-early’gene transcription in vitro. Nature 1985, 317, 179–182. [Google Scholar] [CrossRef]
- Ostler, J.B.; Harrison, K.S.; Schroeder, K.; Thunuguntla, P.; Jones, C. The Glucocorticoid Receptor (GR) Stimulates Herpes Simplex Virus 1 Productive Infection, in Part Because the Infected Cell Protein 0 (ICP0) Promoter Is Cooperatively Transactivated by the GR and Krüppel-Like Transcription Factor 15. J. Virol. 2019, 93, e02063-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostler, J.B.; Thunuguntla, P.; Hendrickson, B.Y.; Jones, C. Transactivation of Herpes Simplex Virus 1 (HSV-1) Infected Cell Protein 4 Enhancer by Glucocorticoid Receptor and Stress-Induced Transcription Factors Requires Overlapping Krüppel-Like Transcription Factor 4/Sp1 Binding Sites. J. Virol. 2021, 95, e01776-20. [Google Scholar] [CrossRef]
- Mangan, S.; Alon, U. Structure and function of the feed-forward loop network motif. Proc. Natl. Acad. Sci. USA 2003, 100, 11980–11985. [Google Scholar] [CrossRef] [Green Version]
- Sasse, S.K.; Zuo, Z.; Kadiyala, V.; Zhang, L.; Pufall, M.A.; Jain, M.K.; Phang, T.L.; Stormo, G.D.; Gerber, A.N. Response element composition governs correlations between binding site affinity and transcription in glucocorticoid receptor feed-forward loops. J. Biol. Chem. 2015, 290, 19756–19769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasse, S.K.; Mailloux, C.M.; Barczak, A.J.; Wang, Q.; Altonsy, M.O.; Jain, M.K.; Haldar, S.M.; Gerber, A.N. The glucocorticoid receptor and KLF15 regulate gene expression dynamics and integrate signals through feed-forward circuitry. Mol. Cell. Biol. 2013, 33, 2104–2115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacks, W.R.; Greene, C.C.; Aschman, D.P.; Schaffer, P.A. Herpes simplex virus type 1 ICP27 is an essential regulatory protein. J. Virol. 1985, 55, 796–805. [Google Scholar] [CrossRef] [Green Version]
- Sandri-Goldin, R.M. The many roles of the highly interactive HSV protein ICP27, a key regulator of infection. Future Microbiol. 2011, 6, 1261–1277. [Google Scholar] [CrossRef]
- Ote, I.; Piette, J.; Sadzot-Delvaux, C. The Varicella-Zoster virus IE4 protein: A conserved member of the herpesviral mRNA export factors family and a potential alternative target in antiherpetic therapies. Biochem. Pharmacol. 2010, 80, 1973–1980. [Google Scholar] [CrossRef]
- Singh, M.; Fraefel, C.; Bello, L.J.; Lawrence, W.C.; Schwyzer, M. Identification and characterization of BICP27, an early protein of bovine herpesvirus 1 which may stimulate mRNA 3′ processing. J. Gen. Virol. 1996, 77 Pt 4, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Holden, V.R.; Smith, R.H.; O’Callaghan, D.J. Regulatory function of the equine herpesvirus 1 ICP27 gene product. J. Virol. 1995, 69, 2786–2793. [Google Scholar] [CrossRef] [Green Version]
- Winkler, M.; Rice, S.A.; Stamminger, T. UL69 of human cytomegalovirus, an open reading frame with homology to ICP27 of herpes simplex virus, encodes a transactivator of gene expression. J. Virol. 1994, 68, 3943–3954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, J.L.; Swaminathan, S.; Silverstein, S.J. The Epstein-Barr virus SM protein is functionally similar to ICP27 from herpes simplex virus in viral infections. J. Virol. 2002, 76, 9420–9433. [Google Scholar] [CrossRef] [Green Version]
- Rutkowski, A.J.; Erhard, F.; L’Hernault, A.; Bonfert, T.; Schilhabel, M.; Crump, C.; Rosenstiel, P.; Efstathiou, S.; Zimmer, R.; Friedel, C.C.; et al. Widespread disruption of host transcription termination in HSV-1 infection. Nat. Commun. 2015, 6, 7126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Liu, L.; Whisnant, A.W.; Hennig, T.; Djakovic, L.; Haque, N.; Bach, C.; Sandri-Goldin, R.M.; Erhard, F.; Friedel, C.C.; et al. Mechanism and consequences of herpes simplex virus 1-mediated regulation of host mRNA alternative polyadenylation. PLoS Genet. 2021, 17, e1009263. [Google Scholar] [CrossRef]
