
Interactions of Influenza and SARS-CoV-2 with the Lung Endothelium: Similarities, Differences, and Implications for Therapy


Abstract
:1. Introduction
2. Virus Background
2.1. Influenza
2.2. SARS-CoV-2
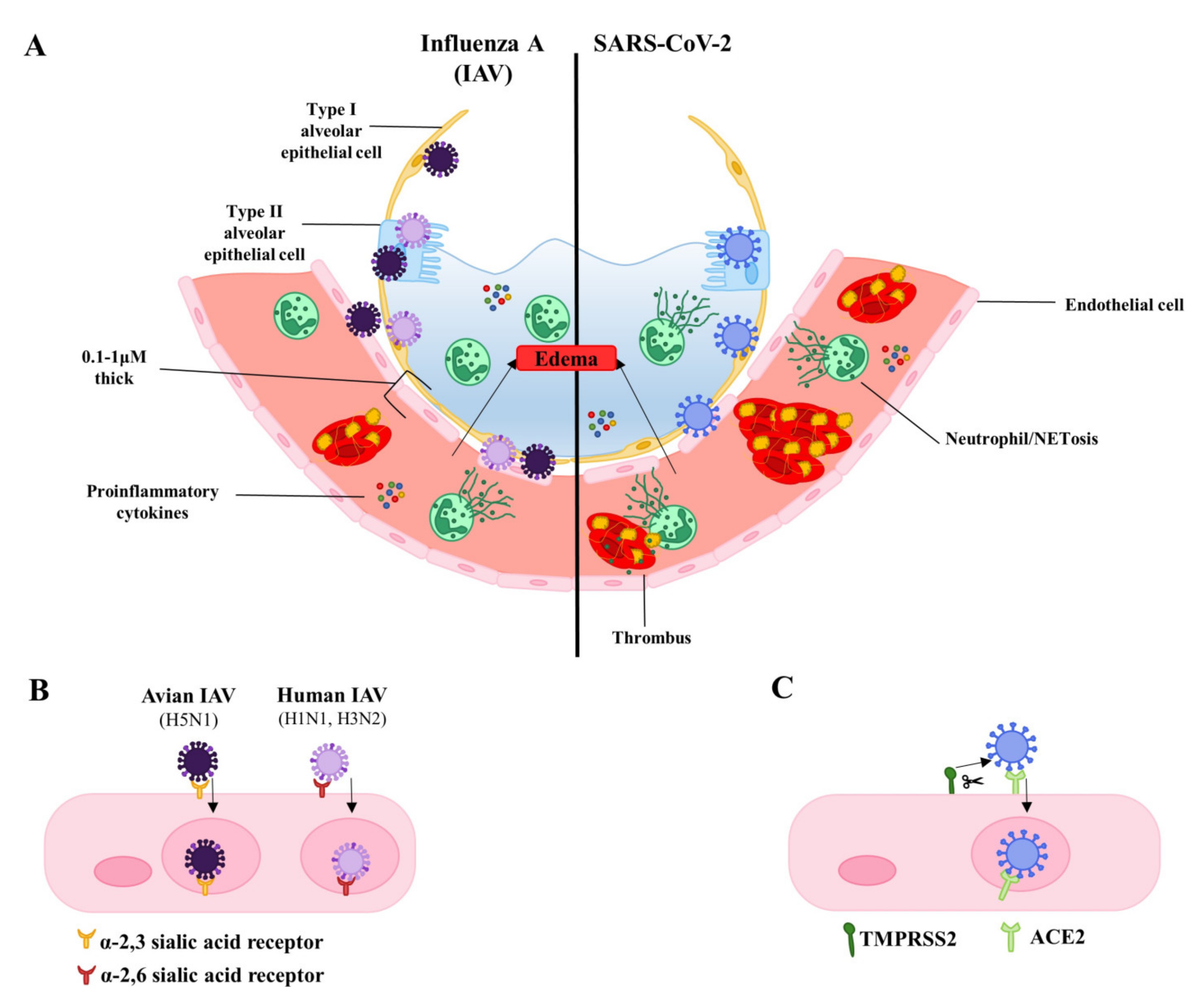
3. Infection of the Lung Endothelium by the Virus: Frequency and Potential Relevance to Pathology
3.1. Cell Death and Endothelial Permeability
3.2. Inflammatory Cell Death
4. Indirect Endothelial Damage and Dysfunction—The Role of Cytokines and Inflammation
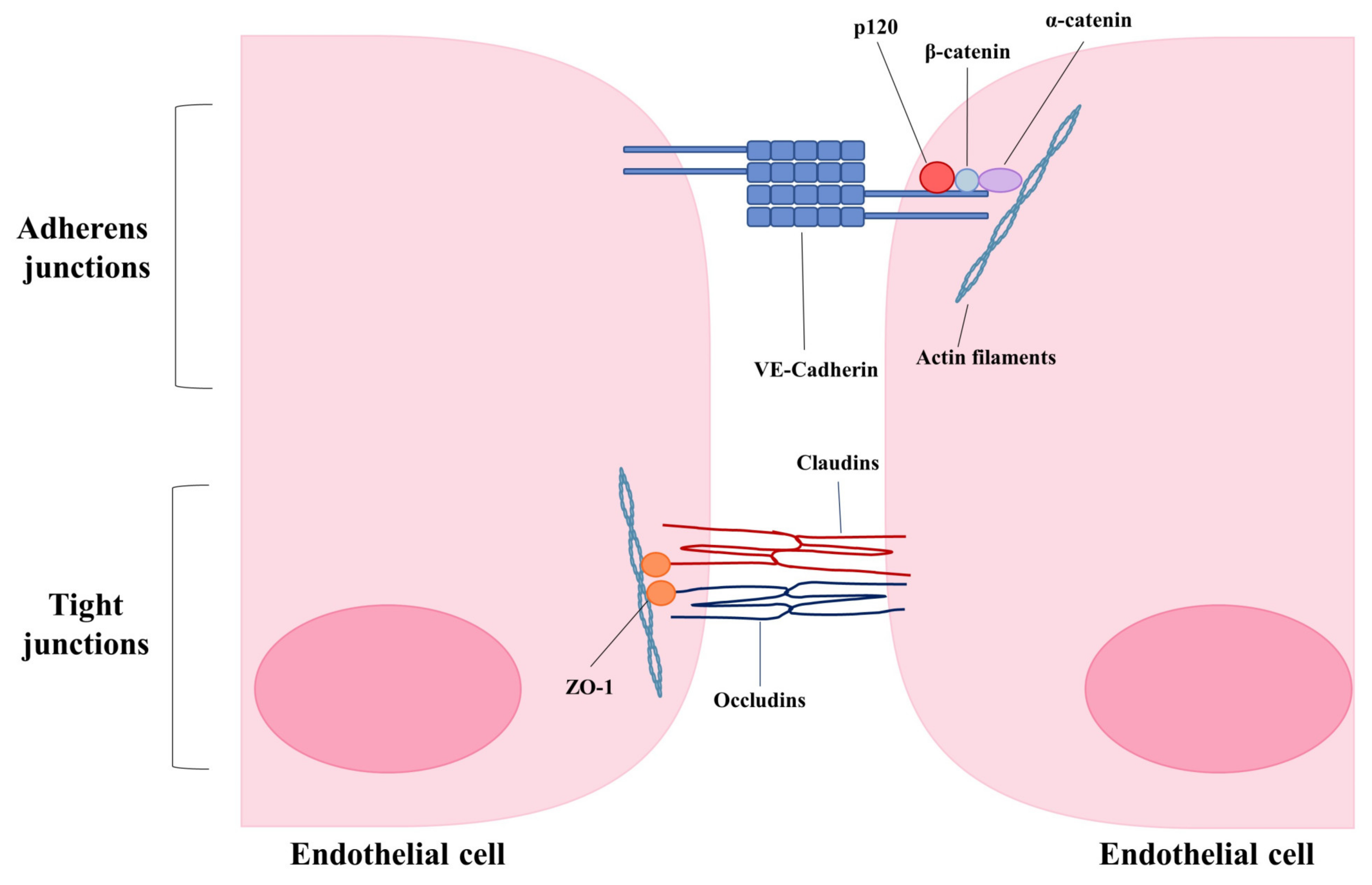
5. Consequences of Endothelial Disruption—Loss of Junctional Integrity
6. Endothelial-Platelet Interactions and Coagulopathy
6.1. Influenza
6.2. SARS-CoV-2
7. Neutrophils and Neutrophil Extracellular Traps
7.1. Influenza
7.2. SARS-CoV-2
8. Implications for Therapy
8.1. Role of Antiviral Drugs
8.2. Improving the Host Response in Severe Influenza and COVID-19: Targeting Endothelial Activation and Inflammation
8.3. Improving the Host Response via Anticoagulation
9. Unanswered Questions
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mehta, D.; Malik, A.B. Signaling Mechanisms Regulating Endothelial Permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef] [PubMed]
- Oslund, K.L.; Baumgarth, N. Influenza-induced innate immunity: Regulators of viral replication, respiratory tract pathology & adaptive immunity. Future Virol. 2011, 6, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Denney, L.; Ho, L.P. The role of respiratory epithelium in host defence against influenza virus infection. Biomed. J. 2018, 41, 218–233. [Google Scholar] [CrossRef] [PubMed]
- Vareille, M.; Kieninger, E.; Edwards, M.R.; Regamey, N. The airway epithelium: Soldier in the fight against respiratory viruses. Clin. Microbiol. Rev. 2011, 24, 210–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanLeuven, J.T.; Ridenhour, B.J.; Gonzalez, A.J.; Miller, C.R.; Miura, T.A. Lung epithelial cells have virus-specific and shared gene expression responses to infection by diverse respiratory viruses. PLoS ONE 2017, 12, e0178408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, S.M.; Darwish, I.; Lee, W.L. Endothelial activation and dysfunction in the pathogenesis of influenza A virus infection. Virulence 2013, 4, 537–542. [Google Scholar] [CrossRef] [Green Version]
- Dou, D.; Revol, R.; Östbye, H.; Wang, H.; Daniels, R. Influenza A virus cell entry, replication, virion assembly and movement. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Prim. 2018, 4. [Google Scholar] [CrossRef]
- McAuley, J.L.; Gilbertson, B.P.; Trifkovic, S.; Brown, L.E.; McKimm-Breschkin, J.L. Influenza virus neuraminidase structure and functions. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Davies, B.E. Pharmacokinetics of oseltamivir: An oral antiviral for the treatment and prophylaxis of influenza in diverse populations. J. Antimicrob. Chemother. 2010, 65 Suppl 2, ii5–ii10. [Google Scholar] [CrossRef] [Green Version]
- Taubenberger, J.K.; Morens, D.M. 1918 Influenza: The Mother of All Pandemics. Emerg. Infect. Dis. 2006, 12, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Li, F.C.K.; Choi, B.C.K.; Sly, T.; Pak, A.W.P. Finding the real case-fatality rate of H5N1 avian influenza. J. Epidemiol. Community Health 2008, 62, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Peiris, J.S.M.; De Jong, M.D.; Guan, Y. Avian influenza virus (H5N1): A threat to human health. Clin. Microbiol. Rev. 2007, 20, 243–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Lian, X.; Su, X.; Wu, W.; Marraro, G.A.; Zeng, Y. From SARS and MERS to COVID-19: A brief summary and comparison of severe acute respiratory infections caused by three highly pathogenic human coronaviruses. Respir. Res. 2020, 21. [Google Scholar] [CrossRef]
- Yang, Y.; Xiao, Z.; Ye, K.; He, X.; Sun, B.; Qin, Z.; Yu, J.; Yao, J.; Wu, Q.; Bao, Z.; et al. SARS-CoV-2: Characteristics and current advances in research. Virol. J. 2020, 17. [Google Scholar] [CrossRef]
- Peiris, J.S.; Yuen, K.Y.; Osterhaus, A.D.M.E.; Stohr, K. The severe acute respiratory syndrome. N. Engl. J. Med. 2003, 349, 2431–2441. [Google Scholar] [CrossRef] [Green Version]
- Memish, Z.A.; Perlman, S.; Van Kerkhove, M.D.; Zumla, A. Middle East respiratory syndrome. Lancet 2020, 395, 1063–1077. [Google Scholar] [CrossRef]
- Petrosillo, N.; Viceconte, G.; Ergonul, O.; Ippolito, G.; Petersen, E. COVID-19, SARS and MERS: Are they closely related? Clin. Microbiol. Infect. 2020, 26, 729–734. [Google Scholar] [CrossRef]
- Cioffi, D.L.; Pandey, S.; Alvarez, D.F.; Cioffi, E.A. Terminal sialic acids are an important determinant of pulmonary endothelial barrier integrity. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 302, 1067–1077. [Google Scholar] [CrossRef] [Green Version]
- Yao, L.; Korteweg, C.; Hsueh, W.; Gu, J. Avian influenza receptor expression in H5N1-infected and noninfected human tissues. FASEB J. 2007, 22, 733–740. [Google Scholar] [CrossRef]
- Zeng, H.; Pappas, C.; Belser, J.A.; Houser, K.V.; Zhong, W.; Wadford, D.A.; Stevens, T.; Balczon, R.; Katz, J.M.; Tumpey, T.M. Human Pulmonary Microvascular Endothelial Cells Support Productive Replication of Highly Pathogenic Avian Influenza Viruses: Possible Involvement in the Pathogenesis of Human H5N1 Virus Infection. J. Virol. 2012, 86, 667–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, S.M.; Mubareka, S.; Lee, W.L. The lung microvascular endothelium as a therapeutic target in severe influenza. Antiviral Res. 2013, 99, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Van Riel, D.; Munster, V.J.; De Wit, E.; Rimmelzwaan, G.F.; Fouchier, R.A.M.; Osterhaus, A.D.M.E.; Kuiken, T. Human and Avian Influenza Viruses Target Different Cells in the Lower Respiratory Tract of Humans and Other Mammals. Am. J. Pathol. 2007, 171, 1215–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Short, K.R.; Kroeze, E.J.B.V.; Reperant, L.A.; Richard, M.; Kuiken, T. Influenza virus and endothelial cells: A species specific relationship. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Tundup, S.; Kandasamy, M.; Perez, J.T.; Mena, N.; Steel, J.; Nagy, T.; Albrecht, R.A.; Manicassamy, B. Endothelial cell tropism is a determinant of H5N1 pathogenesis in mammalian species. PLoS Pathog. 2017, 13, e1006270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ocana-Macchi, M.; Bel, M.; Guzylack-piriou, L.; Ruggli, N.; Liniger, M.; Mccullough, K.C.; Sakoda, Y.; Isoda, N.; Matrosovich, M.; Summerfield, A. Hemagglutinin-Dependent Tropism of H5N1 Avian Influenza Virus for Human Endothelial Cells. J. Virol. 2009, 83, 12947–12955. [Google Scholar] [CrossRef] [Green Version]
