
Emergence of a Distinct Picobirnavirus Genotype Circulating in Patients Hospitalized with Acute Respiratory Illness

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement and Specimens
2.2. Sputum Pre-Treatment
2.3. Extraction
2.4. In Vitro Transcription
2.5. PBV RT-qPCR Assay
2.6. mNGS Library Prep and Sequencing
2.7. mNGS Analysis
2.8. Pairwise Amino Acid Identity
2.9. Phylogenetics
2.10. Phylogeny-Trait Association Analysis
2.11. Statistics
2.12. NCBI Accessions
3. Results
3.1. Discovery of a Novel Picobirnavirus
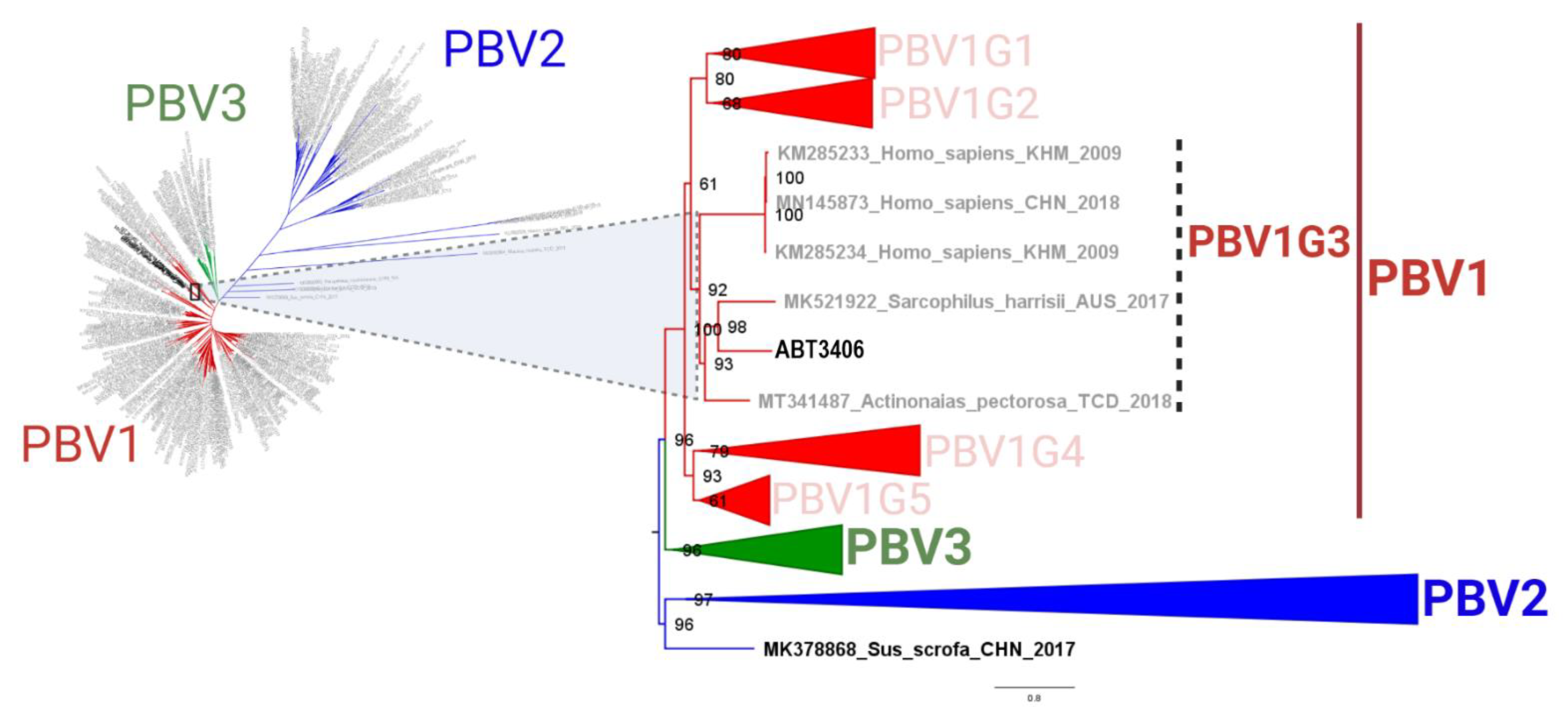
3.2. ABT3406 RdRp Branches with Respiratory PBV Strains
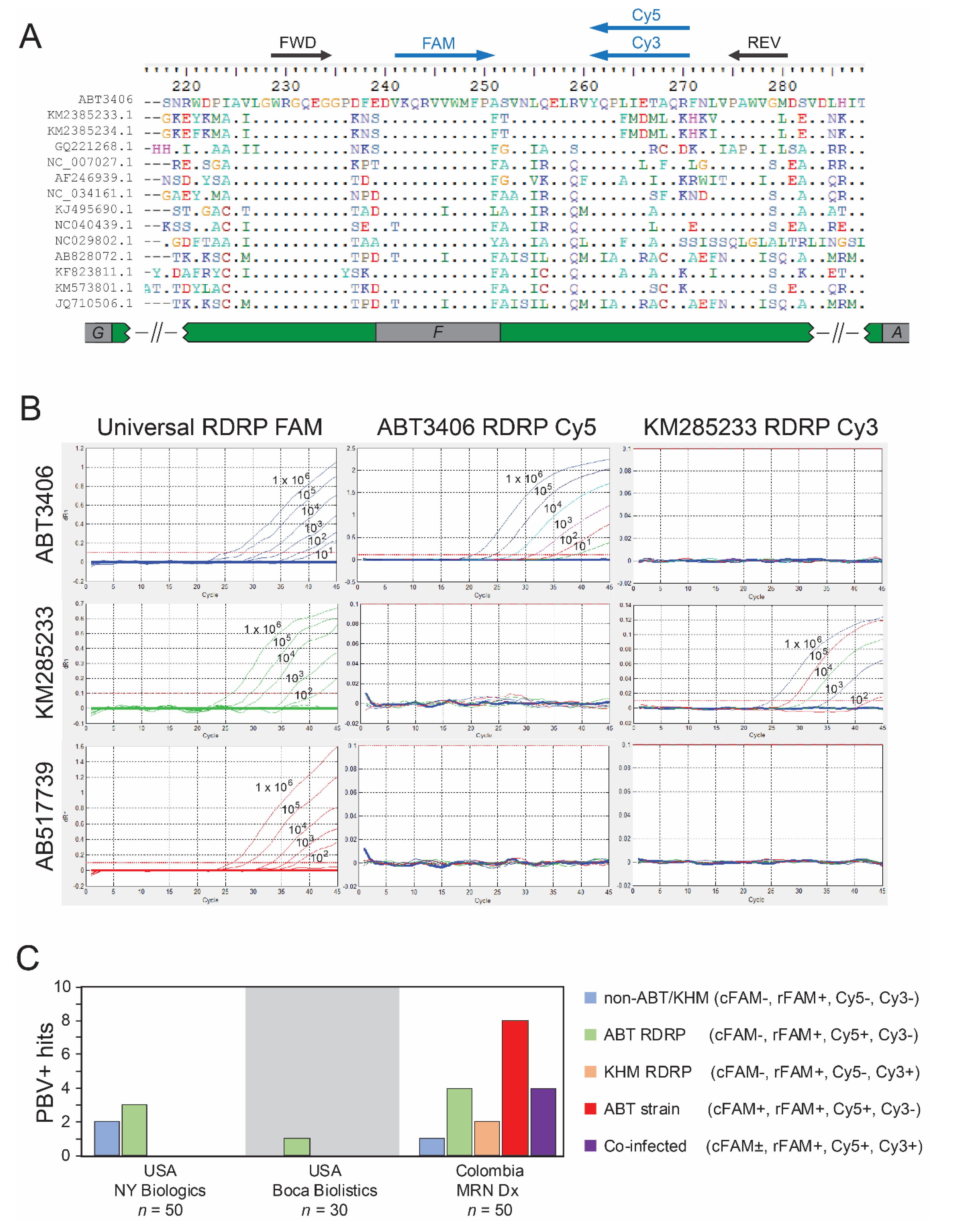
3.3. Quantitative PCR Screen Reveals High Prevalence of PBV among Hospitalized
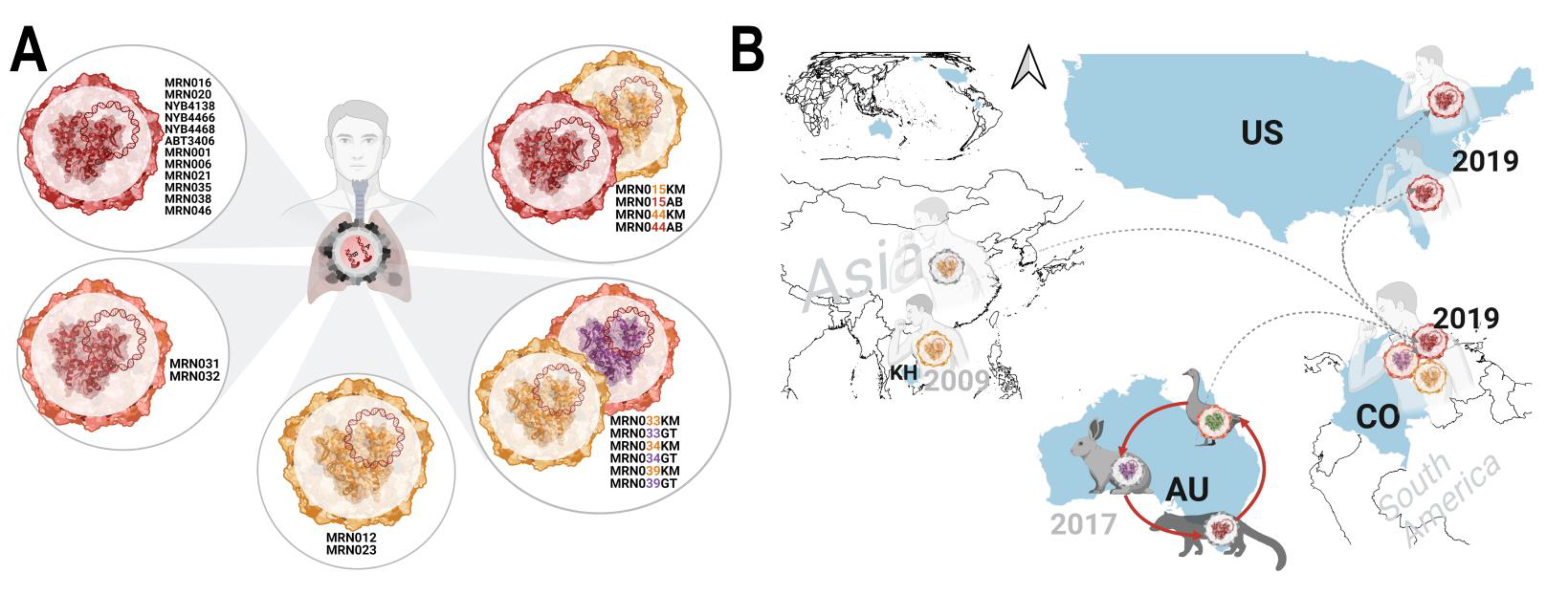
3.4. Emergence of a Distinct PBV Genotype
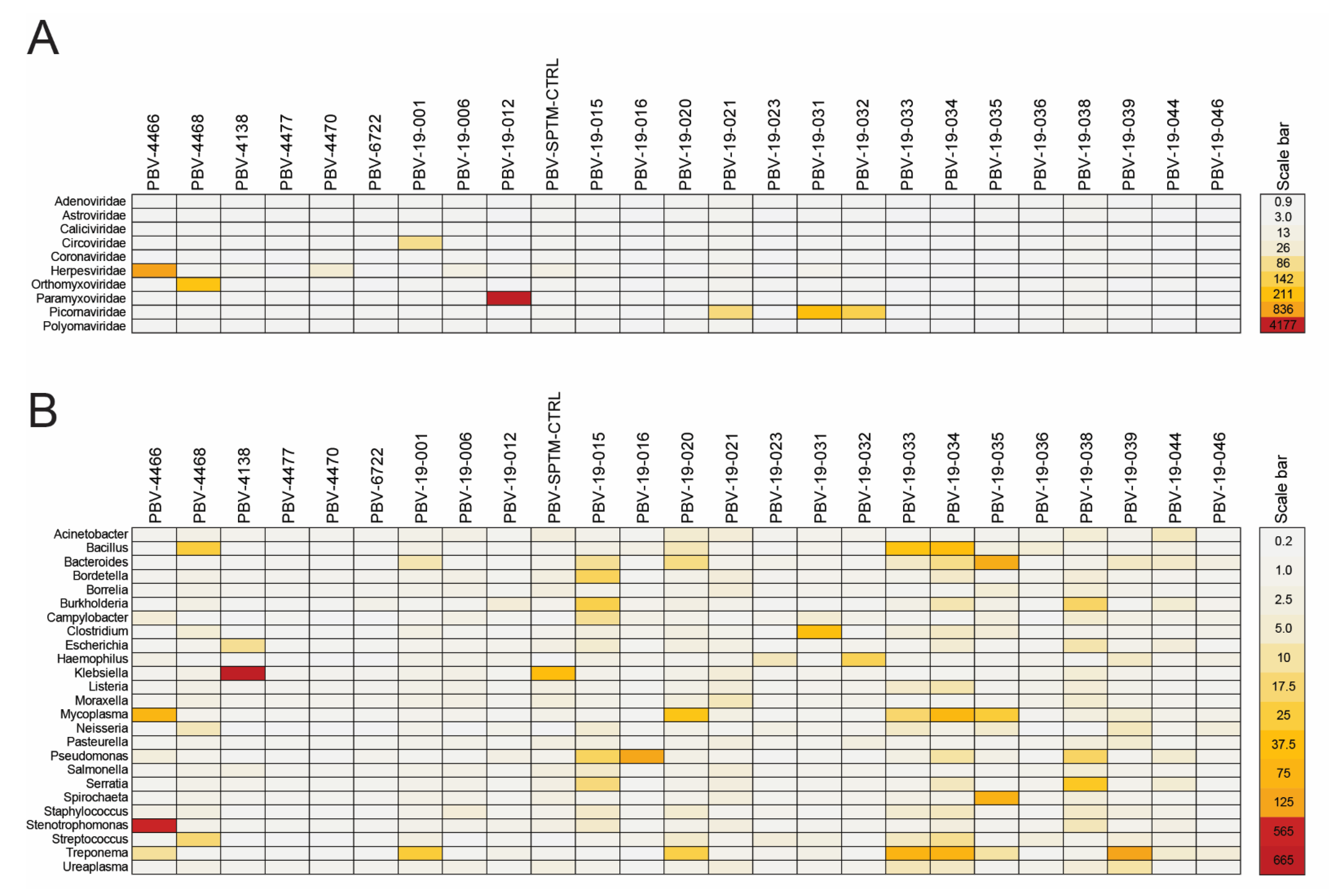
3.5. RdRp and Capsid Are Both Associated with Respiratory Disease
3.6. PBV Is Typically Present as a Co-Infection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sabino, E.C.; Buss, L.F.; Carvalho, M.P.S.; Prete, C.A., Jr.; Crispim, M.A.E.; Fraiji, N.A.; Pereira, R.H.M.; Parag, K.V.; da Silva Peixoto, P.; Kraemer, M.U.G.; et al. Resurgence of COVID-19 in Manaus, Brazil, despite high seroprevalence. Lancet 2021, 397, 452–455. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Lessells, R.J.; Giandhari, J.; Pillay, S.; Msomi, N.; Mlisana, K.; Bhiman, J.N.; von Gottberg, A.; Walaza, S.; et al. Sixteen novel lineages of SARS-CoV-2 in South Africa. Nat. Med. 2021, 27, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, A.; et al. Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.Y. Viral pathogen discovery. Curr. Opin. Microbiol. 2013, 16, 468–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipkin, W.I.; Anthony, S.J. Virus hunting. Virology 2015, 479–480, 194–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greninger, A.L. A decade of RNA virus metagenomics is (not) enough. Virus Res. 2018, 244, 218–229. [Google Scholar] [CrossRef]
- Wilson, M.R.; Naccache, S.N.; Samayoa, E.; Biagtan, M.; Bashir, H.; Yu, G.; Salamat, S.M.; Somasekar, S.; Federman, S.; Miller, S.; et al. Actionable diagnosis of neuroleptospirosis by next-generation sequencing. N. Engl. J. Med. 2014, 370, 2408–2417. [Google Scholar] [CrossRef] [Green Version]
- Dasaraju, P.V.; Liu, C. Infections of the Respiratory System. In Medical Microbiology; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1994; Chapter 93. [Google Scholar]
- Pereira, H.G.; Fialho, A.M.; Flewett, T.H.; Teixeira, J.M.; Andrade, Z.P. Novel viruses in human faeces. Lancet 1988, 2, 103–104. [Google Scholar] [CrossRef]
- Pereira, H.G.; Flewett, T.H.; Candeias, J.A.; Barth, O.M. A virus with a bisegmented double-stranded RNA genome in rat (Oryzomys nigripes) intestines. J. Gen. Virol. 1988, 69 Pt 11, 2749–2754. [Google Scholar] [CrossRef]