- Wang, X.; Hennig, T.; Whisnant, A.W.; Erhard, F.; Prusty, B.K.; Friedel, C.C.; Forouzmand, E.; Hu, W.; Erber, L.; Chen, Y.; et al. Herpes simplex virus blocks host transcription termination via the bimodal activities of ICP27. Nat. Commun. 2020, 11, 293. [Google Scholar] [CrossRef] [Green Version]
- He, T.; Wang, M.; Cheng, A.; Yang, Q.; Wu, Y.; Jia, R.; Liu, M.; Zhu, D.; Chen, S.; Zhang, S.; et al. Host shutoff activity of VHS and SOX-like proteins: Role in viral survival and immune evasion. Virol. J. 2020, 17, 68. [Google Scholar] [CrossRef]
- Taddeo, B.; Zhang, W.; Roizman, B. The herpes simplex virus host shutoff RNase degrades cellular and viral mRNAs made before infection but not viral mRNA made after infection. J. Virol. 2013, 87, 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Taddeo, B.; Zhang, W.; Roizman, B. Role of herpes simplex virus ICP27 in the degradation of mRNA by virion host shutoff RNase. J. Virol. 2010, 84, 10182–10190. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Patel, A.; Krause, P.R. Herpes simplex virus ICP27 regulates alternative pre-mRNA polyadenylation and splicing in a sequence-dependent manner. Proc. Natl. Acad. Sci. USA 2016, 113, 12256–12561. [Google Scholar] [CrossRef] [Green Version]
- da Silva, L.F.; Sinani, D.; Jones, C. ICP27 protein encoded by bovine herpesvirus type 1 (bICP27) interferes with promoter activity of the bovine genes encoding beta interferon 1 (IFN-beta1) and IFN-beta3. Virus Res. 2012, 169, 162–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, L.A.; Li, L.; Sandri-Goldin, R.M. The cellular RNA export receptor TAP/NXF1 is required for ICP27-mediated export of herpes simplex virus 1 RNA, but the TREX complex adaptor protein Aly/REF appears to be dispensable. J. Virol. 2009, 83, 6335–6346. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.A.; Sandri-Goldin, R.M. Efficient nuclear export of herpes simplex virus 1 transcripts requires both RNA binding by ICP27 and ICP27 interaction with TAP/NXF1. J. Virol. 2009, 83, 1184–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, I.H.; Sciabica, K.S.; Sandri-Goldin, R.M. ICP27 interacts with the RNA export factor Aly/REF to direct herpes simplex virus type 1 intronless mRNAs to the TAP export pathway. J. Virol. 2002, 76, 12877–12889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbin-Lickfett, K.A.; Rojas, S.; Li, L.; Cocco, M.J.; Sandri-Goldin, R.M. ICP27 phosphorylation site mutants display altered functional interactions with cellular export factors Aly/REF and TAP/NXF1 but are able to bind herpes simplex virus 1 RNA. J. Virol. 2010, 84, 2212–2222. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Katyal, A. Data on retinoic acid and reduced serum concentration induced differentiation of Neuro-2a neuroblastoma cells. Data Brief 2018, 21, 2435–2440. [Google Scholar] [CrossRef]
- Harrison, K.S.; Zhu, L.; Thunuguntla, P.; Jones, C. Antagonizing the Glucocorticoid Receptor Impairs Explant-Induced Reactivation in Mice Latently Infected with Herpes Simplex Virus 1. J. Virol. 2019, 93, e00418-19. [Google Scholar] [CrossRef] [Green Version]
- Ishii, T.M.; Zerr, P.; Xia, X.M.; Bond, C.T.; Maylie, J.; Adelman, J.P. Site-directed mutagenesis. Methods Enzymol. 1998, 293, 53–71. [Google Scholar] [CrossRef]
- Weiner, M.P.; Costa, G.L.; Schoettlin, W.; Cline, J.; Mathur, E.; Bauer, J.C. Site-directed mutagenesis of double-stranded DNA by the polymerase chain reaction. Gene 1994, 151, 119–123. [Google Scholar] [CrossRef]
- Sawant, L.; Wijesekera, N.; Jones, C. Pioneer transcription factors, progesterone receptor and Krüppel like transcription factor 4, cooperatively stimulate the bovine herpesvirus 1 ICP0 early promoter and productive late protein expression. Virus Res. 2020, 288, 198115. [Google Scholar] [CrossRef]
- Sawant, L.; Thunuguntla, P.; Jones, C. Cooperative activation of bovine herpesvirus 1 productive infection and viral regulatory promoters by androgen receptor and Krüppel-like transcription factors 4 and 15. Virology 2021, 552, 63–72. [Google Scholar] [CrossRef]