- Sumikoshi, M.; Hashimoto, K.; Kawasaki, Y.; Sakuma, H.; Suzutani, T.; Suzuki, H.; Hosoya, M. Human Influenza Virus Infection and Apoptosis Induction in Human Vascular Endothelial Cells. J. Med. Virol. 2008, 80, 1072–1078. [Google Scholar] [CrossRef]
- Ampomah, P.B.; Lim, L.H.K. Influenza A virus - induced apoptosis and virus propagation. Apoptosis 2020, 25, 1–11. [Google Scholar] [CrossRef]
- Zamarin, D.; Garcia-Sastre, A.; Xiao, X.; Wang, R.; Palese, P. Influenza Virus PB1-F2 Protein Induces Cell Death through Mitochondrial ANT3 and VDAC1. PLoS Pathog. 2005, 1, e4. [Google Scholar] [CrossRef]
- Schultz-cherry, S.; Dybdahl-sissoko, N.; Neumann, G.; Kawaoka, Y.; Hinshaw, V.S. Influenza Virus NS1 Protein Induces Apoptosis in Cultured Cells. J. Virol. 2001, 75, 7875–7881. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, M.G.; Gamage, A.; Zyla, R.; Armstrong, S.M.; Advani, S.; Advani, A.; Wang, C.; Lee, L. Influenza Virus Infection Induces Platelet-Endothelial Adhesion Which Contributes to Lung Injury. J. Virol. 2016, 90, 1812–1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogiwara, H.; Yasui, F.; Munekata, K.; Takagi-Kamiya, A.; Munakata, T.; Nomura, N.; Shibasaki, F.; Kuwahara, K.; Sakaguchi, N.; Sakoda, Y.; et al. Histopathological evaluation of the diversity of cells susceptible to H5N1 virulent avian influenza virus. Am. J. Pathol. 2014, 184, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Armstrong, S.M.; Sugiyama, M.G.; Tabuchi, A.; Krauszman, A.; Kuebler, W.M.; Mullen, B.; Advani, S.; Advani, A.; Lee, W.L. Influenza-induced priming and leak of human lung microvascular endothelium upon exposure to staphylococcus aureus. Am. J. Respir. Cell Mol. Biol. 2015, 53, 459–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morens, D.M.; Taubenberger, J.K.; Fauci, A.S. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: Implications for pandemic influenza preparedness. J. Infect. Dis. 2008, 198, 962–970. [Google Scholar] [CrossRef]
- Colmenero, I.; Santonja, C.; Alonso-Riaño, M.; Noguera-Morel, L.; Hernández-Martín, A.; Andina, D.; Wiesner, T.; Rodríguez-Peralto, J.L.; Requena, L.; Torrelo, A. SARS-CoV-2 endothelial infection causes COVID-19 chilblains: Histopathological, immunohistochemical and ultrastructural study of seven paediatric cases. Br. J. Dermatol. 2020, 183, 729–737. [Google Scholar] [CrossRef]
- Paniz-Mondolfi, A.; Bryce, C.; Grimes, Z.; Gordon, R.E.; Reidy, J.; Lednicky, J.; Sordillo, E.M.; Fowkes, M. Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). J. Med. Virol. 2020, 92, 699–702. [Google Scholar] [CrossRef] [Green Version]
- Bradley, B.T.; Maioli, H.; Johnston, R.; Chaudhry, I.; Fink, S.L.; Xu, H.; Najafian, B.; Deutsch, G.; Lacy, J.M.; Williams, T.; et al. Histopathology and ultrastructural findings of fatal COVID-19 infections in Washington State: A case series. Lancet 2020, 396, 320–332. [Google Scholar] [CrossRef]
- Fox, S.E.; Li, G.; Akmatbekov, A.; Harbert, J.L.; Lameira, F.S.; Brown, J.Q.; Vander Heide, R.S. Unexpected features of cardiac pathology in COVID-19 infection. Circulation 2020, 1123–1125. [Google Scholar] [CrossRef]
- Puelles, V.G.; Lütgehetmann, M.; Lindenmeyer, M.T.; Sperhake, J.P.; Wong, M.N.; Allweiss, L.; Chilla, S.; Heinemann, A.; Wanner, N.; Liu, S.; et al. Multiorgan and Renal Tropism of SARS-CoV-2. N. Engl. J. Med. 2020, 383, 590–592. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrier, T.; Erichsen, S.; Schiergens, T.S.; Herrier, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Hamming, I.; Timens, W.; Bulthuis, M.L.C.; Lely, A.T.; Navis, G.J.; Goor, H. Van Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Bertram, S.; Heurich, A.; Lavender, H.; Gierer, S.; Danisch, S.; Soilleux, E.J.; Perin, P.; Lucas, J.M.; Nelson, P.S.; Pohlmann, S.; et al. Influenza and SARS-Coronavirus Activating Proteases TMPRSS2 and HAT Are Expressed at Multiple Sites in Human Respiratory and Gastrointestinal Tracts. PLoS ONE 2012, 7, e35876. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Nao, N.; Shirato, K.; Kawase, M.; Saito, S.; Takayama, I.; Nagata, N.; Sekizuka, T.; Katoh, H.; Kato, F.; et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2- expressing cells. Proc. Natl. Acad. Sci. USA 2020, 117, 7001–7003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.; Vishal, C.C.; Yuan, C.X.X.; Huang, J.L.Y.; Tsz, L.C.; Ng, K. Expression of SARS-CoV-2 receptor ACE2 and TMPRSS2 in human primary conjunctival and pterygium cell lines and in mouse cornea. R. Coll. Ophthalmol. 2020, 34, 1212–1219. [Google Scholar] [CrossRef]
- Maximilian Ackermann, S.E.V.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Arno Vanstapel, C.W.; Stark, H.; Alexandar Tzankov, W.W.L.; Li, V.W.; Mentzer, S.J.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Ren, Y.; Shu, T.; Wu, D.; Mu, J.; Wang, C.; Huang, M.; Han, Y.; Zhang, X.; Zhou, W. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell. Mol. Immunol. 2020, 17, 881–883. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Y.; Guan, Z.; Li, H.; Ye, M.; Chen, X.; Shen, J.; Zhou, Y.; Shi, Z.; Zhou, P.; et al. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal Transduct. Target. Ther. 2020, 5. [Google Scholar] [CrossRef]
