
The A179L Gene of African Swine Fever Virus Suppresses Virus-Induced Apoptosis but Enhances Necroptosis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Plasmid DNA
2.3. Cell Lines and Viruses
2.4. Western Blot
2.5. Flow Cytometry
2.6. Statistical Analysis
3. Results
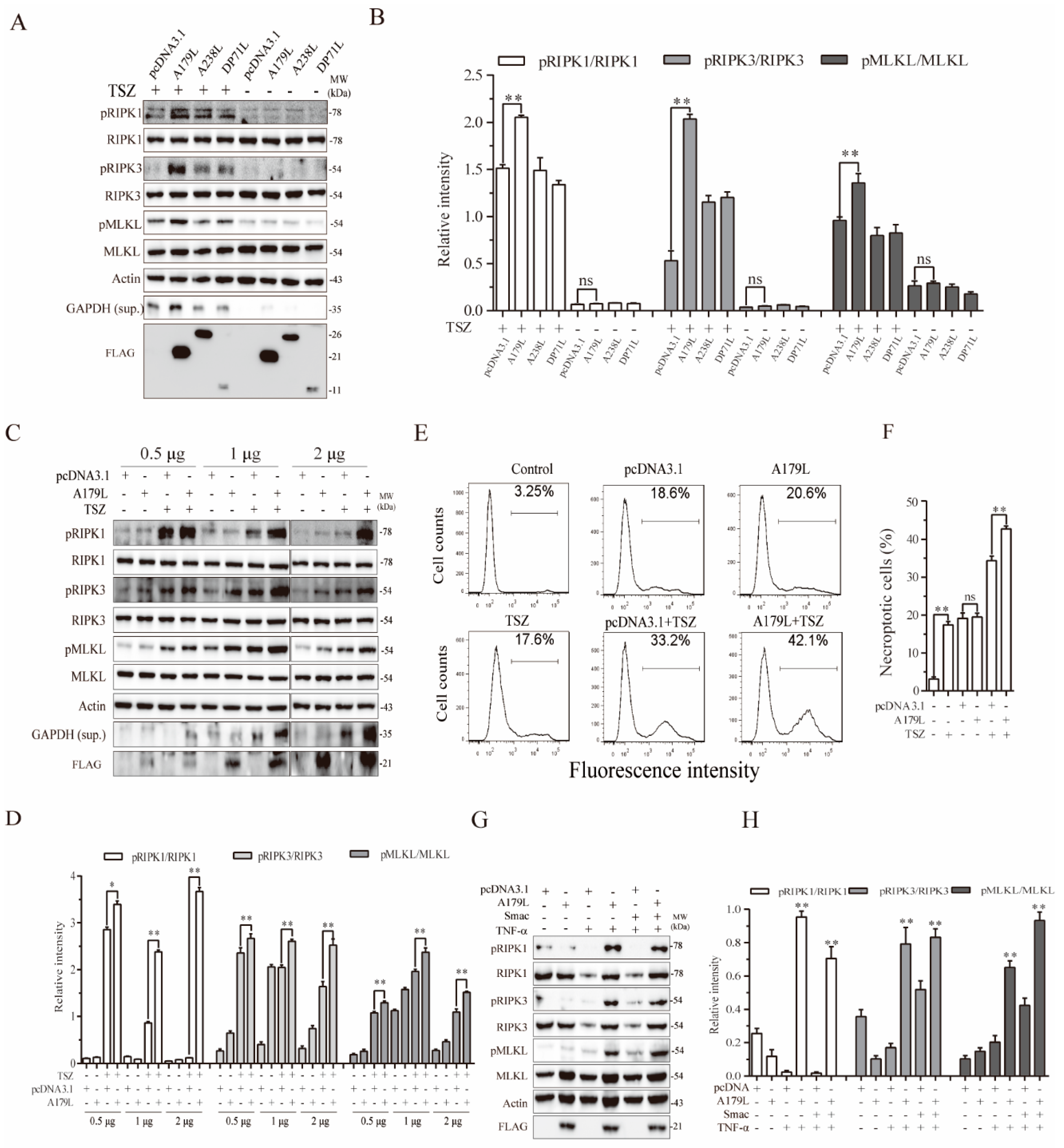
3.1. A179L Enhances TSZ-Induced Necroptosis
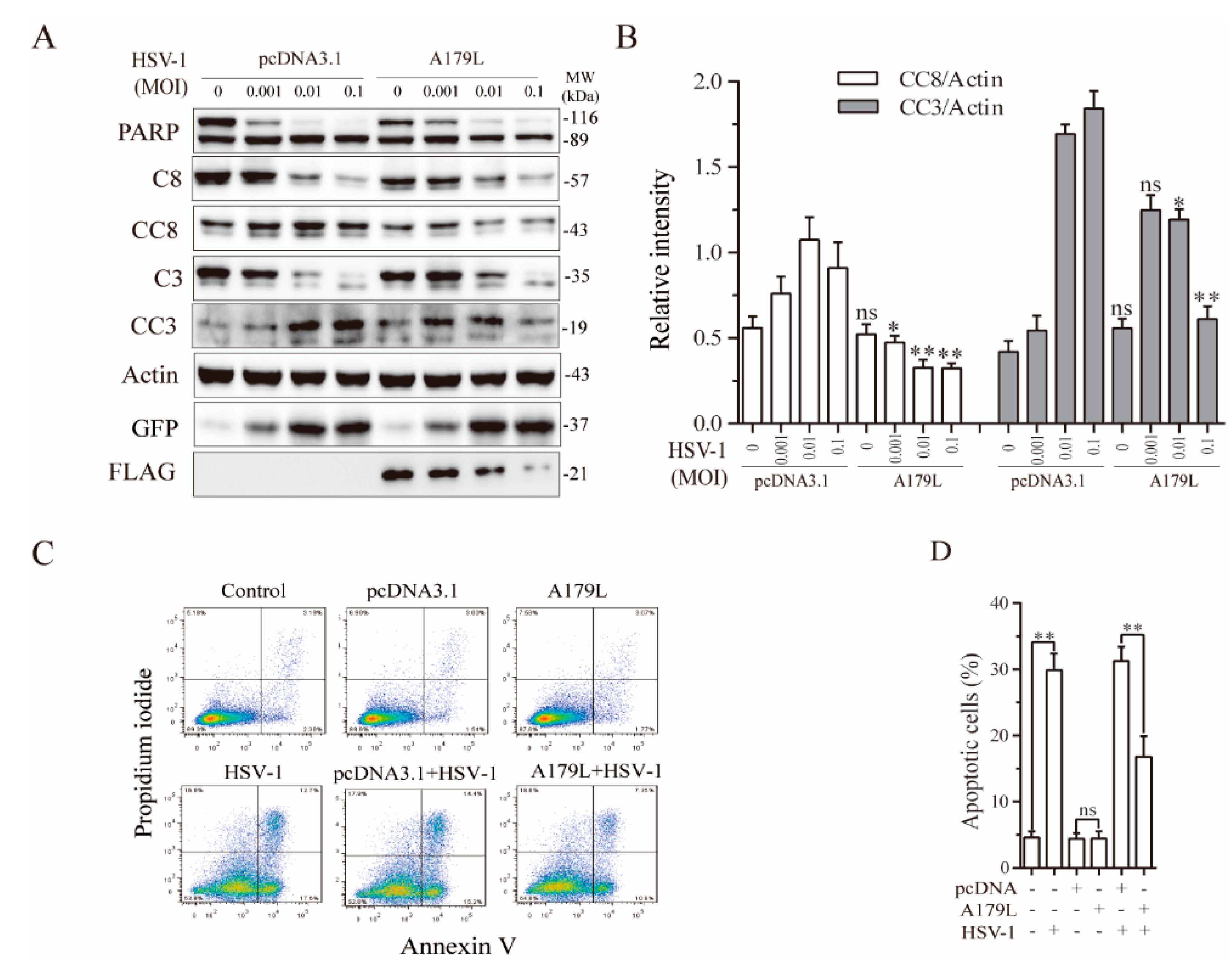
3.2. A179L Suppresses HSV-1-Induced Apoptosis
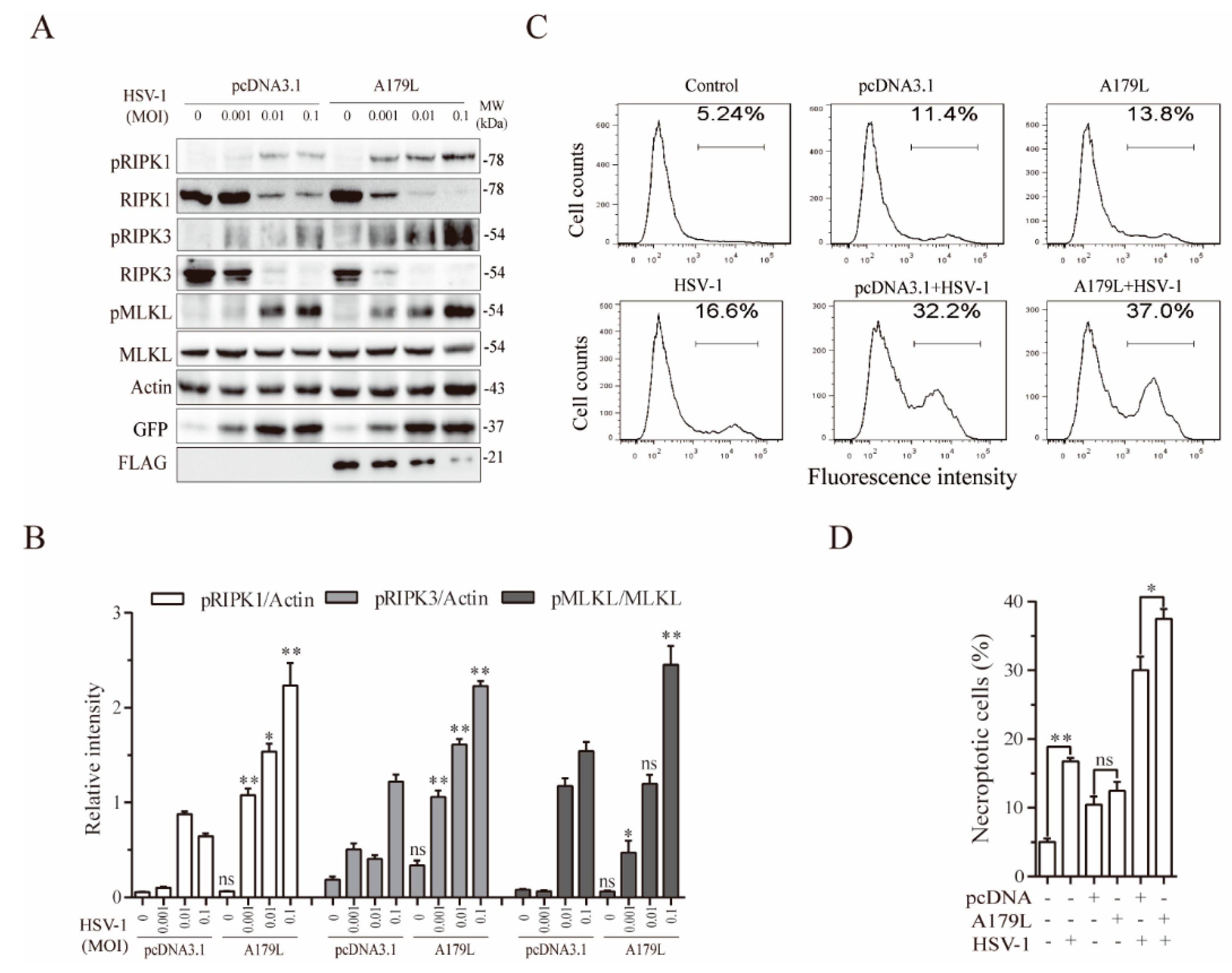
3.3. A179L Enhances HSV-1-Induced Necroptosis
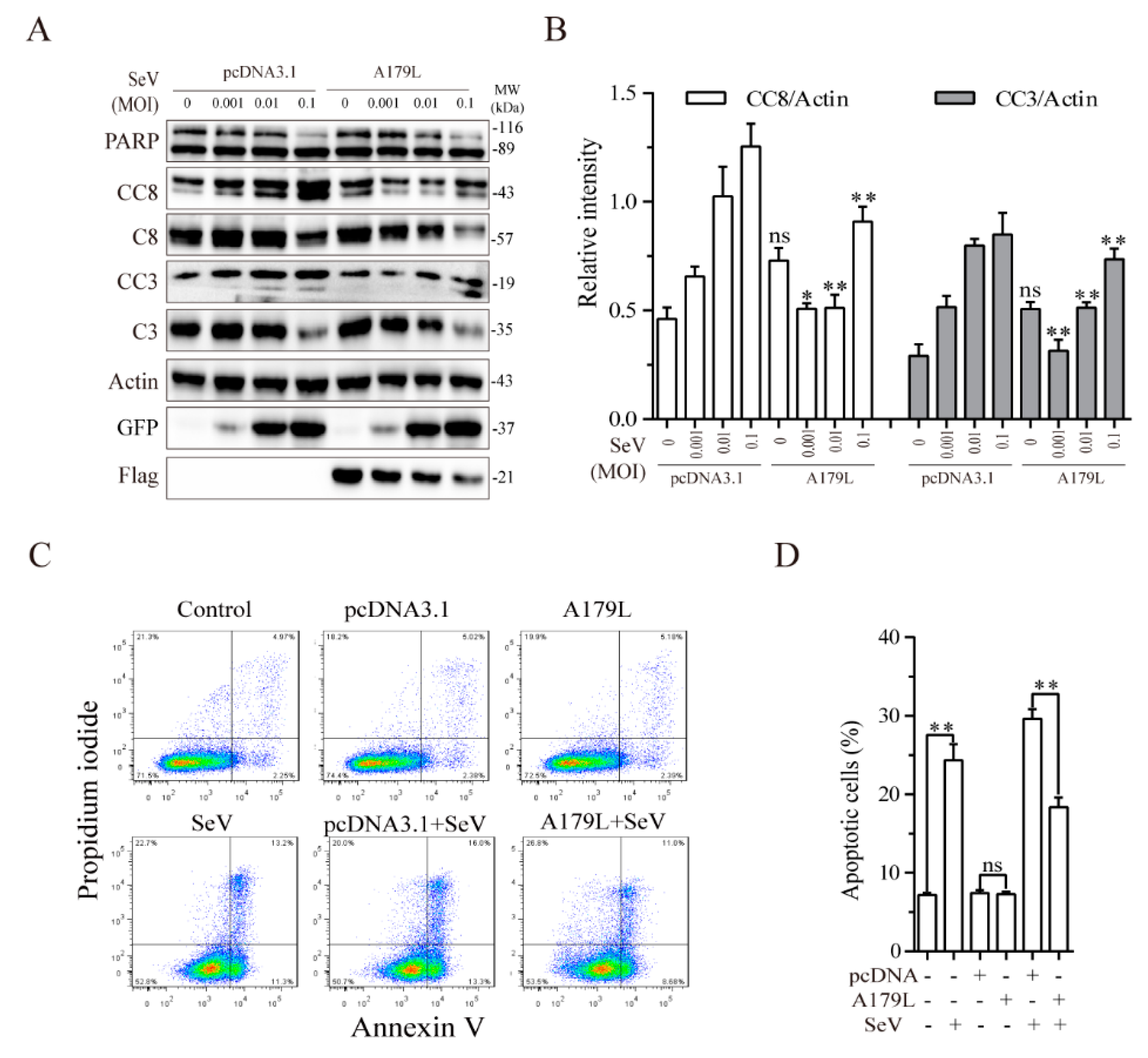
3.4. A179L Suppresses Sendai Virus-Induced Apoptosis
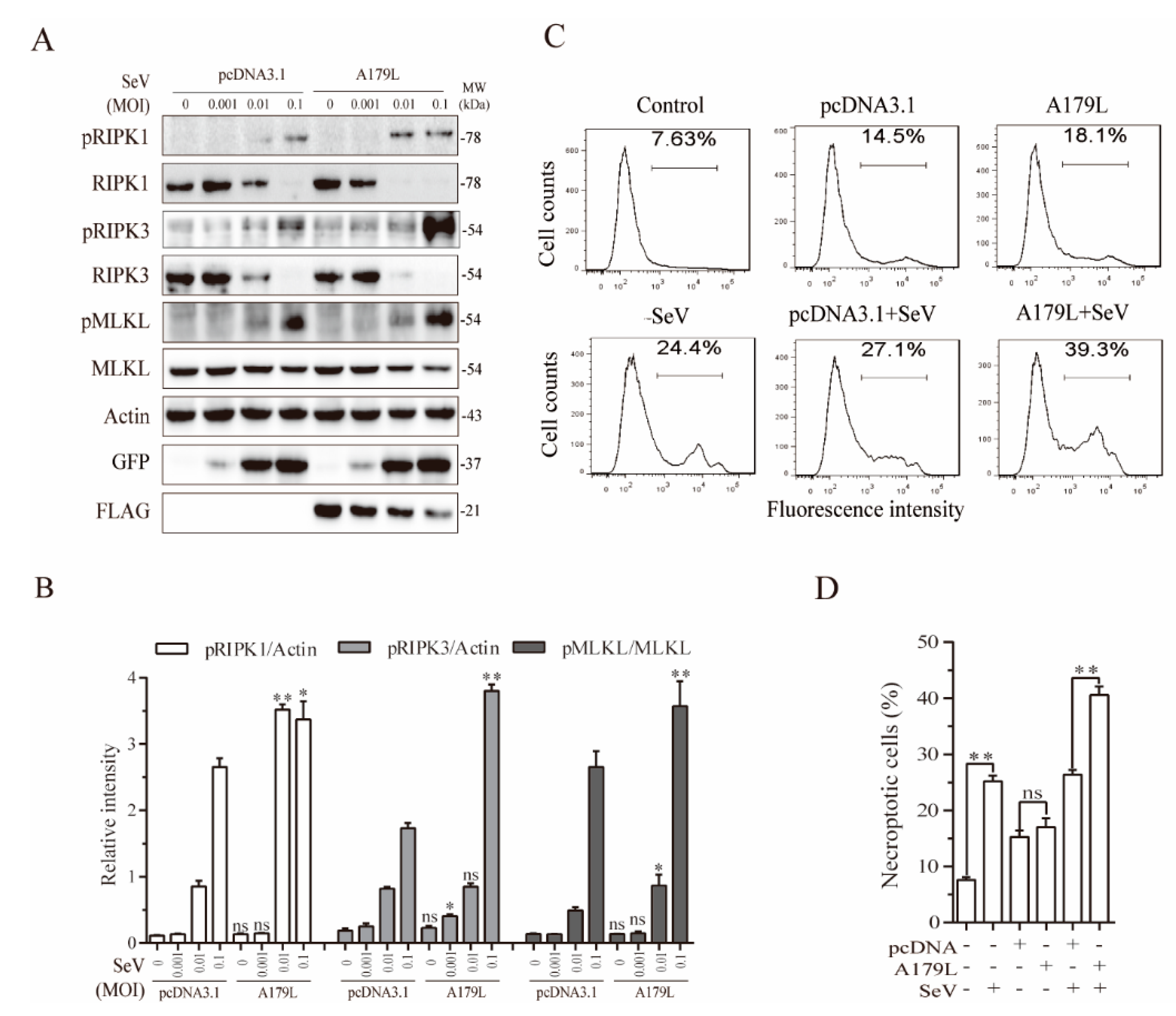
3.5. A179L Enhances Sendai Virus-Induced Necroptosis
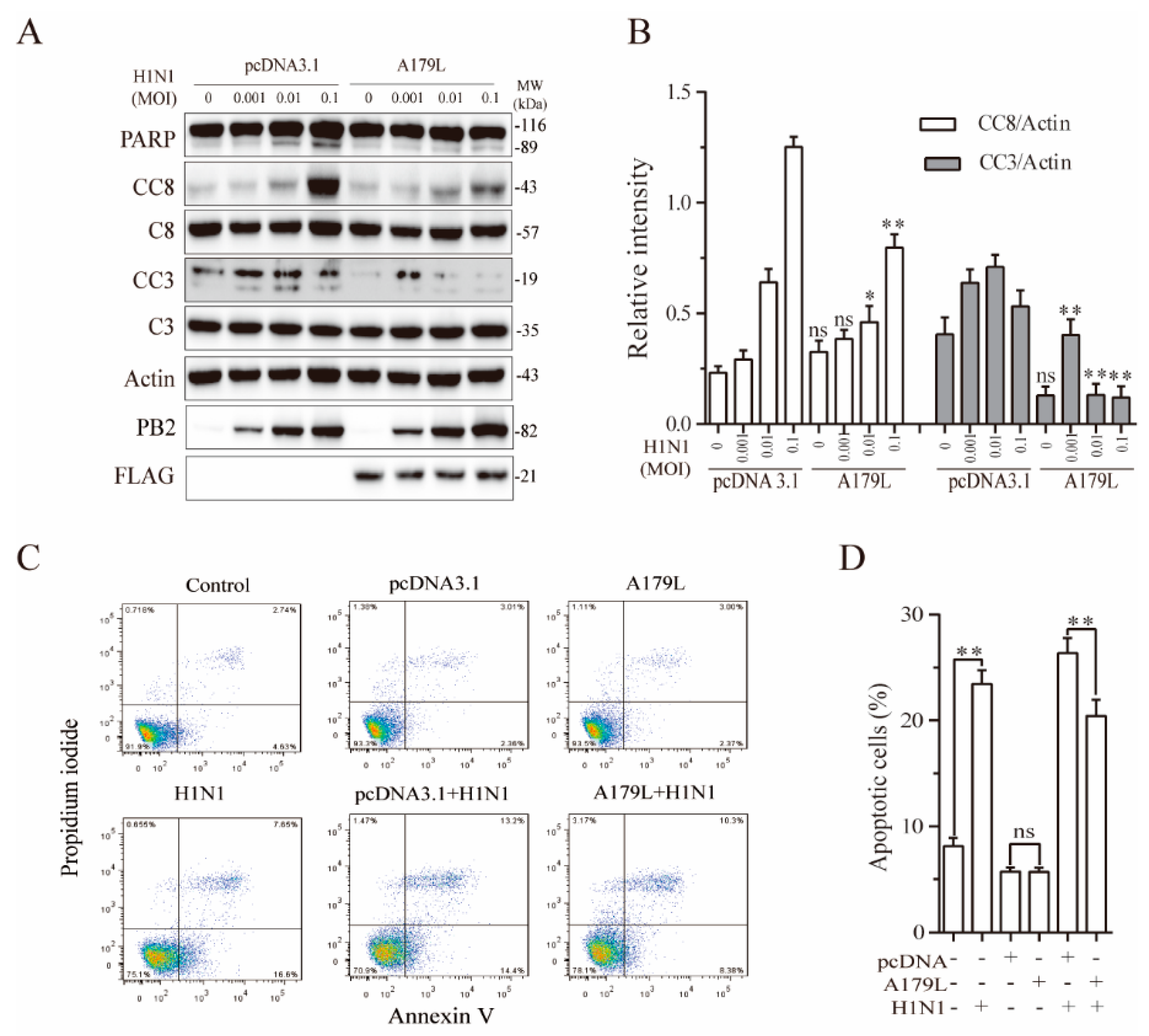
3.6. A179L Suppresses Influenza Virus-Induced Apoptosis
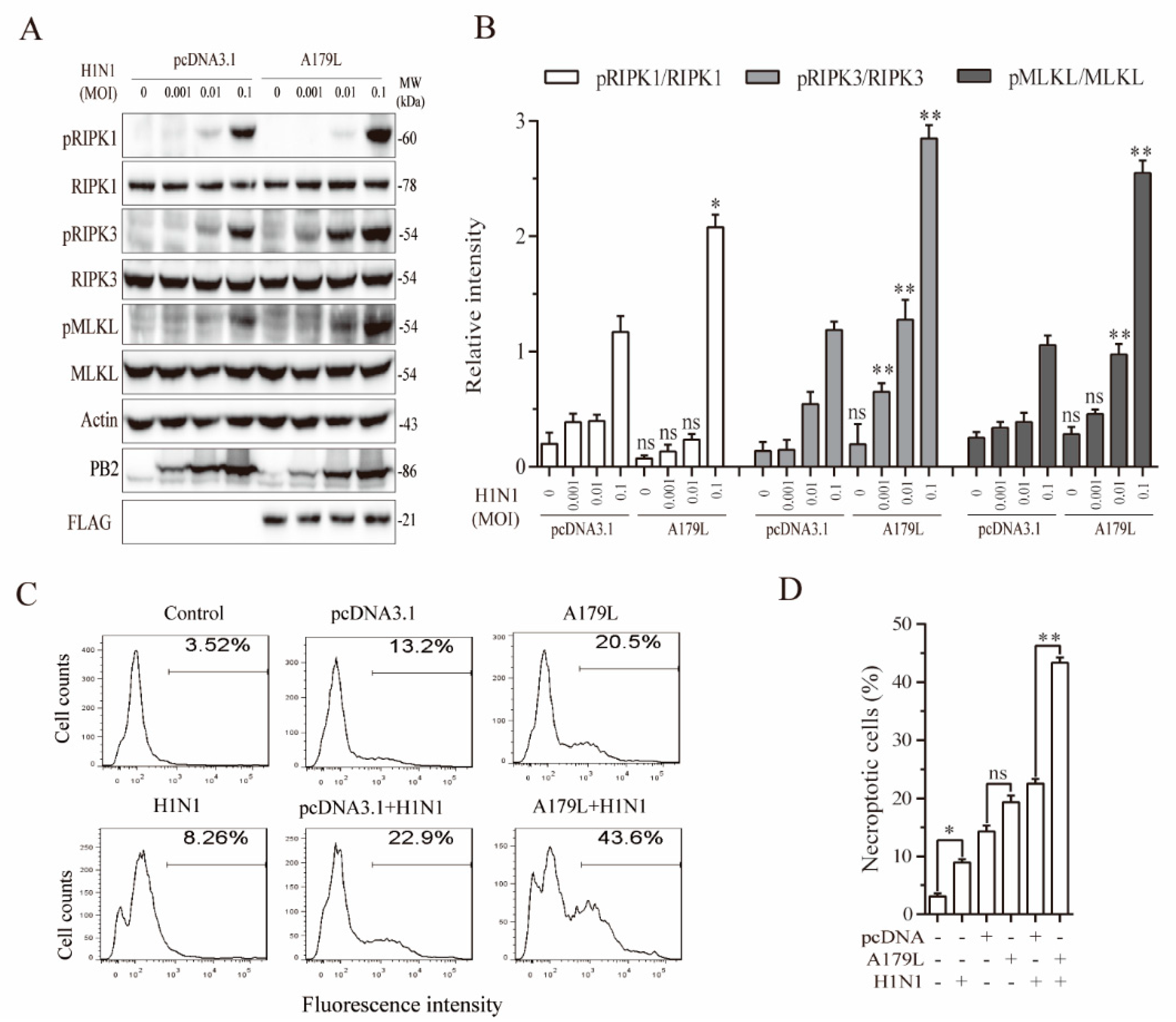
3.7. A179L Enhances Influenza Virus-Induced Necroptosis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galindo, I.; Alonso, C. African Swine Fever Virus: A Review. Viruses 2017, 9, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, D.; Liu, R.; Zhang, X.; Li, F.; Wang, J.; Zhang, J.; Liu, X.; Wang, L.; Zhang, J.; Wu, X.; et al. Replication and virulence in pigs of the first African swine fever virus isolated in China. Emerg. Microbes Infect. 2019, 8, 438–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karger, A.; Pérez-Núñez, D.; Urquiza, J.; Hinojar, P.; Alonso, C.; Freitas, F.B.; Revilla, Y.; Le Potier, M.-F.; Montoya, M. An Update on African Swine Fever Virology. Viruses 2019, 11, 864. [Google Scholar] [CrossRef] [Green Version]