- El-Mayet, F.S.; Sawant, L.; Thunuguntla, P.; Jones, C. Combinatorial effects of the glucocorticoid receptor and Krüppel-like transcription factor 15 on bovine herpesvirus 1 transcription and productive infection. J. Virol. 2017, 91, e00904–e00917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-mayet, F.S.; Sawant, L.; Thunuguntla, P.; Zhao, J.; Jones, C. Two Pioneer Transcription Factors, Krüppel-Like Transcription Factor 4 and Glucocorticoid Receptor, Cooperatively Transactivate the Bovine Herpesvirus 1 ICP0 Early Promoter and Stimulate Productive Infection. J. Virol. 2020, 94, 1670–1689. [Google Scholar] [CrossRef] [PubMed]
- Whitlow, Z.W.; Kristie, T.M. Recruitment of the Transcriptional Coactivator HCF-1 to Viral Immediate-Early Promoters during Initiation of Reactivation from Latency of Herpes Simplex Virus Type 1. J. Virol. 2009, 83, 9591–9595. [Google Scholar] [CrossRef] [Green Version]
- Kristie, T.M.; Roizman, B. Differentiation and DNA contact points of host proteins binding at the cis site for virion-mediated induction of alpha genes of herpes simplex virus 1. J. Virol. 1988, 62, 1145–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristie, T.M.; Roizman, B. Host cell proteins bind to the cis-acting site required for virion-mediated induction of herpes simplex virus 1 alpha genes. Proc. Natl. Acad. Sci. USA 1987, 84, 71–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremblay, R.G.; Sikorska, M.; Sandhu, J.K.; Lanthier, P.; Ribecco-Lutkiewicz, M.; Bani-Yaghoub, M. Differentiation of mouse Neuro 2A cells into dopamine neurons. J. Neurosci. Methods 2010, 186, 60–67. [Google Scholar] [CrossRef]
- Bardon, S.; Vignon, F.; Chalbos, D.; Rochefort, H. RU486, a progestin and glucocorticoid antagonist, inhibits the growth of breast cancer cells via the progesterone receptor. J. Clin. Endocrinol. Metab. 1985, 60, 692–697. [Google Scholar] [CrossRef]
- Pandit, S.; Geissler, W.; Harris, G.; Sitlani, A. Allosteric effects of dexamethasone and RU486 on glucocorticoid receptor-DNA interactions. J. Biol. Chem. 2002, 277, 1538–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, A.J.; Bechberger, J.; Lidington, D.; Galipeau, J.; Laird, D.W.; Naus, C.C. Neuronal Differentiation and Growth Control of Neuro-2a Cells After Retroviral Gene Delivery of Connexin. J. Biol. Chem. 2000, 275, 34407–34414. [Google Scholar] [CrossRef] [Green Version]
- Sinani, D.; Frizzo da Silva, L.; Jones, C. A bovine herpesvirus 1 protein expressed in latently infected neurons (ORF2) promotes neurite sprouting in the presence of activated Notch1 or Notch3. J. Virol. 2013, 87, 1183–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thunuguntla, P.; El-Mayet, F.S.; Jones, C. Bovine herpesvirus 1 can efficiently infect the human (SH-SY5Y) but not the mouse neuroblastoma cell line (Neuro-2A). Virus Res. 2017, 232, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Zhu, L.; Wijesekera, N.; Jones, C. Specific Akt family members impair stress mediated transactivation of viral promoters and enhance neuronal differentiation: Important functions for maintaining latency. J. Virol. 2020, 94, e00901–e00920. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jones, C. Regulation of Notch-mediated transcription by a bovine herpesvirus 1 encoded protein (ORF2) that is expressed in latently infected sensory neurons. J. Neurovirol. 2016, 22, 518–528. [Google Scholar] [CrossRef]
- Liu, Z.N.; Cidlowski, J.A. Glucocorticoid receptor isoforms generate transcription specificity. Trends Cell Biol. 2006, 16, 301–307. [Google Scholar] [CrossRef]
- El-mayet, F.; El-Habbaa, A.S.; El-Bagoury, G.F.; Sharawi, S.S.A.; El-Nahas, E.M.; Jones, C. Transcription Factors Have the Potential to Synergistically Stimulate Bovine Herpesvirus 1 Transcription and Reactivation from Latency. In Transcriptional and Post-Transcriptional Regulation; Kais, G., Ed.; Intech: Rejeka, Croatia, 2018; Volume 1, pp. 36–54. [Google Scholar]
- Richard-Foy, H.; Hagera, G.L. Sequence-specific positioning of nucleosomes over the steroid-inducible MMTV promoter. EMBO J. 1987, 6, 2321–2328. [Google Scholar] [CrossRef]