- Ganesh, B.; Masachessi, G.; Mladenova, Z. Animal picobirnavirus. Virusdisease 2014, 25, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Banyai, K.; Martella, V.; Bogdan, A.; Forgach, P.; Jakab, F.; Meleg, E.; Biro, H.; Melegh, B.; Szucs, G. Genogroup I picobirnaviruses in pigs: Evidence for genetic diversity and relatedness to human strains. J. Gen. Virol. 2008, 89, 534–539. [Google Scholar] [CrossRef]
- Ribeiro Silva, R.; Bezerra, D.A.; Kaiano, J.H.; Oliveira Dde, S.; Silvestre, R.V.; Gabbay, Y.B.; Ganesh, B.; Mascarenhas, J.D. Genogroup I avian picobirnavirus detected in Brazilian broiler chickens: A molecular epidemiology study. J. Gen. Virol. 2014, 95, 117–122. [Google Scholar] [CrossRef] [Green Version]
- Smits, S.L.; Poon, L.L.; van Leeuwen, M.; Lau, P.N.; Perera, H.K.; Peiris, J.S.; Simon, J.H.; Osterhaus, A.D. Genogroup I and II picobirnaviruses in respiratory tracts of pigs. Emerg. Infect. Dis. 2011, 17, 2328–2330. [Google Scholar] [CrossRef]
- Duquerroy, S.; Da Costa, B.; Henry, C.; Vigouroux, A.; Libersou, S.; Lepault, J.; Navaza, J.; Delmas, B.; Rey, F.A. The picobirnavirus crystal structure provides functional insights into virion assembly and cell entry. EMBO J. 2009, 28, 1655–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, Y.S.; Kumar, N.; Sharma, K.; Dhama, K.; Shabbir, M.Z.; Ganesh, B.; Kobayashi, N.; Banyai, K. Epidemiology, phylogeny, and evolution of emerging enteric Picobirnaviruses of animal origin and their relationship to human strains. Biomed Res. Int. 2014, 2014, 780752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesh, B.; Nagashima, S.; Ghosh, S.; Nataraju, S.M.; Rajendran, K.; Manna, B.; Ramamurthy, T.; Niyogi, S.K.; Kanungo, S.; Sur, D.; et al. Detection and molecular characterization of multiple strains of Picobirnavirus causing mixed infection in a diarrhoeic child: Emergence of prototype Genogroup II-like strain in Kolkata, India. Int. J. Mol. Epidemiol. Genet. 2011, 2, 61–72. [Google Scholar] [PubMed]
- Ghosh, S.; Malik, Y.S. The True Host/s of Picobirnaviruses. Front. Vet. Sci. 2020, 7, 615293. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, S.R.; Wang, D. Extensive conservation of prokaryotic ribosomal binding sites in known and novel picobirnaviruses. Virology 2018, 516, 108–114. [Google Scholar] [CrossRef]
- Haga, I.R.; Martins, S.S.; Hosomi, S.T.; Vicentini, F.; Tanaka, H.; Gatti, M.S. Identification of a bisegmented double-stranded RNA virus (Picobirnavirus) in faeces of giant anteaters (Myrmecophaga tridactyla). Vet. J. 1999, 158, 234–236. [Google Scholar] [CrossRef] [PubMed]
