
Innate Immune Sensing of Viruses and Its Consequences for the Central Nervous System


Abstract
:1. Introduction
2. Viral Recognition and Antiviral Response
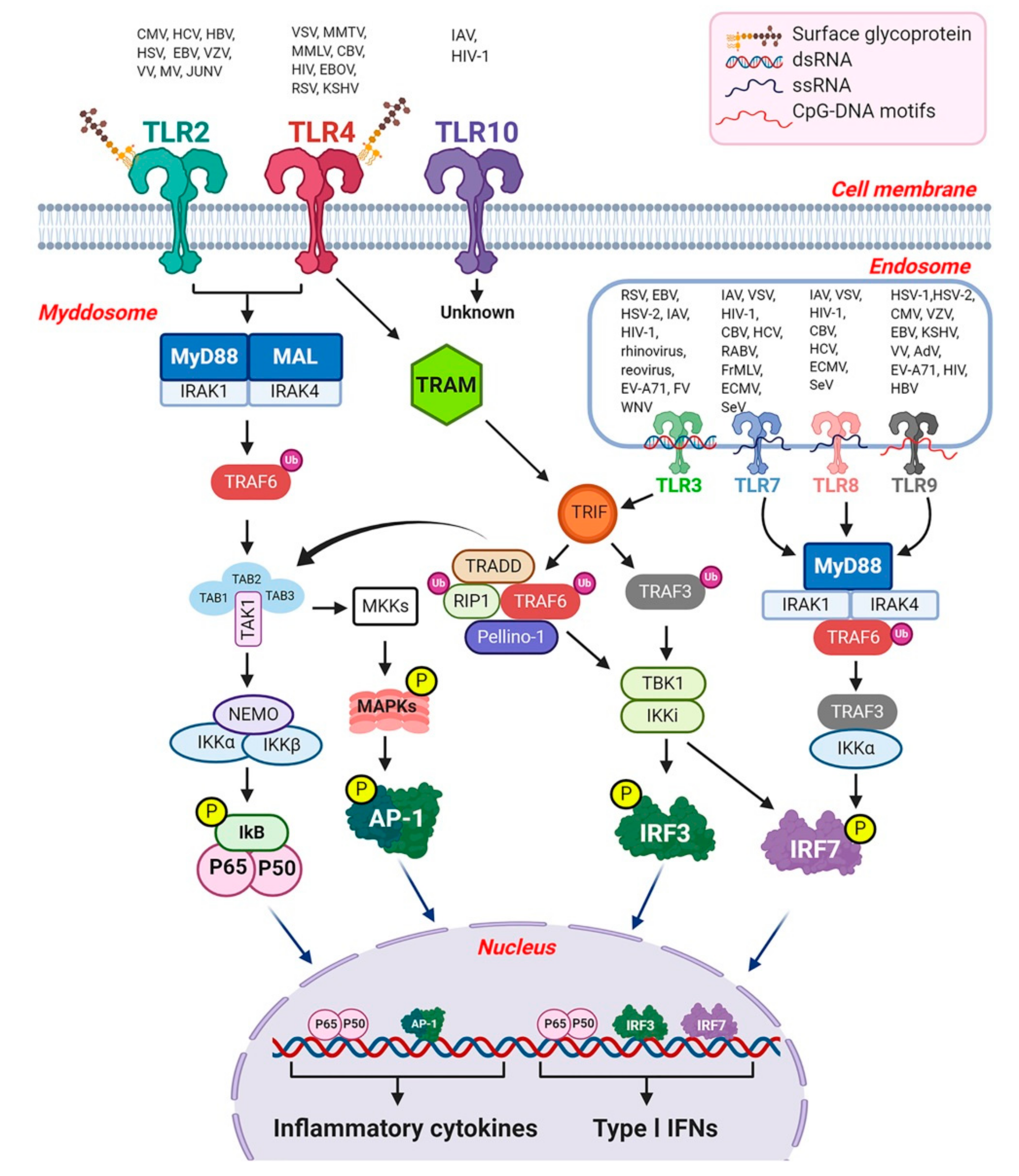
3. Toll-Like Receptors
3.1. Recognition of Viruses by TLRs
3.2. Toll-Like Receptor Signaling
3.2.1. The MyD88-Dependent Pathway
3.2.2. The TRIF-Dependent Pathway
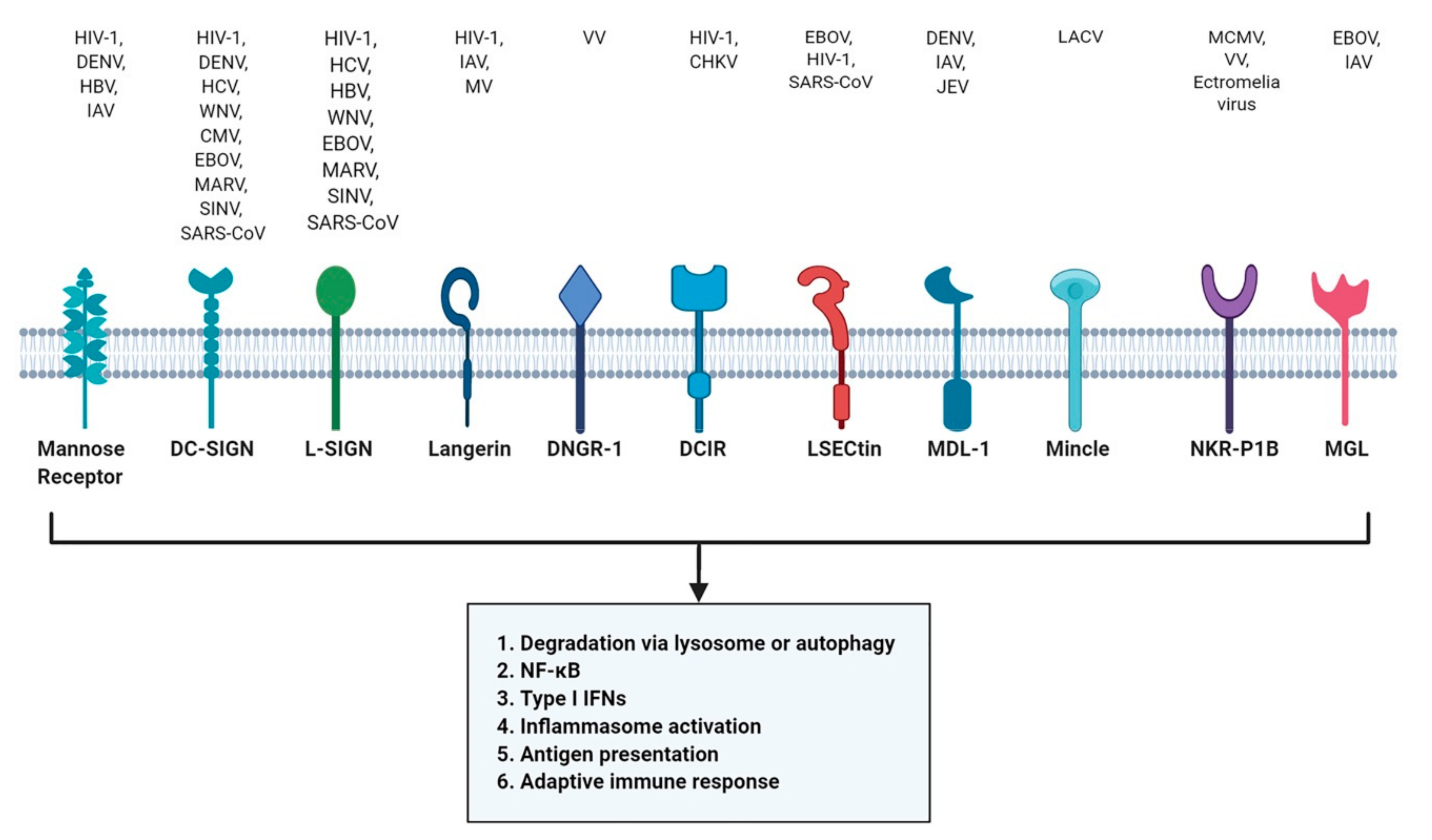
4. C-Type Lectin Receptors (CLR)
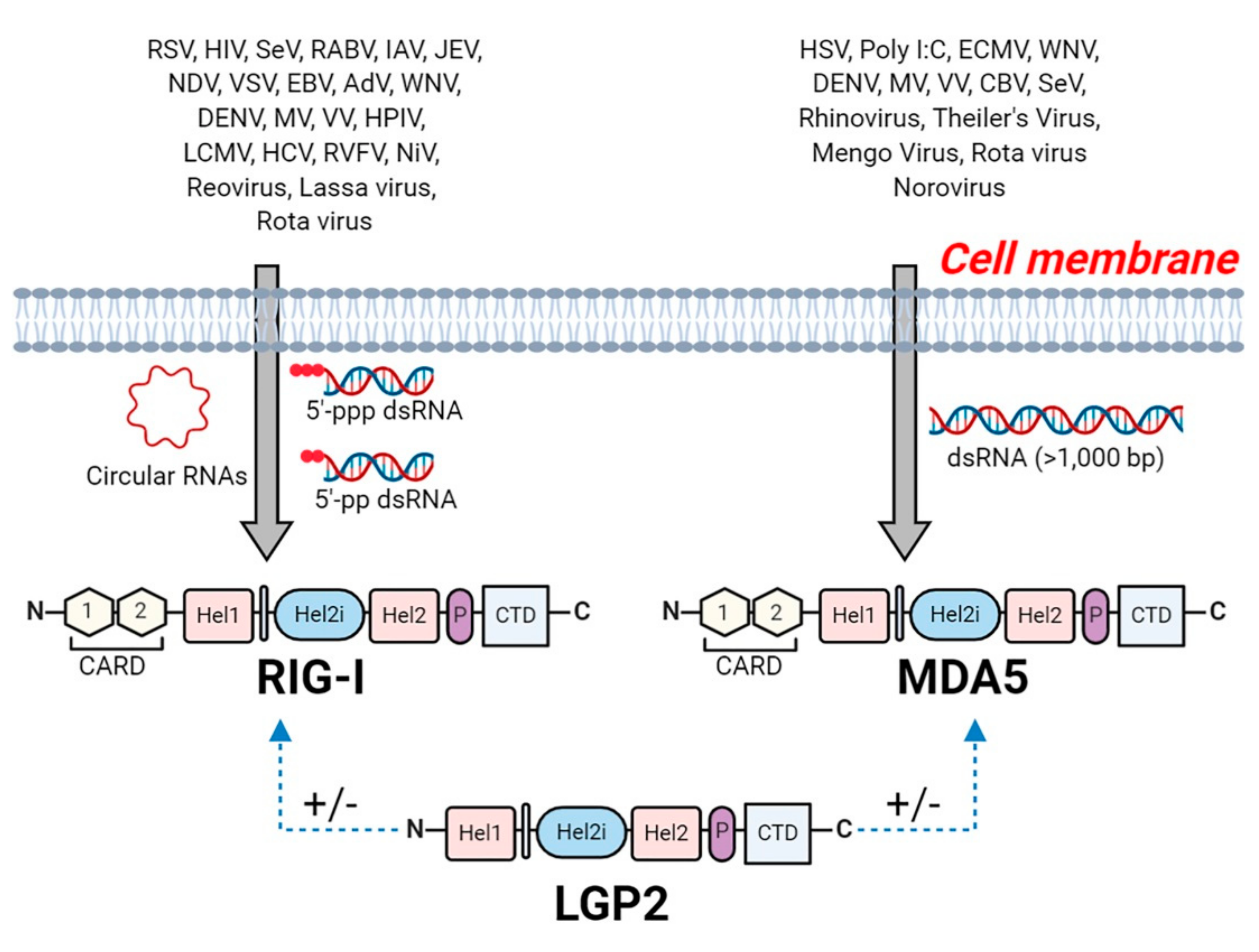
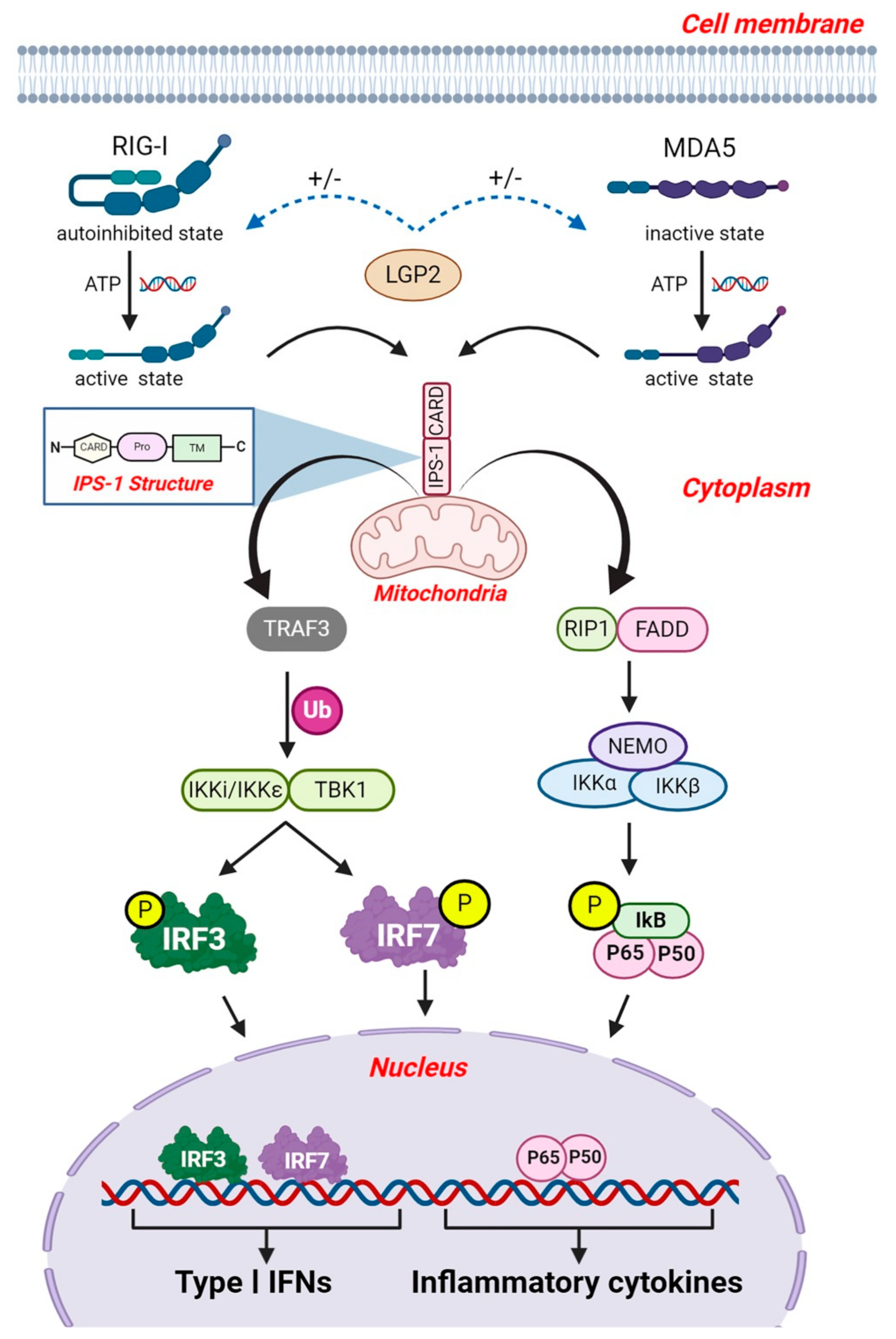