- Blome, S.; Franzke, K.; Beer, M. African swine fever–A review of current knowledge. Virus Res. 2020, 287, 198099. [Google Scholar] [CrossRef]
- Correia, S.; Ventura, S.; Parkhouse, R.M. Identification and utility of innate immune system evasion mechanisms of ASFV. Virus Res. 2013, 173, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.; Islam, M.; Nash, R.; Reis, A. African swine fever virus evasion of host defences. Virus Res. 2019, 266, 25–33. [Google Scholar] [CrossRef]
- Li, D.; Zhang, J.; Yang, W.; Li, P.; Ru, Y.; Kang, W.; Li, L.; Ran, Y.; Zheng, H. African swine fever virus protein MGF-505-7R promotes virulence and pathogenesis by inhibiting JAK1- and JAK2-mediated signaling. J. Biol. Chem. 2021, 297. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, C.; Sánchez, E.G.; Pérez-Núñez, D.; Nogal, M.; de Leon, P.; Carrascosa, L.; Nieto, R.; Soler, A.; Arias, M.L.; Revilla, Y. African swine fever virus (ASFV) protection mediated by NH/P68 and NH/P68 recombinant live-attenuated viruses. Vaccine 2018, 36, 2694–2704. [Google Scholar] [CrossRef]
- Zhang, Y.; Ke, J.; Zhang, J.; Yang, J.; Yue, H.; Zhou, X.; Qi, Y.; Zhu, R.; Miao, F.; Li, Q.; et al. ASFV bearing an I226R gene-deletion elicits a robust immunity in pigs to African swine fever. J. Virol. 2021. [Google Scholar] [CrossRef]