- Masachessi, G.; Martinez, L.C.; Giordano, M.O.; Barril, P.A.; Isa, B.M.; Ferreyra, L.; Villareal, D.; Carello, M.; Asis, C.; Nates, S.V. Picobirnavirus (PBV) natural hosts in captivity and virus excretion pattern in infected animals. Arch. Virol. 2007, 152, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Banyai, K.; Jakab, F.; Reuter, G.; Bene, J.; Uj, M.; Melegh, B.; Szucs, G. Sequence heterogeneity among human picobirnaviruses detected in a gastroenteritis outbreak. Arch. Virol. 2003, 148, 2281–2291. [Google Scholar] [CrossRef]
- Bhattacharya, R.; Sahoo, G.C.; Nayak, M.K.; Ghosh, S.; Dutta, P.; Bhattacharya, M.K.; Mitra, U.; Gangopadhyay, D.; Dutta, S.; Niyogi, S.K.; et al. Molecular epidemiology of human astrovirus infections in Kolkata, India. Infect. Genet. Evol. 2006, 6, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, R.; Sahoo, G.C.; Nayak, M.K.; Rajendran, K.; Dutta, P.; Mitra, U.; Bhattacharya, M.K.; Naik, T.N.; Bhattacharya, S.K.; Krishnan, T. Detection of Genogroup I and II human picobirnaviruses showing small genomic RNA profile causing acute watery diarrhoea among children in Kolkata, India. Infect. Genet. Evol. 2007, 7, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Giordano, M.O.; Martinez, L.C.; Rinaldi, D.; Guinard, S.; Naretto, E.; Casero, R.; Yacci, M.R.; Depetris, A.R.; Medeot, S.I.; Nates, S.V. Detection of picobirnavirus in HIV-infected patients with diarrhea in Argentina. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1998, 18, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Smits, S.L.; van Leeuwen, M.; Schapendonk, C.M.; Schurch, A.C.; Bodewes, R.; Haagmans, B.L.; Osterhaus, A.D. Picobirnaviruses in the human respiratory tract. Emerg. Infect. Dis. 2012, 18, 1539–1540. [Google Scholar] [CrossRef]
- Cummings, M.J.; Tokarz, R.; Bakamutumaho, B.; Kayiwa, J.; Byaruhanga, T.; Owor, N.; Namagambo, B.; Wolf, A.; Mathema, B.; Lutwama, J.J.; et al. Precision surveillance for viral respiratory pathogens: Virome capture sequencing for the detection and genomic characterization of severe acute respiratory infection in Uganda. Clin. Infect. Dis. 2019, 7, 1118–1125. [Google Scholar] [CrossRef]
- Li, C.X.; Li, W.; Zhou, J.; Zhang, B.; Feng, Y.; Xu, C.P.; Lu, Y.Y.; Holmes, E.C.; Shi, M. High resolution metagenomic characterization of complex infectomes in paediatric acute respiratory infection. Sci. Rep. 2020, 10, 3963. [Google Scholar] [CrossRef] [Green Version]
- Stokell, J.R.; Khan, A.; Steck, T.R. Mechanical homogenization increases bacterial homogeneity in sputum. J. Clin. Microbiol. 2014, 52, 2340–2345. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, J.; Olivo, A.; Laeyendecker, O.; Forberg, K.; Ndembi, N.; Mbanya, D.; Kaptue, L.; Quinn, T.C.; Cloherty, G.A.; Rodgers, M.A.; et al. Universal Target Capture of HIV Sequences From NGS Libraries. Front. Microbiol. 