- Tan, Y.; Tan, T.H.P.; Lee, M.J.; Tham, P.; Gunalan, V.; Druce, J.; Birch, C.; Catton, M.; Fu, N.Y.; Yu, V.C.; et al. Induction of Apoptosis by the Severe Acute Respiratory Syndrome Coronavirus 7a Protein Is Dependent on Its Interaction with the Bcl-Xl Protein. J. Virol. 2007, 81, 6346–6355. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.; Ma, C.; Chan, W.; Yin, H.; Chan, E. The SARS-Coronavirus Membrane protein induces apoptosis through modulating the Akt survival pathway. Arch. Biochem. Biophys. 2007, 459, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2010, 7, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mcauley, J.L.; Tate, M.D.; Mackenzie-kludas, C.J.; Pinar, A.; Zeng, W.; Stutz, A.; Latz, E.; Brown, L.E.; Mansell, A. Activation of the NLRP3 Inflammasome by IAV Virulence Protein PB1-F2 Contributes to Severe Pathophysiology and Disease. PLoS Pathog. 2013, 9, e1003392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; Mcelvania-, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P.-Y. The NLRP3 Inflammasome Mediates in vivo Innate Immunity to Influenza A Virus through Recognition of Viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef] [Green Version]
- Thomas, P.G.; Dash, P.; Aldridge, J.R.; Ellebedy, A.H.; Reynolds, C.; Funk, A.J.; Martin, W.J.; Lamkanfi, M.; Webby, R.J.; Boyd, K.L.; et al. Article The Intracellular Sensor NLRP3 Mediates Key Innate and Healing Responses to Influenza A Virus via the Regulation of Caspase-1. Immunity 2009, 30, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Carlos, J.; Vaccari, D.R.; Dietrich, W.D.; Keane, R.W.; Pablo, J.; Vaccari, D.R. The Inflammasome in Times of COVID-19. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Yap, J.K.Y.; Moriyama, M.; Iwasaki, A. Inflammasomes and Pyroptosis as Therapeutic Targets for COVID-19. J. Immunol. 2020. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.; Wang, X.; Hu, B.; Zhang, L.; Zhang, W.; Guo, H.; Jiang, R.; Liu, M.; Chen, Y.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579. [Google Scholar] [CrossRef] [Green Version]
- Soares, V.C.; De Azevedo, I.G.; Fintelman-rodrigues, N.; Carolina, Q.; Mattos, M.; De Freitas, C.S.; Temerozo, J.R.; Hottz, E.D.; Barreto, E.A.; Miranda, M.; et al. SARS-CoV-2 induces inflammasome-dependent pyroptosis and downmodulation of HLA-DR in human monocytes. MedRxiv 2020, in press. [Google Scholar] [CrossRef]
- Nagashima, S.; Mendes, M.C.; Paula, A.; Martins, C.; Borges, N.H.; Godoy, T.M.; Flavia, A.; Miggiolaro, S.; Dezidério, S.; Machado-souza, C.; et al. Endothelial Dysfunction and Thrombosis in Patients With COVID-19—Brief Report. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2404–2407. [Google Scholar] [CrossRef]
- Perico, L.; Renia, L.; Casiraghi, F.; Ng, L.F.P.; Renia, L.; Remuzzi, G. Immunity, endothelial injury and complement-induced coagulopathy in COVID-19. Nat. Rev. Nephrol. 2020. [Google Scholar] [CrossRef]
- Brandes, M.; Klauschen, F.; Kuchen, S.; Germain, R.N. A systems analysis identifies a feed-forward inflammatory circuit to lethal influenza infection. Cell 2014, 154, 197–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jong, M.D.; Simmons, C.P.; Thanh, T.T.; Hien, V.M.; Smith, G.J.D.; Nguyen, T.; Chau, B.; Hoang, D.M.; Chau, N.V.V.; Khanh, T.H.; et al. Fatal outcome of human influenza A ( H5N1 ) is associated with high viral load and hypercytokinemia. Nat. Med. 2006, 12, 1203–1207. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.G.N.; Liu, F.; Verin, A.D.; Birukova, A.; Dechert, M.A.; Gerthoffer, W.T.; Bamburg, J.R.; English, D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J. Clin. Invest. 2001, 108, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Teijaro, J.R.; Walsh, K.B.; Cahalan, S.; Fremgen, D.M.; Roberts, E.; Scott, F.; Martinborough, E.; Peach, R.; Oldstone, M.B.A.; Rosen, H. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 2011, 146, 980–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, Q.; Yang, K.; Wang, W.; Jiang, L.; Song, J. Clinical predictors of mortality due to COVID - 19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020, 46, 846–848. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhao, B.; Qu, Y.; Chen, Y.; Xiong, J.; Feng, Y.; Men, D.; Huang, Q.; Liu, Y.; Yang, B.; et al. Detectable serum SARS-CoV-2 viral load (RNAaemia) is closely correlated with drastically elevated interleukin 6 (IL-6) level in critically ill COVID-19 patients. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Zegeye, M.M.; Lindkvist, M.; Fälker, K.; Kumawat, A.K.; Paramel, G.; Grenegård, M.; Sirsjö, A.; Ljungberg, L.U. Activation of the JAK / STAT3 and PI3K / AKT pathways are crucial for IL-6 trans- response in human vascular endothelial cells. Cell Commun. Signal. 2018, 16. [Google Scholar] [CrossRef]
- Didion, S.P. Cellular and Oxidative Mechanisms Associated with Interleukin-6 Signaling in the Vasculature. Int. J. Mol. Sci. 2017, 18, 2563. [Google Scholar] [CrossRef] [Green Version]
- Rose-john, S. IL-6 Trans-Signaling via the Soluble IL-6 Receptor: Importance for the Pro-Inflammatory Activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237–1247. [Google Scholar] [CrossRef]
- Chen, W.; Yuan, H.; Cao, W.; Wang, T.; Chen, W.; Yu, H.; Fu, Y.; Jiang, B.; Zhou, H.; Guo, H.; et al. Blocking interleukin-6 trans-signaling protects against renal fibrosis by suppressing STAT3 activation. Theranostics 2019, 9, 3980–3991. [Google Scholar] [CrossRef] [PubMed]
- Greenhill, C.J.; Rose-john, S.; Lissilaa, R.; Ferlin, W.; Ernst, M.; Hertzog, P.J.; Mansell, A.; Jenkins, B.J. IL-6 Trans -Signaling Modulates TLR4-Dependent Inflammatory Responses via STAT3. J. Immunol. 2011, 186, 1199–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuett, H.; Waetzig, G.H.; Annema, W.; Luchtefeld, M.; Hillmer, A.; Bavendiek, U.; Von Felden, J.; Divchev, D.; Kempf, T.; Wollert, K.C.; et al. Transsignaling of Interleukin-6 Crucially Contributes to Atherosclerosis in Mice. Arter. Thromb. Vasc. Biol. 2011, 32, 281–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atreya, R.; Mudter, J.; Finotto, S.; Mullberg, J.; Jostock, T.; Wirtz, S.; Schutz, M.; Bartsch, B.; Holtmann, M.; Becker, C.; et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: Evidence in Crohn disease and experimental colitis in vivo. Nat. Med. 2000, 6, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-john, S.; Treiber, M.; Kloppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akia, S.; et al. Stat3/Socs3 Activation by IL-6 Transsignaling Promotes Progression of Pancreatic Intraepithelial Neoplasiaand Development of Pancreatic Cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, S.; Hara, T.; Mitsuyama, K.; Yamamoto, M.; Tsuruta, O.; Sata, M.; Scheller, J.; Rose-John, S.; Kado, S.; Takada, T. Essential Roles of IL-6 Trans -Signaling in Colonic Epithelial Cells, Induced by the IL-6/Soluble− IL-6 Receptor Derived from Lamina Propria Macrophages, on the Development of Colitis-Associated Premalignant Cancer in a Murine Model. J. Immunol. 2009, 184, 1543–1551. [Google Scholar] [CrossRef] [Green Version]