- Li, D.; Yang, W.; Li, L.; Li, P.; Ma, Z.; Zhang, J.; Qi, X.; Ren, J.; Ru, Y.; Niu, Q.; et al. African Swine Fever Virus MGF-505-7R Negatively Regulates cGAS-STING-Mediated Signaling Pathway. J. Immunol. 2021, 206, 1844–1857. [Google Scholar] [CrossRef]
- Borca, M.V.; Ramirez-Medina, E.; Silva, E.; Vuono, E.; Rai, A.; Pruitt, S.; Holinka, L.G.; Velazquez-Salinas, L.; Zhu, J.; Gladue, D.P. Development of a Highly Effective African Swine Fever Virus Vaccine by Deletion of the I177L Gene Results in Sterile Immunity against the Current Epidemic Eurasia Strain. J. Virol. 2020, 94, e02017-19. [Google Scholar] [CrossRef]
- Li, J.; Song, J.; Kang, L.; Huang, L.; Zhou, S.; Hu, L.; Zheng, J.; Li, C.; Zhang, X.; He, X.; et al. pMGF505-7R determines pathogenicity of African swine fever virus infection by inhibiting IL-1beta and type I IFN production. PLoS Pathog. 2021, 17, e1009733. [Google Scholar] [CrossRef]
- Cackett, G.; Matelska, D.; Sýkora, M.; Portugal, R.; Malecki, M.; Bähler, J.; Dixon, L.; Werner, F. The African Swine Fever Virus Transcriptome. J. Virol. 2020, 94, e00119-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orzalli, M.H.; Kagan, J.C. Apoptosis and Necroptosis as Host Defense Strategies to Prevent Viral Infection. Trends Cell Biol. 2017, 27, 800–809. [Google Scholar] [CrossRef]
- Imre, G. Cell death signalling in virus infection. Cell. Signal. 2020, 76, 109772. [Google Scholar] [CrossRef] [PubMed]