2018, 9, 2150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naccache, S.N.; Federman, S.; Veeraraghavan, N.; Zaharia, M.; Lee, D.; Samayoa, E.; Bouquet, J.; Greninger, A.L.; Luk, K.C.; Enge, B.; et al. A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identification from next-generation sequencing of clinical samples. Genome Res. 2014, 24, 1180–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, L.J.; Cloherty, G.A.; Berg, M.G. Understanding the Genetic Diversity of Picobirnavirus: A Classification Update Based on Phylogenetic and Pairwise Sequence Comparison Approaches. Viruses 2021, 13, 1476. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Ota, R.; Waddell, P.J.; Hasegawa, M.; Shimodaira, H.; Kishino, H. Appropriate likelihood ratio tests and marginal distributions for evolutionary tree models with constraints on parameters. Mol. Biol. Evol. 2000, 17, 798–803. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.; Rambaut, A.; Pybus, O.G. Correlating viral phenotypes with phylogeny: Accounting for phylogenetic uncertainty. Infect. Genet. Evol. 2008, 8, 239–246. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Costa, B.; Duquerroy, S.; Tarus, B.; Delmas, B. Picobirnaviruses encode a protein with repeats of the ExxRxNxxxE motif. Virus Res. 2011, 158, 251–256. [Google Scholar] [CrossRef]
- Wille, M.; Shi, M.; Klaassen, M.; Hurt, A.C.; Holmes, E.C. Virome heterogeneity and connectivity in waterfowl and shorebird communities. ISME J. 2019, 13, 2603–2616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, B.I.; Fang, Z.Y.; Glass, R.I.; Monroe, S.S. Cloning of human picobirnavirus genomic segments and development of an RT-PCR detection assay. Virology 2000, 277, 316–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, P.C.; Teng, J.L.; Bai, R.; Wong, A.Y.; Martelli, P.; Hui, S.W.; Tsang, A.K.; Lau, C.C.; Ahmed, S.S.; Yip, C.C.; et al. High Diversity of Genogroup I Picobirnaviruses in Mammals. Front. Microbiol. 2016, 7, 1886. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Teng, J.L.L.; Bai, R.; Tang, Y.; Wong, A.Y.P.; Li, K.S.M.; Lam, C.S.F.; Fan, R.Y.Y.; Lau, S.K.P.; Yuen, K.Y. Novel Picobirnaviruses in Respiratory and Alimentary Tracts of Cattle and Monkeys with Large Intra- and Inter-Host Diversity. Viruses 2019, 11, 574. [Google Scholar] [CrossRef] [Green Version]
- Legoff, J.; Resche-Rigon, M.; Bouquet, J.; Robin, M.; Naccache, S.N.; Mercier-Delarue, S.; Federman, S.; Samayoa, E.; Rousseau, C.; Piron, P.; et al. The eukaryotic gut virome in hematopoietic stem cell transplantation: New clues in enteric graft-versus-host disease. Nat. Med. 2017, 23, 1080–1085. [Google Scholar] [CrossRef]
- van Leeuwen, M.; Williams, M.M.; Koraka, P.; Simon, J.H.; Smits, S.L.; Osterhaus, A.D. Human picobirnaviruses identified by molecular screening of diarrhea samples. J. Clin. Microbiol. 2010, 48, 1787–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakuda, M.; Pongsuwanna, Y.; Taniguchi, K. Complete nucleotide sequences of two RNA segments of human picobirnavirus. J. Virol. Methods 2005, 126, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Collier, A.M.; Lyytinen, O.L.; Guo, Y.R.; Toh, Y.; Poranen, M.M.; Tao, Y.J. Initiation of RNA Polymerization and Polymerase Encapsidation by a Small dsRNA Virus. PLoS Pathog. 2016, 12, e1005523. [Google Scholar] [CrossRef] [Green Version]