- Desai, T.R.; Leeper, N.J.; Hynes, K.L.; Gewertz, B.L. Interleukin-6 Causes Endothelial Barrier Dysfunction via the Protein Kinase C Pathway. J. Surg. Res. 2002, 104, 118–123. [Google Scholar] [CrossRef]
- Lo, C.; Chen, M.; Hsiao, M.; Wang, S.; Chen, C.; Hsiao, S.; Chang, J.-S.; Lai, T.-C.; Rose-John, S.; Kuo, M.-L.; et al. IL-6 Trans-Signaling in Formation and Progression of Malignant Ascites in Ovarian Cancer. Cancer Res. 2011, 71, 424–435. [Google Scholar] [CrossRef] [Green Version]
- Kobasa, D.; Jones, S.M.; Shinya, K.; Kash, J.C.; Copps, J.; Ebihara, H.; Hatta, Y.; Kim, J.H.; Halfmann, P.; Hatta, M.; et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nat. Lett. 2007, 445. [Google Scholar] [CrossRef]
- Leisman, D.E.; Ronner, L.; Pinotti, R.; Taylor, M.D.; Sinha, P.; Calfee, C.S.; Hirayama, A.V.; Mastroiani, F.; Turtle, C.J.; Harhay, M.O.; et al. Cytokine elevation in severe and critical COVID-19: A rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir. Med. 2020, 8, 1233–1244. [Google Scholar] [CrossRef]
- Mudd, P.A.; Crawford, J.C.; Turner, J.S.; Souquette, A.; Reynolds, D.; Bender, D.; Bosanquet, J.P.; Anand, N.J.; Striker, D.A.; Martin, R.S.; et al. Distinct inflammatory profiles distinguish COVID-19 from influenza with limited contributions from cytokine storm. Sci. Adv. 2020, 19. [Google Scholar] [CrossRef]
- Ong, E.Z.; Chan, Y.F.Z.; Leong, W.Y.; Lee, N.M.Y.; Kalimuddin, S.; Haja Mohideen, S.M.; Chan, K.S.; Tan, A.T.; Bertoletti, A.; Ooi, E.E.; et al. A Dynamic Immune Response Shapes COVID-19 Progression. Cell Host Microbe 2020, 27, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 2015, 15, 692–704. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, M.G.; Armstrong, S.M.; Wang, C.; Hwang, D.; Leong-Poi, H.; Advani, A.; Advani, S.; Zhang, H.; Szaszi, K.; Tabuchi, A.; et al. The Tie2-agonist vasculotide rescues mice from influenza virus infection. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Filewod, N.C.; Lee, W.L. Inflammation without vascular leakage: Science fiction no longer? Am. J. Respir. Crit. Care Med. 2019, 200, 1472–1476. [Google Scholar] [CrossRef] [Green Version]
- Xiao, K.; Garner, J.; Buckley, K.M.; Vincent, P.A.; Chiasson, C.M.; Dejana, E.; Faundez, V.; Kowalczyk, A.P. p120-Catenin Regulates Clathrin-dependent Endocytosis of VE-Cadherin. Mol. Biol. Cell 2005, 16, 5141–5151. [Google Scholar] [CrossRef] [Green Version]
- Brasch, J.; Harrison, O.J.; Ahlsen, G.; Carnally, S.M.; Henderson, R.M.; Honig, B.; Shapiro, L. Structure and binding mechanism of vascular endothelial cadherin, a divergent classical cadherin. J. Mol. Biol. 2011, 408, 57–73. [Google Scholar] [CrossRef] [Green Version]
- Leckband, D.; Sivasankar, S. Mechanism of homophilic cadherin adhesion. Curr. Opin. Cell Biol. 2000, 12, 587–592. [Google Scholar] [CrossRef]
- Herron, C.R.; Lowery, A.M.; Hollister, P.R.; Reynolds, A.B.; Vincent, P.A. p120 regulates endothelial permeability independently of its NH 2 terminus and Rho binding. Am. J. Physiol. Heart Circ. Physiol. 2010, 300, 36–48. [Google Scholar] [CrossRef] [Green Version]
- Gavard, J.; Gutkind, J.S. VEGF controls endothelial-cell permeability by promoting the β -arrestin-dependent endocytosis of VE-cadherin. Nat. Cell Biol. 2006, 8, 1223–1234. [Google Scholar] [CrossRef]
- Zhu, W.; London, N.R.; Gibson, C.C.; Davis, C.T.; Tong, Z.; Sorensen, L.K.; Shi, D.S.; Guo, J.; Grossmann, A.H.; Thomas, K.R.; et al. Interleukin receptor activates a MYD88-ARNO-ARF6 cascade to disrupt vascular stability. Nature 2013, 492, 252–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- London, N.R.; Zhu, W.; Bozza, F.A.; Smith, M.C.P.; Greif, D.M.; Sorensen, L.K.; Chen, L.; Kaminoh, Y.; Aubrey, C.; Passi, S.F.; et al. Targeting Robo4-Dependent slit signaling to survive the cytokine storm in sepsis and influenza. Sci. Transl. Med. 2010, 2, 23ra19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parikh, S.M.; Mammoto, T.; Schultz, A.; Yuan, H.T.; Christiani, D.; Karumanchi, S.A.; Sukhatme, V.P. Excess circulating angiopoietin-2 may contribute to pulmonary vascular leak in sepsis in humans. PLoS Med. 2006, 3, 356–370. [Google Scholar] [CrossRef] [PubMed]
- Saharinen, P.; Eklund, L.; Alitalo, K. Therapeutic targeting of the angiopoietin-TIE pathway. Nat. Rev. Drug Discov. 2017, 16, 635–661. [Google Scholar] [CrossRef] [PubMed]
- Gavard, J.; Patel, V.; Gutkind, J.S. Article Angiopoietin-1 Prevents VEGF-Induced Endothelial Permeability by Sequestering Src through mDia. Dev. Cell 2008, 14, 25–36. [Google Scholar] [CrossRef] [Green Version]
- David, S.; Ghosh, C.C.; Kümpers, P.; Shushakova, N.; Van Slyke, P.; Khankin, E.V.; Karumanchi, S.A.; Dumont, D.; Parikh, S.M.; Ev, K.; et al. Effects of a synthetic PEG-ylated Tie-2 agonist peptide on endotoxemic lung injury and mortality. Am J Physiol Lung Cell Mol Physiol 2011, 300, 851–862. [Google Scholar] [CrossRef] [Green Version]
- Rübig, E.; Stypmann, J.; Van Slyke, P.; Dumont, D.J.; Spieker, T.; Buscher, K.; Reuter, S.; Goerge, T.; Pavenstädt, H.; Kümpers, P. The Synthetic Tie2 Agonist Peptide Vasculotide Protects Renal Vascular Barrier Function In Experimental Acute Kidney Injury. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, S.M.; Wang, C.; Tigdi, J.; Si, X.; Dumpit, C.; Charles, S.; Gamage, A.; Moraes, T.J.; Lee, W.L. Influenza Infects Lung Microvascular Endothelium Leading to Microvascular Leak: Role of Apoptosis and Claudin-5. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [Green Version]