- Nogusa, S.; Thapa, R.J.; Dillon, C.P.; Liedmann, S.; Oguin, T.H., 3rd; Ingram, J.P.; Rodrigues, D.A.; Kosoff, R.; Sharma, S.; Sturm, O.; et al. RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell Host Microbe 2016, 20, 13–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thapa, R.J.; Ingram, J.P.; Ragan, K.B.; Nogusa, S.; Boyd, D.; Benitez, A.; Sridharan, H.; Kosoff, R.; Shubina, M.; Landsteiner, V.J.; et al. DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell Host Microbe 2016, 20, 674–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkin-Smith, G.K.; Duan, M.; Chen, W.; Poon, I.K.H. The induction and consequences of Influenza A virus-induced cell death. Cell Death Dis. 2018, 9, 1002. [Google Scholar] [CrossRef]
- Ampomah, P.B.; Lim, L.H.K. Influenza A virus-induced apoptosis and virus propagation. Apoptosis 2019, 25, 1–11. [Google Scholar] [CrossRef]
- He, S.; Han, J. Manipulation of Host Cell Death Pathways by Herpes Simplex Virus. Curr. Top Microbiol. Immunol. 2020, 1–19. [Google Scholar] [CrossRef]
- Xia, X.; Lei, L.; Wang, S.; Hu, J.; Zhang, G. Necroptosis and its role in infectious diseases. Apoptosis 2020, 25, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Nailwal, H.; Chan, F.K.-M. Necroptosis in anti-viral inflammation. Cell Death Differ. 2018, 26, 4–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schock, S.N.; Chandra, N.V.; Sun, Y.; Irie, T.; Kitagawa, Y.; Gotoh, B.; Coscoy, L.; Winoto, A. Induction of necroptotic cell death by viral activation of the RIG-I or STING pathway. Cell Death Differ. 2017, 24, 615–625. [Google Scholar] [CrossRef]