- Patton, J.T.; Silvestri, L.S.; Tortorici, M.A.; Vasquez-Del Carpio, R.; Taraporewala, Z.F. Rotavirus genome replication and morphogenesis: Role of the viroplasm. Curr. Top. Microbiol. Immunol. 2006, 309, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Ooms, L.S.; Kobayashi, T.; Dermody, T.S.; Chappell, J.D. A post-entry step in the mammalian orthoreovirus replication cycle is a determinant of cell tropism. J. Biol. Chem. 2010, 285, 41604–41613. [Google Scholar] [CrossRef] [Green Version]
- Ng, T.F.; Kondov, N.O.; Deng, X.; Van Eenennaam, A.; Neibergs, H.L.; Delwart, E. A metagenomics and case-control study to identify viruses associated with bovine respiratory disease. J. Virol. 2015, 89, 5340–5349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valle, M.C.; Martinez, L.C.; Ferreyra, L.J.; Giordano, M.O.; Isa, M.B.; Pavan, J.V.; De Boccardo, G.; Massari, P.U.; Nates, S.V. Viral agents related to diarrheic syndrome in kidney transplanted patients. Medicina 2001, 61, 179–182. [Google Scholar]
- Boros, A.; Polgar, B.; Pankovics, P.; Fenyvesi, H.; Engelmann, P.; Phan, T.G.; Delwart, E.; Reuter, G. Multiple divergent picobirnaviruses with functional prokaryotic Shine-Dalgarno ribosome binding sites present in cloacal sample of a diarrheic chicken. Virology 2018, 525, 62–72. [Google Scholar] [CrossRef]
- Tang, J.; Ochoa, W.F.; Li, H.; Havens, W.M.; Nibert, M.L.; Ghabrial, S.A.; Baker, T.S. Structure of Fusarium poae virus 1 shows conserved and variable elements of partitivirus capsids and evolutionary relationships to picobirnavirus. J. Struct. Biol. 2010, 172, 363–371. [Google Scholar] [CrossRef] [Green Version]
- Kleymann, A.; Becker, A.; Malik, Y.S.; Kobayashi, N.; Ghosh, S. Detection and Molecular Characterization of Picobirnaviruses (PBVs) in the Mongoose: Identification of a Novel PBV Using an Alternative Genetic Code. Viruses 2020, 12, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yinda, C.K.; Ghogomu, S.M.; Conceicao-Neto, N.; Beller, L.; Deboutte, W.; Vanhulle, E.; Maes, P.; Van Ranst, M.; Matthijnssens, J. Cameroonian fruit bats harbor divergent viruses, including rotavirus H, bastroviruses, and picobirnaviruses using an alternative genetic code. Virus Evol. 2018, 4, vey008. [Google Scholar] [CrossRef] [Green Version]