- Michalick, L.; Weidenfeld, S.; Grimmer, B.; Fatykhova, D.; Solymosi, P.D.; Behrens, F.; Dohmen, M.; Brack, M.C.; Schulz, S.; Thomasch, E.; et al. Plasma mediators in patients with severe COVID-19 cause lung endothelial barrier failure. Eur. Respir. J. 2020. [Google Scholar] [CrossRef]
- Buzhdygan, T.P.; Deore, B.J.; Baldwin-leclair, A.; Bullock, T.A.; Mcgary, H.M.; Khan, J.A.; Razmpour, R.; Hale, J.F.; Galie, P.A.; Potulaa, R.; et al. The SARS-CoV-2 spike protein alters barrier function in 2D static and 3D microfluidic in-vitro models of the human blood—brain barrier. Neurobiol. Dis. 2020, 146. [Google Scholar] [CrossRef]
- Visseren, F.L.J.; Bouwman, J.J.M.; Bouter, K.P.; Diepersloot, R.J.A.; De Groot, P.G.; Erkelens, D.W. Procoagulant Activity of Endothelial Cells after Infection with respiratory viruses. Thromb Haemost 2000, 84, 319–324. [Google Scholar] [PubMed]
- Zelaya, H.; Tada, A.; Vizoso-pinto, M.G.; Salva, S.; Kanmani, P.; Aguero, G.; Alvarez, S.; Kitzawa, H.; Villena, J. Nasal priming with immunobiotic Lactobacillus rhamnosus modulates inflammation—Coagulation interactions and reduces influenza virus-associated pulmonary damage. Inflamm. Res. 2015, 64, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Keller, T.T.; Van Der Sluijs, K.F.; De Kruif, M.D.; Gerdes, V.E.A.; Meijers, J.C.M.; Florquin, S.; Van Der Poll, T.; Van Gorp, E.C.M.; Brandjes, D.P.M.; Bu, H.R.; et al. Effects on Coagulation and Fibrinolysis Induced by Influenza in Mice With a Reduced Capacity to Generate Activated Protein C and a Deficiency in Plasminogen Activator Inhibitor Type 1. Circ. Res. 2006, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Boilard, E.; Par, G.; Rousseau, M.; Cloutier, N.; Dubuc, I.; Tania, L.; Borgeat, P.; Flamand, L. Influenza virus H 1 N 1 activates platelets through FcgRIIA signaling and thrombin generation. Platelets Thrombopoiesis 2014, 123, 2854–2863. [Google Scholar] [CrossRef]
- Le, V.B.; Schneider, J.G.; Boergeling, Y.; Berri, F.; Ducatez, M.; Guerin, J.-L.; Adrian, I.; Errazuriz-Cerda, E.; Frasquilho, S.; Antunes, L.; et al. Platelet Activation and Aggregation Promote Lung Inflammation and Influenza Virus Pathogenesis. Am. J. Respir. Crit. Care Med. 2015, 191, 804–819. [Google Scholar] [CrossRef] [PubMed]
- Dimakakos, E.; Grapsa, D.; Vathiotis, I.; Papaspiliou, A.; Panagiotarakou, M.; Manolis, E.; Syrigos, K. H1N1-Induced Venous Thromboembolic Events? Results of a single-institution case series. Open Forum Infect. Dis. 2016, 3. [Google Scholar] [CrossRef] [Green Version]
- Gill, J.R.; Sheng, Z.-M.; Ely, S.F.; Jr, D.G.G.; Beasley, M.B.; Suh, J.; Deshpande, C.; Mollura, D.J.; Morens, D.M.; Bray, M.; et al. Pulmonary Pathologic Findings of Fatal 2009 Pandemic Influenza A/H1N1 viral infections. Arch. Pathol. Lab. Med. 2010, 134. [Google Scholar] [CrossRef]
- Bunce, P.E.; High, S.M.; Nadjafi, M.; Stanley, K.; Liles, C.; Christian, M.D. Pandemic H1N1 Influenza Infection and Vascular Thrombosis. Clin. Infect. Dis. 2011, 52, 14–17. [Google Scholar] [CrossRef] [Green Version]
- Van Wissen, M.; Keller, T.T.; Ronkes, B.; Gerdes, V.E.A.; Zaaijer, H.L.; Van Gorp, E.C.M.; Brandjes, D.P.M.; Levi, M.; Büller, H.R. Influenza infection and risk of acute pulmonary embolism. Thromb. J. 2007, 5. [Google Scholar] [CrossRef] [Green Version]
- Hariri, L.P.; North, C.M.; Shih, A.R.; Israel, R.A.; Maley, J.H.; Villalba, J.A.; Vinarsky, V.; Rubin, J.; Okin, J.A.; Sclafani, A.; et al. Lung Histopathology in Coronavirus Disease 2019 as Compared With Severe Acute Respiratory Syndrome and H1N1 Influenza. Chest 2020. [Google Scholar] [CrossRef]
- Nicolai, L.; Leunig, A.; Brambs, S.; Kaiser, R.; Joppich, M.; Hoffknecht, M.; Gold, C.; Engel, A.; Polewka, V.; Muenchhoff, M.; et al. Vascular neutrophilic inflammation and immunothrombosis distinguish severe COVID-19 from influenza pneumonia. J. Thromb. Haemost. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bilaloglu, S.; Aphinyanaphongs, Y.; Jones, S.; Iturrate, E.; Hochman, J.; Berger, J.S. Thrombosis in Hospitalized Patients with COVID-19 in a New York City Health System. JAMA J. Am. Med. Assoc. 2020, 324, 799–801. [Google Scholar] [CrossRef] [PubMed]
- Bao, C.; Tao, X.; Cui, W.; Yi, B.; Pan, T.; Young, K.H.; Qian, W. SARS -CoV-2 induced thrombocytopenia as an important biomarker significantly correlated with abnormal coagulation function, increased intravascular blood clot risk and mortality in COVID - 19 patients. Exp. Hematol. Oncol. 2020, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Zhou, Q.; Xu, J. Mechanism of thrombocytopenia in COVID-19 patients. Ann. Hematol. 2020, 99, 1205–1208. [Google Scholar] [CrossRef] [Green Version]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020, 7, e575–e582. [Google Scholar] [CrossRef]
- Helms, J.; Tacquard, C.; Severac, F.; Leonard-Lorant, I.; Ohana, M.; Delabranche, X.; Merdji, H.; Jehl, R.C.; Schenck, M.; Gandet, F.F.; et al. High risk of thrombosis in patients with severe SARS - CoV - 2 infection: A multicenter prospective cohort study. Intensive Care Med. 2020, 46, 1089–1098. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y.; Wang, X.; Yang, L.; Li, H.; Wang, Y.; Liu, M.; Zhao, X.; Xie, Y.; Yang, Y.; et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J. Hematol. Oncol. 2020, 13, 1–22. [Google Scholar] [CrossRef]
- Yang, Y.; Tang, H. Aberrant coagulation causes a hyper-inflammatory response in severe influenza pneumonia. Cell. Mol. Immunol. 2016, 13, 432–442. [Google Scholar] [CrossRef]
- Colotta, F.; Sciacca, F.L.; Sironi, M.; Luini, W.; Rabiet, M.J.; Mantovani, A. Expression of Monocyte Chemotactic Protein-1 by Monocytes and Endothelial Cells Exposed to Thrombin. Am. J. Physiol. 1994, 144, 975–985. [Google Scholar]
- Johnson, K.; Choi, Y.; Degroot, E.; Samuels, I.; Aarden, L. Potential Mechanisms for a Proinflammatory Vascular Cytokine Response to Coagulation Activation. J. Immunol. 1998, 160, 5130–5135. [Google Scholar]