- Dixon, L.K.; Sanchez-Cordon, P.J.; Galindo, I.; Alonso, C. Investigations of Pro- and Anti-Apoptotic Factors Affecting African Swine Fever Virus Replication and Pathogenesis. Viruses 2017, 9, 241. [Google Scholar] [CrossRef] [Green Version]
- Hernaez, B.; Díaz-Gil, G.; García-Gallo, M.; Quetglas, J.I.; Rodriguez-Crespo, J.I.; Dixon, L.; Escribano, J.M.; Alonso, C. The African swine fever virus dynein-binding protein p54 induces infected cell apoptosis. FEBS Lett. 2004, 569, 224–228. [Google Scholar] [CrossRef] [Green Version]
- Brun, A.; Rivas, C.; Esteban, M.; Escribano, J.M.; Alonso, C. African swine fever virus gene A179L, a viral homologue of bcl-2, protects cells from programmed cell death. Virology 1996, 225, 227–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revilla, Y.; Cebrián, A.; Baixeras, E.; Martínez, C.; Viñuela, E.; Salas, M.L. Inhibition of Apoptosis by the African Swine Fever Virus Bcl-2 Homologue: Role of the BH1 Domain. Virology 1997, 228, 400–404. [Google Scholar] [CrossRef] [Green Version]
- Galindo, I.; Hernaez, B.; Diaz-Gil, G.; Escribano, J.M.; Alonso, C. A179L, a viral Bcl-2 homologue, targets the core Bcl-2 apoptotic machinery and its upstream BH3 activators with selective binding restrictions for Bid and Noxa. Virology 2008, 375, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Banjara, S.; Caria, S.; Dixon, L.K.; Hinds, M.G.; Kvansakul, M. Structural Insight into African Swine Fever Virus A179L-Mediated Inhibition of Apoptosis. J. Virol. 2017, 91, e02228-16. [Google Scholar] [CrossRef] [Green Version]