- Abbas, A.A.; Taylor, L.J.; Dothard, M.I.; Leiby, J.S.; Fitzgerald, A.S.; Khatib, L.A.; Collman, R.G.; Bushman, F.D. Redondoviridae, a Family of Small, Circular DNA Viruses of the Human Oro-Respiratory Tract Associated with Periodontitis and Critical Illness. Cell Host Microbe 2019, 26, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allander, T.; Tammi, M.T.; Eriksson, M.; Bjerkner, A.; Tiveljung-Lindell, A.; Andersson, B. Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proc. Natl. Acad. Sci. USA 2005, 102, 12891–12896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, M.O.; Martinez, L.C.; Masachessi, G.; Barril, P.A.; Ferreyra, L.J.; Isa, M.B.; Valle, M.C.; Massari, P.U.; Nates, S.V. Evidence of closely related picobirnavirus strains circulating in humans and pigs in Argentina. J. Infect. 2011, 62, 45–51. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Capsid-FAM | RdRp-FAM | RdRp-CY3 | RdRp-CY5 | PBV Segment 1 | PBV Capsid Only | PBV Segment 2-RdRp | NGS Reads | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample ID | Country | Year | Ct | Ct | Ct | Ct | NGS Reads | % Coverage | Closest Reference | NGS Reads | % Coverage | NGS Reads | % Coverage | Closest Reference | Total |
| NYB4466 | USA-NY | 2018 | −1 | 29.41 | −1 | −1 | 3394 | 99% | ABT3406 | 2539 | 100% | 2616 | 97% | ABT3406 | 8,048,648 |
| NYB4468 | USA-NY | 2018 | −1 | 33.36 | −1 | 30.34 | 35 | 62% | ABT3406 | 28 | 70% | 35 | 67% | ABT3406 | 14,782,666 |
| NYB4138 | USA-NY | 2018 | −1 | 26.4 | −1 | 23.87 | 201 | 96% | ABT3406 | 143 | 97% | 266 | 94% | ABT3406 | 7,321,340 |
| MRN001 | COL | 2019 | 26.53 | 30.78 | −1 | 24.06 | 178 | 100% | ABT3406 | 139 | 100% | 288 | 97% | ABT3406 | 3,749,428 |
| MRN006 | COL | 2019 | 22.18 | 28.33 | −1 | 20.42 | 2229 | 100% | ABT3406 | 1655 | 100% | 2905 | 100% | ABT3406 | 3,541,114 |
| MRN012 | COL | 2019 | −1 | 23.36 | 22.78 | −1 | 111 | 42% | KY928792 | 111 | 50% | 215 | 96% | KY928727 | 9,517,318 |
| MRN012 | COL | 2019 | −1 | 23.36 | 22.78 | −1 | 3202 | 100% | MRN012 | 2190 | 100% | 3020 | 100% | KM285233 | 9,517,318 |
| MRN015 | COL | 2019 | 24.69 | 23.1 | 24.15 | 25.5 | 7518 | 100% | ABT3406 | 5425 | 100% | 5793 | 99% | ABT3406 | 4,685,264 |
| MRN015 | COL | 2019 | 24.69 | 23.1 | 24.15 | 25.5 | 15,340 | 100% | MRN012 | 10,584 | 100% | 11,053 | 100% | KM285233 | 4,685,264 |
| MRN016 | COL | 2019 | −1 | 28.71 | −1 | 25.56 | 675 | 100% | ABT3406 | 586 | 100% | 812 | 100% | ABT3406 | 14,733,470 |
| MRN020 | COL | 2019 | −1 | 35.51 | −1 | −1 | 169 | 87% | ABT3406 | 131 | 89% | 72 | 67% | ABT3406 | 34,621,664 |
| MRN021 | COL | 2019 | 35.59 | 35.25 | −1 | 31.49 | 36 | 54% | ABT3406 | 22 | 54% | 72 | 92% | ABT3406 | 34,481,740 |
| MRN023 | COL | 2019 | −1 | 25.12 | 24.22 | −1 | 3233 | 100% | MRN012 | 2533 | 100% | 3925 | 100% | KM285233 | 8,746,944 |
| MRN031 | COL | 2019 | −1 | 32.26 | −1 | 30.16 | 25 | 62% | MK204396 | 22 | 71% | 20 | 47% | ABT3406 | 6,175,024 |
| MRN032 | COL | 2019 | −1 | 27.69 | −1 | 25.94 | 66 | 92% | MK204396 | 56 | 89% | 57 | 95% | ABT3406 | 5,853,846 |
| MRN033 | COL | 2019 | −1 | 25.35 | 26.91 | 34.56 | 28 | 48% | MK204396 | 14 | 40% | 307 | 100% | MH933835 | 16,786,708 |