- De Jonge, E.; Friederich, P.W.; Vlasuk, G.P.; Rote, W.E.; Vroom, M.B.; Levi, M.; Van Der Poll, T. Activation of Coagulation by Administration of Recombinant Factor VIIa Elicits Interleukin 6 (IL-6) and IL-8 Release in Healthy Human Subjects. Clin. Diagn. Lab. Immunol. 2003, 10, 495–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueno, A.; Murakami, K.; Yamanouchi, K.; Watanabe, M.; Kondo, T. Thrombin stimulates production of interleukin-8 in human umbilical vein endothelial cells. Immunology 1996, 88, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Perrone, L.A.; Plowden, J.K.; García-Sastre, A.; Katz, J.M.; Tumpey, T.M. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog. 2008, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanchana, S.; Kanchana, S.; Vijitsopa, T.; Thammakumpee, K.; Yamwong, S.; Sawanyawisuth, K. Clinical Factors Predictive of Pneumonia Caused by Pandemic 2009 H1N1 Influenza Virus. Am. J. Trop. Med. Hyg. 2013, 88, 461–463. [Google Scholar] [CrossRef] [Green Version]
- Narasaraju, T.; Yang, E.; Samy, R.P.; Ng, H.H.; Poh, W.P.; Liew, A.-A.; Phoon, M.C.; van Rooijen, N.; Chow, V.T. Excessive Neutrophils and Neutrophil Extracellular Traps Contribute to Acute Lung Injury of Influenza Pneumonitis. Immunopathol. Infect. Dis. 2011, 179, 199–210. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Middleton, E.A.; He, X.-Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136. [Google Scholar] [CrossRef]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil Extracellular Traps Directly Induce Epithelial and Endothelial Cell Death: A Predominant Role of Histones. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Zhu, L.; Liu, L.; Zhang, Y.; Pu, L.; Liu, J.; Li, X.; Chen, Z.; Hao, Y.; Wang, B.; Han, J.; et al. High Level of Neutrophil Extracellular Traps Correlates With Poor Prognosis of Severe Influenza A Infection. J. Infect. Dis. 2018, 217, 428–437. [Google Scholar] [CrossRef]
- Jing, L.; Li, S.; Liu, J.; Liang, B.; Wang, X.; Wang, H.; Li, W.; Tong, Q.; Yi, J.; Zhao, L.; et al. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. EBioMedicine 2020, 55, 2352–3964. [Google Scholar] [CrossRef]
- Wang, J.; Li, Q.; Yin, Y.; Zhang, Y.; Cao, Y.; Lin, X.; Huang, L.; Hoffmann, D.; Lu, M.; Qiu, Y. Excessive Neutrophils and Neutrophil Extracellular Traps in COVID-19. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Leppkesa, M.; Knopfb, J.; Naschberger, E.; Lindemann, A.; Singh, J.; Herrmann, I.; Sturzl, M.; Staats, L.; Mahajan, A.; Schauer, C.; et al. Vascular occlusion by neutrophil extracellular traps in COVID-19. EBioMedicine 2020, 58. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Kanthi, Y.; Knight, J.S.; Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [Green Version]
- Veras, F.; Pontelli, M.; Silva, C.; Toller-Kawahisa, J.; de Lima, M.; Nascimento, D.; Schneider, A.; Caetité, D.; Rosales, R.; Colón, D.; et al. SARS-CoV-2–triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020, 217, e20201129. [Google Scholar] [CrossRef] [PubMed]
- Schurink, B.; Roos, E.; Radonic, T.; Barbe, E.; Bouman, C.S.C.; De Boer, H.H.; De Bree, G.J.; Bulle, E.B.; Aronica, E.M.; Florquin, S.; et al. Viral presence and immunopathology in patients with lethal COVID-19: A prospective autopsy cohort study. The Lancet Microbe 2020, 1, e290–e299. [Google Scholar] [CrossRef]
- Radermecker, C.; Detrembleur, N.; Guiot, J.; Cavalier, E.; Henket, M.; D’Emal, C.; Vanwinge, C.; Cataldo, D.; Oury, C.; Delvenne, P.; et al. Neutrophil extracellular traps infiltrate the lung airway, interstitial, and vascular compartments in severe COVID-19. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Davidson, S. Treating influenza infection, from now and into the future. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Baker, J.; Block, S.L.; Matharu, B.; Macutkiewicz, L.B.; Wildum, S.; Dimonaco, S.; Collinson, N.; Clinch, B.; Peidra, P.A. Baloxavir Marboxil Single-dose Treatment in influenza infected children. Antimicrob. Rep. 2020, 39, 700–705. [Google Scholar] [CrossRef]
- Yang, T. Baloxavir Marboxil: The First Cap-Dependent Endonuclease Inhibitor for the Treatment of Influenza. Ann. Pharmacother. 2019, 53, 754–759. [Google Scholar] [CrossRef]
- Hussain, M.; Galvin, H.D.; Haw, T.Y.; Nutsford, A.N.; Husain, M. Drug resistance in influenza A virus: The epidemiology and management. Infect. Drug Resist. 2017, 10, 121–134. [Google Scholar] [CrossRef] [Green Version]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the treatment of covid19 final report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Sheahan, T.P.; Sims, A.C.; Graham, R.L.; Menachery, V.D.; Gralinski, L.E.; Case, J.B.; Leist, S.R.; Pyrc, K.; Feng, J.Y.; Trantcheva, I.; et al. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Pizzorno, A.; Padey, B.; Julien, T.; Trouillet-Assant, S.; Traversier, A.; Errazuriz-Cerda, E.; Fouret, J.; Dubois, J.; Gaymard, A.; Lescur, F.-X.; et al. Characterization and Treatment of SARS-CoV-2 in Nasal and Bronchial Human Airway Epithelia. Cell Rep. Med. 2020, 1. [Google Scholar] [CrossRef]
- Williamson, B.N.; Feldmann, F.; Schwarz, B.; Meade-White, K.; Porter, D.P.; Schulz, J.; van Doremalen, N.; Leighton, I.; Yinda, C.K.; Pérez-Pérez, L.; et al. Clinical benefit of remdesivir in rhesus macaques infected with SARS-CoV-2. Nature 2020, 585, 273–276. [Google Scholar] [CrossRef] [PubMed]
- De Wit, E.; Feldmann, F.; Cronin, J.; Jordan, R.; Okumura, A.; Thomas, T.; Scott, D.; Cihlar, T.; Feldmann, H. Prophylactic and therapeutic remdesivir (GS-5734) treatment in the rhesus macaque model of MERS-CoV infection. Proc. Natl. Acad. Sci. USA 2020, 117, 6771–6776. [Google Scholar] [CrossRef] [Green Version]
- Pan, H.; Peto, R.; Abdool Karim, Q.; Alejandria, M.; Henao-Restrepo, A.-M.; Hernández García, C.K.M.-P.; Malekzadeh, R.; Murthy, S.; Preziosi, M.-P.; Reddy, S.; et al. Repurposed antiviral drugs for COVID-19—Interim WHO SOLIDARITY trial results. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Spinner, C.D.; Gottlieb, R.L.; Criner, G.J.; Arribas López, J.R.; Cattelan, A.M.; Soriano Viladomiu, A.; Ogbuagu, O.; Malhotra, P.; Mullane, K.M.; Castagna, A.; et al. Effect of Remdesivir vs Standard Care on Clinical Status at 11 Days in Patients With Moderate COVID-19. JAMA 2020, 324, 1048–1057. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef]