- Hernaez, B.; Cabezas, M.; Munoz-Moreno, R.; Galindo, I.; Cuesta-Geijo, M.A.; Alonso, C. A179L, a new viral Bcl2 homolog targeting Beclin 1 autophagy related protein. Curr. Mol. Med. 2013, 13, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Banjara, S.; Shimmon, G.L.; Dixon, L.K.; Netherton, C.L.; Hinds, M.G.; Kvansakul, M. Crystal Structure of African Swine Fever Virus A179L with the Autophagy Regulator Beclin. Viruses 2019, 11, 789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimmon, G.L.; Hui, J.Y.K.; Wileman, T.E.; Netherton, C.L. Autophagy impairment by African swine fever virus. J. Gen. Virol. 2021, 102, 001637. [Google Scholar] [CrossRef] [PubMed]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef]
- Zhang, F.; Moon, A.; Childs, K.; Goodbourn, S.; Dixon, L.K. The African swine fever virus DP71L protein recruits the protein phosphatase 1 catalytic subunit to dephosphorylate eIF2alpha and inhibits CHOP induction but is dispensable for these activities during virus infection. J. Virol. 2010, 84, 10681–10689. [Google Scholar] [CrossRef] [Green Version]
- Barber, C.; Netherton, C.; Goatley, L.; Moon, A.; Goodbourn, S.; Dixon, L. Identification of residues within the African swine fever virus DP71L protein required for dephosphorylation of translation initiation factor eIF2α and inhibiting activation of pro-apoptotic CHOP. Virology 