| MRN033 | COL | 2019 | −1 | 25.35 | 26.91 | 34.56 | 1880 | 99% | MRN012 | 1439 | 100% | 1361 | 100% | KM285233 | 16,786,708 |
| MRN034 | COL | 2019 | −1 | 26.41 | 27.39 | 35.75 | 12 | 30% | MK204396 | 10 | 33% | 957 | 100% | MH933835 | 9,571,150 |
| MRN034 | COL | 2019 | −1 | 26.41 | 27.39 | 35.75 | 2627 | 100% | MRN012 | 2071 | 100% | 1315 | 100% | KM285233 | 9,571,150 |
| MRN035 | COL | 2019 | 35.84 | 29.3 | −1 | 26.64 | 458 | 100% | ABT3406 | 431 | 100% | 437 | 92% | ABT3406 | 8,335,936 |
| MRN038 | COL | 2019 | 26.57 | 30.67 | −1 | 29.08 | 352 | 100% | ABT3406 | 300 | 100% | 280 | 97% | ABT3406 | 1,585,992 |
| MRN039 | COL | 2019 | 34.43 | 21.56 | 25.45 | 29.43 | 545 | 100% | MK204396 | 363 | 100% | 417 | 89% | MH933835 | 4,737,986 |
| MRN039 | COL | 2019 | 34.43 | 21.56 | 25.45 | 29.43 | 1416 | 97% | MRN012 | 1372 | 100% | 2387 | 99% | KM285233 | 4,737,986 |
| MRN044 | COL | 2019 | 22.58 | 31.48 | −1 | 26.95 | 5395 | 99% | ABT3406 | 4823 | 98% | 5603 | 100% | ABT3406 | 8,691,792 |
| MRN044 | COL | 2019 | 22.58 | 31.48 | −1 | 26.95 | 40 | 56% | MRN012 | 36 | 67% | 109 | 70% | KM285233 | 8,691,792 |
| MRN046 | COL | 2019 | 19.61 | 27.14 | −1 | 23.29 | 2490 | 99% | ABT3406 | 1786 | 99% | 2302 | 100% | ABT3406 | 4,679,714 |
| Patient ID | Age | Sex | RdRp FAM Ct | Log Titer cp/mL | Sputum Color | Viral Coinfection | Bacterial Coinfection | Medical Diagnosis | Symptoms |
|---|---|---|---|---|---|---|---|---|---|
| NYB4466 | 52 | M | 29.41 | 4.34 | Yellow | HHV-4 | Stenotrophomonas, Mycoplasma | n.d. | n.d. |
| NYB4468 | 40 | F | 33.36 | 3.15 | Clear | Influenza, Strep phage | Streptococcus pneumonia, Bacillus | n.d. | n.d. |
| NYB4138 | 71 | M | 26.4 | 5.25 | Brown/yellow | Klebsiella phage | Klebsiella pneumonia | n.d. | n.d. |
| NYB4477 | 51 | F | 36.26 | 2.28 | Brown/yellow | none | None | n.d. | n.d. |
| NYB4470 | 62 | F | 34.6 | 2.23 | Yellow | HHV-1, coronavirus OC43 | None | n.d. | n.d. |
| BBL46722 | 57 | F | 35.04 | 2.10 | Brown | None | None | n.d. | n.d. |
| MRN001 | 32 | F | 30.78 | 3.38 | Brown | None | Treponema, Bacteroides | Myco TB+ | Multidrug resistance, 2nd phase of treatment, fever, weight loss, chest pain, pain with breathing |
| MRN006 | 36 | M | 28.33 | 4.12 | Clear | None | none | Pulmonary TB | Smear microscopy (+), BARR (++), chest pains, fever, weight loss, fatigue |
| MRN012 | 18 | F | 23.36 | 5.62 | Clear | Respirovirus 3 | none | Pulmonary TB | Smear microscopy (−), Radiography (+), chest pains, cough, fever, weight loss |
| Sptm Ctrl | 36.34 | 1.71 | n.d. | None | Klebsiella pneumonia | n.d. | n.d. | ||
| MRN015 | 36 | M | 23.1 | 5.96 | Clear | None | Bordetalla, Burkholderia, Pseudomonas, Serretia | Pulmonary TB | Smear microscopy (+), BARR++, severe cough, fever, weight loss, chills, fatigue |
| MRN016 | 28 | F | 28.71 | 4.27 | Clear | Pseudomonas phage | Pseudomonas aeruginosa | Pulmonary TB | Smear microscopy (+), BARR+++, 2nd phase of treatment |