- Simonovich, V.A.; Pratx, L.D.B.; Scibona, P.; Beruto, M.V.; Vallone, M.G.; Vázquez, C.; Savoy, N.; Giunta, D.H.; Pérez, L.G.; Sánchez, M.L.; et al. A Randomized Trial of Convalescent Plasma in Covid-19 Severe Pneumonia. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Mahase, E. Covid-19: FDA authorises neutralising antibody bamlanivimab for non-admitted patients. BMJ 2020, 371. [Google Scholar] [CrossRef] [PubMed]
- Tleyjeh, I.M.; Kashour, Z.; Damlaj, M.; Riaz, M.; Tlayjeh, H.; Altannir, M.; Altannir, Y.; Al-tannir, M.; Tleyjeh, R.; Hassett, L.; et al. Efficacy and safety of tocilizumab in COVID-19 patients: A living systematic review and meta-analysis. Clin. Microbiol. Infect. 2020. [Google Scholar] [CrossRef] [PubMed]
- Biran, N.; Ip, A.; Ahn, J.; Go, R.C.; Wang, S.; Mathura, S.; Sinclaire, B.A.; Bednarz, U.; Marafelias, M.; Hansen, E.; et al. Tocilizumab among patients with COVID-19 in the intensive care unit: A multicentre observational study. Lancet Rheumatol. 2020, 2, e603–e612. [Google Scholar] [CrossRef]
- Somers, E.C.; Eschenauer, G.A.; Troost, J.P.; Golob, J.L.; Gandhi, T.N.; Wang, L.; Zhou, N.; Petty, L.A.; Baang, J.H.; Dillman, N.O.; et al. Tocilizumab for Treatment of Mechanically Ventilated Patients With COVID-19. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Stone, J.H.; Frigault, M.J.; Serling- Boyd, N.J.; Fernandes, A.D.; Harvey, L.; Foulkes, A.S.; Horick, N.K.; Healy, B.C.; Shah, R.; Bensaci, A.M.; et al. Efficacy of Tocilizumab in Patients Hospitalized with Covid-19. N. Engl. J. Med. 2020, 383, 2333–2334. [Google Scholar] [CrossRef]
- Gordon, A.C.; Mouncey, P.R.; Al-Beidh, F.; Rowan, K.M.; Nichol, A.D.; Arabi, Y.M.; Annane, D.; Beane, A.; van Bentum-Puijk, W.; Berry, L.R.; et al. Interleukin-6 Receptor Antagonists in Critically Ill Patients with Covid-19—Preliminary report. medRxiv 2021, in press. [Google Scholar] [CrossRef]
- Wang, R.; Simoneau, C.R.; Kulsuptrakul, J.; Bouhaddou, M.; Travisano, K.A.; Hayashi, J.M.; Carlson-Stevermer, J.; Zengel, J.R.; Richards, C.M.; Fozouni, P.; et al. Genetic Screens Identify Host Factors for SARS-CoV-2 and Common Cold Coronaviruses. Cell 2020, 184, 106–119. [Google Scholar] [CrossRef]
- Wei, J.; Alfajaro, M.M.; DeWeirdt, P.C.; Hanna, R.E.; Lu-Culligan, W.J.; Cai, W.L.; Strine, M.S.; Zhang, S.M.; Graziano, V.R.; Schmitz, C.O.; et al. Genome-wide CRISPR Screens Reveal Host Factors Critical for SARS-CoV-2 Infection. Cell 2020, 184, 76–91. [Google Scholar] [CrossRef]
- Schneider, W.M.; Luna, J.M.; Hoffmann, H.H.; Sánchez-Rivera, F.J.; Leal, A.A.; Ashbrook, A.W.; Le Pen, J.; Ricardo-Lax, I.; Michailidis, E.; Peace, A.; et al. Genome-Scale Identification of SARS-CoV-2 and Pan-coronavirus Host Factor Networks. Cell 2021, 120–132. [Google Scholar] [CrossRef]
- Daniloski, Z.; Jordan, T.X.; Wessels, H.H.; Hoagland, D.A.; Kasela, S.; Legut, M.; Maniatis, S.; Mimitou, E.P.; Lu, L.; Geller, E.; et al. Identification of Required Host Factors for SARS-CoV-2 Infection in Human Cells. Cell 2020, 92–105. [Google Scholar] [CrossRef]
- RECOVERY Collaborative Group. Dexamethasone in Hospitalized Patients with Covid-19—Preliminary Report. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Zielinska, K.A.; Van Moortel, L.; Opdenakker, G.; De Bosscher, K.; Van Den Steen, P.E. Endothelial Response to Glucocorticoids in Inflammatory Diseases. Front. Immunol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Romero, I.A.; Radewicz, K.; Jubin, E.; Michel, C.C.; Greenwood, J.; Couraud, P.; Adamson, P. Changes in cytoskeletal and tight junctional proteins correlate with decreased permeability induced by dexamethasone in cultured rat brain endothelial cells. Neurosci. Lett. 2003, 344, 112–116. [Google Scholar] [CrossRef]
- Blecharz, K.G.; Drenckhahn, D.; Forster, C.Y. Glucocorticoids increase VE-cadherin expression and cause cytoskeletal rearrangements in murine brain endothelial cEND cells. J. Cereb. Blood Flow Metab. 2008, 28, 1139–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, T.; Qiao, J.; Zhao, L.; He, G.; Li, K.; Wang, J.; Tian, Y.; Wang, H. Effect of dexamethasone on acute respiratory distress syndrome induced by the H5N1 virus in mice. Eur. Respir. J. 2009, 33, 852–860. [Google Scholar] [CrossRef] [Green Version]
- Thomas, B.J.; Porritt, R.A.; Hertzog, P.J.; Bardin, P.G.; Tate, M.D. Glucocorticosteroids enhance replication of respiratory viruses: Effect of adjuvant interferon. Sci. Rep. 2014, 2014. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.; Leo, Y.-S.; Cao, B.; Chan, P.K.S.; Kyaw, W.M.; Uyeki, T.M.; Tam, W.W.S.; Cheung, C.S.K.; Yung, I.M.H.; Li, H.; et al. Neuraminidase inhibitors, superinfection and corticosteroids affect survival of influenza patients. Eur. Respir. J. 2015, 45, 1642–1652. [Google Scholar] [CrossRef]
- Xi, X.; Xu, Y.; Jiang, L.; Li, A.; Duan, J.; Du, B. Hospitalized adult patients with 2009 influenza A ( H1N1 ) in Beijing, China: Risk factors for hospital mortality. BMC Infect. Dis. 2010, 10. [Google Scholar] [CrossRef] [Green Version]
- Brun-Buisson, C.; Richard, J.-C.M.; Mercat, A.; Thiebaut, A.C.M.; Brochard, L. Early Corticosteroids in Severe Influenza A / H1N1 Pneumonia and Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2011, 183, 1200–1206. [Google Scholar] [CrossRef]
- Li, H.; Li, S.Y.; Yao, G.; Yan, Z.X.; Zhang, W.; Wei, H.J.; Meng, C.; Yu, L.K.; Wang, C. Effect of low-to-moderate-dose corticosteroids on mortality of hospitalized adolescents and adults with influenza A ( H1N1 ) pdm09 viral pneumonia. Influ. Other Respir. Viruses 2017, 11, 345–354. [Google Scholar] [CrossRef]
- Sepulveda, J.; Westblade, L.F.; Whittier, S.; Satlin, M.J.; Greendyke, W.G.; Aaron, J.G.; Zucker, J.; Dietz, D.; Sobieszczyk, M.; Choi, J.J.; et al. Bacteremia and blood culture utilization during covid-19 surge in New York City. J. Clin. Microbiol. 2020, 58, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rawson, T.M.; Zhu, N.; Ranganathan, N.; Gilchrist, M.; Satta, G.; Cooke, G.; Holmes, A. Bacterial and fungal co-infection in individuals with coronavirus: A rapid review to support COVID-19 antimicrobial prescribing. Clin. Infect. Dis. 2020, 71, 2459–2468. [Google Scholar] [CrossRef] [PubMed]