- Galliher-Beckley, A.J.; Williams, J.G.; Cidlowski, J.A. Ligand-Independent Phosphorylation of the Glucocorticoid Receptor Integrates Cellular Stress Pathways with Nuclear Receptor Signaling. Mol. Cell. Biol. 2011, 31, 4663–4675. [Google Scholar] [CrossRef] [Green Version]
- O’Malley, B.; Schrader, W.T.; Mani, S.; Smith, C.; Weigel, N.L.; Conneely, O.M.; Clark, J.H. An alternative ligand-independent pathway for activation of steroid receptors. Recent Prog. Horm. Res. 1995, 50, 333–347. [Google Scholar]
- Rooney, J.T.; Straus, S.E.; Mannix, M.L.; Wohlenberg, C.R.; Banks, S.; Jagannath, S.; Brauer, J.E.; Notkins, A.L. UV light-induced reactivation of herpes simplex virus type 2 and prevention by acyclovir. J. Infect. Dis. 1992, 166, 500–506. [Google Scholar] [CrossRef]
- Jones, C. Alphaherpesvirus Latency: Its Role in Disease and Survival of the Virus in Nature. In Advances in Virus Research; Maramorosch, K., Murphy, F.A., Shatkin, A.J., Eds.; Academic Press: Cambridge, MD, USA, 1998; Volume 51, pp. 81–133. [Google Scholar]
- Kushnir, A.S.; Davido, D.J.; Schaffer, P.A. Role of nuclear factor Y in stress-induced activation of the herpes simplex virus type 1 ICP0 promoter. J. Virol. 2010, 84, 188–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennesch, M.A.; Picard, D. Minireview: Tipping the Balance: Ligand-Independent Activation of Steroid Receptors. Mol. Endocrinol. 2015, 29, 349–363. [Google Scholar] [CrossRef] [Green Version]
- Davies, L.; Karthikeyan, N.; Lynch, J.T.; Sial, E.-A.; Gkourtsa, A.; Demonacos, C.; Krstic-Demonacos, M. Cross Talk of Signaling Pathways in the Regulation of the Glucocorticoid Receptor Function. Mol. Endocrinol. 2018, 22, 1331–1344. [Google Scholar] [CrossRef] [Green Version]
- Skobowiat, C.; Sayre, R.M.; Dowdy, J.C.; Slominski, A.T. Ultraviolet radiation regulates cortisol activity in a waveband dependent manner in human skin ex-vivo. Br. J. Dermatol. 2013, 168, 595–601. [Google Scholar] [CrossRef] [Green Version]
- Eickelberg, O.; Roth, M.; Lorx, R.; Bruce, V.; Rudiger, J.; Johnson, M.; Blocki, L.-H. Ligand-independent Activation of the Glucocorticoid Receptor by B2-Adrenergic Receptor Agonists in Primary Human Lung Fibroblasts and Vascular Smooth Muscle Cells. J. Biol. Chem. 1999, 274, 1005–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory action of glucocorticoids—New mechanisms of old drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [Green Version]
- Erlandsson, A.C.; Bladh, L.G.; Stierna, P.; Yucel-Lindberg, T.; Hammarsten, O.; Modéer, T.; Harmenberg, J.; Wikström, A.C. Herpes simplex virus type 1 infection and glucocorticoid treatment regulate viral yield, glucocorticoid receptor and NF-kappaB levels. J. Endocrinol. 2002, 175, 165–176. [Google Scholar] [CrossRef] [Green Version]
- El-Mayet, F.S.; Harrison, K.S.; Jones, C. Regulation of Krüppel-Like Factor 15 Expression by Herpes Simplex Virus Type 1 or Bovine Herpesvirus 1 Productive Infection. Viruses 2021, 13, 1148. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ostler, J.B.; Jones, C. Stress Induced Transcription Factors Transactivate the Herpes Simplex Virus 1 Infected Cell Protein 27 (ICP27) Transcriptional Enhancer. Viruses 2021, 13, 2296. https://doi.org/10.3390/v13112296
Ostler JB, Jones C. Stress Induced Transcription Factors Transactivate the Herpes Simplex Virus 1 Infected Cell Protein 27 (ICP27) Transcriptional Enhancer. Viruses. 2021; 13(11):2296. https://doi.org/10.3390/v13112296
Chicago/Turabian StyleOstler, Jeffery B., and Clinton Jones. 2021. "Stress Induced Transcription Factors Transactivate the Herpes Simplex Virus 1 Infected Cell Protein 27 (ICP27) Transcriptional Enhancer" Viruses 13, no. 11: 2296. https://doi.org/10.3390/v13112296
APA StyleOstler, J. B., & Jones, C. (2021). Stress Induced Transcription Factors Transactivate the Herpes Simplex Virus 1 Infected Cell Protein 27 (ICP27) Transcriptional Enhancer. Viruses, 13(11), 2296. https://doi.org/10.3390/v13112296