| MRN020 | 83 | M | 35.51 | 2.22 | Clear | None | Mycoplasma, Treponema, Streptococcus | Negative | Coughing up blood, fatigue, weight loss |
| MRN021 | 68 | F | 35.25 | 2.30 | Yellow/clear | Enterovirus D | None | Negative | Fever, night sweats, pain when breathing |
| MRN023 | 60 | M | 25.12 | 5.35 | Brown/yellow | None | Haemophilus parainfluenza | Negative | Night sweats, fever, coughing up blood |
| MRN031 | 86 | M | 32.26 | 3.20 | Clear | Rhinovirus A | Clostridium | Smear(−), Rad(+) | Fatigue, fever, night sweats |
| MRN032 | 71 | M | 27.69 | 4.58 | Yellow | Rhinovirus A, Haemophilus virus | Haemophilus influenzae | Pneumonia | Chest pains, coughing up blood, pain when breathing |
| MRN033 | 52 | M | 25.35 | 5.28 | Clear | None | Bacillus, Treponema | Chronic bronchitis (−) | Chest pains, Chest pains when coughing |
| MRN034 | 61 | M | 26.41 | 4.96 | Clear | None | Bacillus, Mycoplasma, Treponema | EPOC BARR(+) | Night sweats |
| MRN035 | 68 | M | 29.3 | 4.09 | Clear | None | Spirochaeta, Treponema, Bacteroides, Mycoplasma | Pulmonary TB, Barr(++) | Coughing up blood, fatigue, chest pains |
| MRN036 | 71 | F | 34.19 | 2.62 | Yellow | None | None | Bronchopneumonia (−) | Chest pains, hest pains when coughing |
| MRN038 | 69 | M | 30.67 | 3.68 | Clear | None | Burkholderia, Serretia, Pseudomonas | EPOC BARR (−) | Fatigue, night sweats |
| MRN039 | 20 | M | 21.56 | 6.42 | Clear | None | Treponema | Chronic bronchitis (−) | Chest pains when coughing and breathing |
| MRN044 | 72 | M | 31.48 | 3.44 | Clear | None | None | EPOC BARR (−) | Night sweats, chills, fatigue |
| MRN046 | 72 | F | 27.14 | 4.74 | Clear | None | None | Pneumonia (−) | Chest pains when coughing, weight loss, chills |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berg, M.G.; Forberg, K.; Perez, L.J.; Luk, K.-C.; Meyer, T.V.; Cloherty, G.A. Emergence of a Distinct Picobirnavirus Genotype Circulating in Patients Hospitalized with Acute Respiratory Illness. Viruses 2021, 13, 2534. https://doi.org/10.3390/v13122534
Berg MG, Forberg K, Perez LJ, Luk K-C, Meyer TV, Cloherty GA. Emergence of a Distinct Picobirnavirus Genotype Circulating in Patients Hospitalized with Acute Respiratory Illness. Viruses. 2021; 13(12):2534. https://doi.org/10.3390/v13122534
Chicago/Turabian StyleBerg, Michael G., Kenn Forberg, Lester J. Perez, Ka-Cheung Luk, Todd V. Meyer, and Gavin A. Cloherty. 2021. "Emergence of a Distinct Picobirnavirus Genotype Circulating in Patients Hospitalized with Acute Respiratory Illness" Viruses 13, no. 12: 2534. https://doi.org/10.3390/v13122534
APA StyleBerg, M. G., Forberg, K., Perez, L. J., Luk, K. -C., Meyer, T. V., & Cloherty, G. A. (2021). Emergence of a Distinct Picobirnavirus Genotype Circulating in Patients Hospitalized with Acute Respiratory Illness. Viruses, 13(12), 2534. https://doi.org/10.3390/v13122534