- Boltz, D.A.; Aldridge, J.R.; Webster, R.G.; Govorkova, E.A. Drugs in development for influenza. Drugs 2010, 70, 1349–1362. [Google Scholar] [CrossRef] [PubMed]
- Yip, T.F.; Selim, A.S.M.; Lian, I.; Lee, S.M.Y. Advancements in host-based interventions for influenza treatment. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Pulavendran, S.; Rudd, J.M.; Maram, P.; Thomas, P.G.; Akhilesh, R.; Malayer, J.R.; Chow, V.T.K.; Teluguakula, N. Combination therapy targeting platelet activation and virus replication protects mice against lethal influenza pneumonia. Am. J. Respir. Cell Mol. Biol. 2019, 61, 689–701. [Google Scholar] [CrossRef]
- Ashar, H.K.; Mueller, N.C.; Rudd, J.M.; Snider, T.A.; Achanta, M.; Prasanthi, M.; Pulavendran, S.; Thomas, P.G.; Ramachandran, A.; Malayer, J.R.; et al. The Role of Extracellular Histones in Influenza Virus Pathogenesis. Am. J. Pathol. 2018, 188, 135–148. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Ying, Z.H.; Yu, C.H.; Zhang, H.H.; Yu, W.Y.; Wu, X.N. Isofraxidin ameliorated influenza viral inflammation in rodents via inhibiting platelet aggregation. Int. Immunopharmacol. 2020, 84. [Google Scholar] [CrossRef]
- Troxell Klingenbjerg, P.M. Effect of Aspirin on Development of ARDS in At-Risk Patients Presenting to the Emergency Department. J. Emerg. Med. 2016, 51, 339–340. [Google Scholar] [CrossRef]
- Tang, N.; Bai, H.; Chen, X.; Gong, J.; Li, D.; Sun, Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 2020, 18, 1094–1099. [Google Scholar] [CrossRef]
- Paranjpe, I.; Fuster, V.; Lala, A.; Russak, A.J.; Glicksberg, B.S.; Levin, M.A.; Charney, A.W.; Narula, J.; Fayad, Z.A.; Bagiella, E.; et al. Association of Treatment Dose Anticoagulation With In-Hospital Survival Among Hospitalized Patients With COVID-19. J. Am. Coll. Cardiol. 2020, 76, 122–124. [Google Scholar] [CrossRef]
- Lemos, A.C.B.; do Espírito Santo, D.A.; Salvetti, M.C.; Gilio, R.N.; Agra, L.B.; Pazin-Filho, A.; Miranda, C.H. Therapeutic versus prophylactic anticoagulation for severe COVID-19: A randomized phase II clinical trial (HESACOVID). Thromb. Res. 2020, 196, 359–366. [Google Scholar] [CrossRef] [PubMed]
- NIH ACTIV Trial of Blood Thinners Pauses Enrollment of Critically ill COVID-19 Patients. Available online: https://www.nih.gov/news-event (accessed on 28 December 2020).
- Ni, Y.N.; Chen, G.; Sun, J.; Liang, B.M.; Liang, Z.A. The effect of corticosteroids on mortality of patients with influenza pneumonia: A systematic review and meta-analysis. Crit. Care 2019, 23, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.W.; Fan, L.C.; Miao, X.Y.; Mao, B.; Li, M.H.; Lu, H.W.; Liang, S.; Xu, J.F. Corticosteroids for the treatment of human infection with influenza virus: A systematic review and meta-analysis. Clin. Microbiol. Infect. 2015, 21, 956–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamieson, A.M.; Yu, S.; Annicelli, C.H.; Medzhitov, R. Influenza virus-induced glucocorticoids compromise innate host defense against a secondary bacterial infection. Cell Host Microbe 2010, 7, 103–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermine, O.; Mariette, X.; Tharaux, P.L.; Resche-Rigon, M.; Porcher, R.; Ravaud, P. Effect of Tocilizumab vs Usual Care in Adults Hospitalized with COVID-19 and Moderate or Severe Pneumonia: A Randomized Clinical Trial. JAMA Intern. Med. 2020. [Google Scholar] [CrossRef]
- Sellers, S.A.; Hagan, R.S.; Hayden, F.G.; Fischer, W.A. The hidden burden of influenza: A review of the extra-pulmonary complications of influenza infection. Influ. Other Respir Viruses 2017, 11, 372–393. [Google Scholar] [CrossRef]
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Abbreviation | Full Name |
|---|---|
| ACE2 | Angiotensin converting enzyme 2 |
| ANG | Angiopoietin |
| ARDS | Acute respiratory distress syndrome |
| COVID-19 | Coronavirus disease |
| IAV | Influenza A virus |
| ICAM-1 | Intercellular adhesion molecule 1 |
| IL | Interleukin |
| MERS-CoV | Middle Eastern respiratory syndrome coronavirus |
| NET | Neutrophil extracellular traps |
| NLRP3 | NLR family pyrin domain containing 3 |
| SARS-CoV | Severe acute respiratory syndrome coronavirus |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| TMPRSS2 | Transmembrane protease serine 2 |
| VCAM-1 | Vascular cell adhesion molecule 1 |
| VE-cadherin | Vascular endothelial cadherin |
| VEGF | Vascular endothelial growth factor |
| vWF | von Willebrand Factor |
| ZO-1 | Zonula occludens 1 |
| Influenza | SARS-CoV-2 | |
|---|---|---|
| 1. Receptor | Sialic acid | ACE2/TMPRSS2 |
| 2. Infects lung endothelial cells? |
|
|
| 3. Complications by bacterial superinfection | Frequent | Not frequent |
| 4. Role for steroids? | Unclear, potentially harmful | Beneficial in severe cases |
| 5a. Other host modulating drugs-barrier integrity |
|
|
| 5b. Other host modulating drugs-blockade of cytokines |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Latreille, E.; Lee, W.L. Interactions of Influenza and SARS-CoV-2 with the Lung Endothelium: Similarities, Differences, and Implications for Therapy. Viruses 2021, 13, 161. https://doi.org/10.3390/v13020161
Latreille E, Lee WL. Interactions of Influenza and SARS-CoV-2 with the Lung Endothelium: Similarities, Differences, and Implications for Therapy. Viruses. 2021; 13(2):161. https://doi.org/10.3390/v13020161
Chicago/Turabian StyleLatreille, Elyse, and Warren L. Lee. 2021. "Interactions of Influenza and SARS-CoV-2 with the Lung Endothelium: Similarities, Differences, and Implications for Therapy" Viruses 13, no. 2: 161. https://doi.org/10.3390/v13020161
APA StyleLatreille, E., & Lee, W. L. (2021). Interactions of Influenza and SARS-CoV-2 with the Lung Endothelium: Similarities, Differences, and Implications for Therapy. Viruses, 13(2), 161. https://doi.org/10.3390/v13020161