2017, 504, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Saveljeva, S.; Mc Laughlin, S.L.; Vandenabeele, P.; Samali, A.; Bertrand, M.J. Endoplasmic reticulum stress induces ligand-independent TNFR1-mediated necroptosis in L929 cells. Cell Death Dis. 2015, 6, e1587. [Google Scholar] [CrossRef]
- Vanlangenakker, N.; Bertrand, M.; Bogaert, P.; Vandenabeele, P.; Berghe, T.V. TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis. 2011, 2, e230. [Google Scholar] [CrossRef]
- Guo, X.; Yin, H.; Chen, Y.; Li, L.; Li, J.; Liu, Q. TAK1 regulates caspase 8 activation and necroptotic signaling via multiple cell death checkpoints. Cell Death Dis. 2016, 7, e2381. [Google Scholar] [CrossRef] [Green Version]
- Brune, W.; Andoniou, C.E. Die Another Day: Inhibition of Cell Death Pathways by Cytomegalovirus. Viruses 2017, 9, 249. [Google Scholar] [CrossRef] [Green Version]
- Balachandran, S.; Rall, G.F. Benefits and Perils of Necroptosis in Influenza Virus Infection. J. Virol. 2020, 94, e01101-19. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Yin, C.; Boyd, D.F.; Quarato, G.; Ingram, J.P.; Shubina, M.; Ragan, K.B.; Ishizuka, T.; Crawford, J.C.; Tummers, B.; et al. Influenza Virus Z-RNAs Induce ZBP1-Mediated Necroptosis. Cell 2020, 180, 1115–1129.e13. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Harding, A.T.; Sweeney, C.; Miao, D.; Swan, G.; Zhou, C.; Jiang, Z.; Fitzgerald, K.A.; Hammer, G.; Bergo, M.O.; et al. Control of antiviral innate immune response by protein geranylgeranylation. Sci. Adv. 2019, 5, eaav7999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalaoui, N.; Boyden, S.E.; Oda, H.; Wood, G.M.; Stone, D.L.; Chau, D.; Liu, L.; Stoffels, M.; Kratina, T.; Lawlor, K.E.; et al. Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature 2020, 577, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Wickliffe, K.E.; Dugger, D.L.; Maltzman, A.; Roose-Girma, M.; Dohse, M.; Kőműves, L.; Webster, J.D.; Dixit, V.M. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature 2019, 574, 428–431. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, J.; Liu, W.; Zhang, M.; Sun, J.; Xu, X. The A179L Gene of African Swine Fever Virus Suppresses Virus-Induced Apoptosis but Enhances Necroptosis. Viruses 2021, 13, 2490. https://doi.org/10.3390/v13122490
Shi J, Liu W, Zhang M, Sun J, Xu X. The A179L Gene of African Swine Fever Virus Suppresses Virus-Induced Apoptosis but Enhances Necroptosis. Viruses. 2021; 13(12):2490. https://doi.org/10.3390/v13122490
Chicago/Turabian StyleShi, Jun, Wei Liu, Miao Zhang, Jing Sun, and Xiulong Xu. 2021. "The A179L Gene of African Swine Fever Virus Suppresses Virus-Induced Apoptosis but Enhances Necroptosis" Viruses 13, no. 12: 2490. https://doi.org/10.3390/v13122490
APA StyleShi, J., Liu, W., Zhang, M., Sun, J., & Xu, X. (2021). The A179L Gene of African Swine Fever Virus Suppresses Virus-Induced Apoptosis but Enhances Necroptosis. Viruses, 13(12), 2490. https://doi.org/10.3390/v13122490