5. RIG-I Like Receptors (RLRs)
5.1. Recognition of Viral RNA by RLRs
5.2. RLR Signaling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PRRs/Receptor | Localization | Adapter | Viruses PAMPS | Viruses | Reference |
|---|---|---|---|---|---|
| RIG-I | Cytoplasm | IPS-1 | 5′ppp dsRNA, 5′pp dsRNA, Circular RNA, ssRNA, dsRNA, Virus-encoded RNA | Newcastle disease virus | [142] |
| West Nile Virus | [164] | ||||
| Sendai virus | [142] | ||||
| Vesicular stomatitis virus | [142] | ||||
| Epstein-Barr virus | [144] | ||||
| Adenoviruses | [143] | ||||
| Dengue virus | [165] | ||||
| Influenza A virus | [141] | ||||
| Japanese encephalitis virus | [141] | ||||
| Rabies Virus | [180] | ||||
| Ebola virus | [181,182] | ||||
| Nipah Virus | [182] | ||||
| Lassa Virus | [182] | ||||
| Rift valley fever virus | [182] | ||||
| Rota virus | [183] | ||||
| Measles virus | [184] | ||||
| Vaccinia virus | [146] | ||||
| Human immunodeficiency virus | [185] | ||||
| Human parainfluenza virus | [186] | ||||
| Hepatitis C virus | [187] | ||||
| Reovirus | [188] | ||||
| Lymphocytic choriomeningitis virus | [189] | ||||
| MDA5 | Cytoplasm | IPS-1 | dsRNA, RNA (>1000 bp) | Encephalomyocarditis picornavirus | [159] |
| Theiler’s virus | [141] | ||||
| Mengo virus | [141] | ||||
| Murine norovirus | [160] | ||||
| West Nile virus | [164] | ||||
| Dengue virus | [165] | ||||
| Sendai virus | [190] | ||||
| Rotavirus | [183] | ||||
| Measles virus | [184] | ||||
| Vaccinia virus | [146] | ||||
| Coxsackie B virus | [191] | ||||
| Herpes simplex virus | [192] | ||||
| Rhinovirus | [163] |
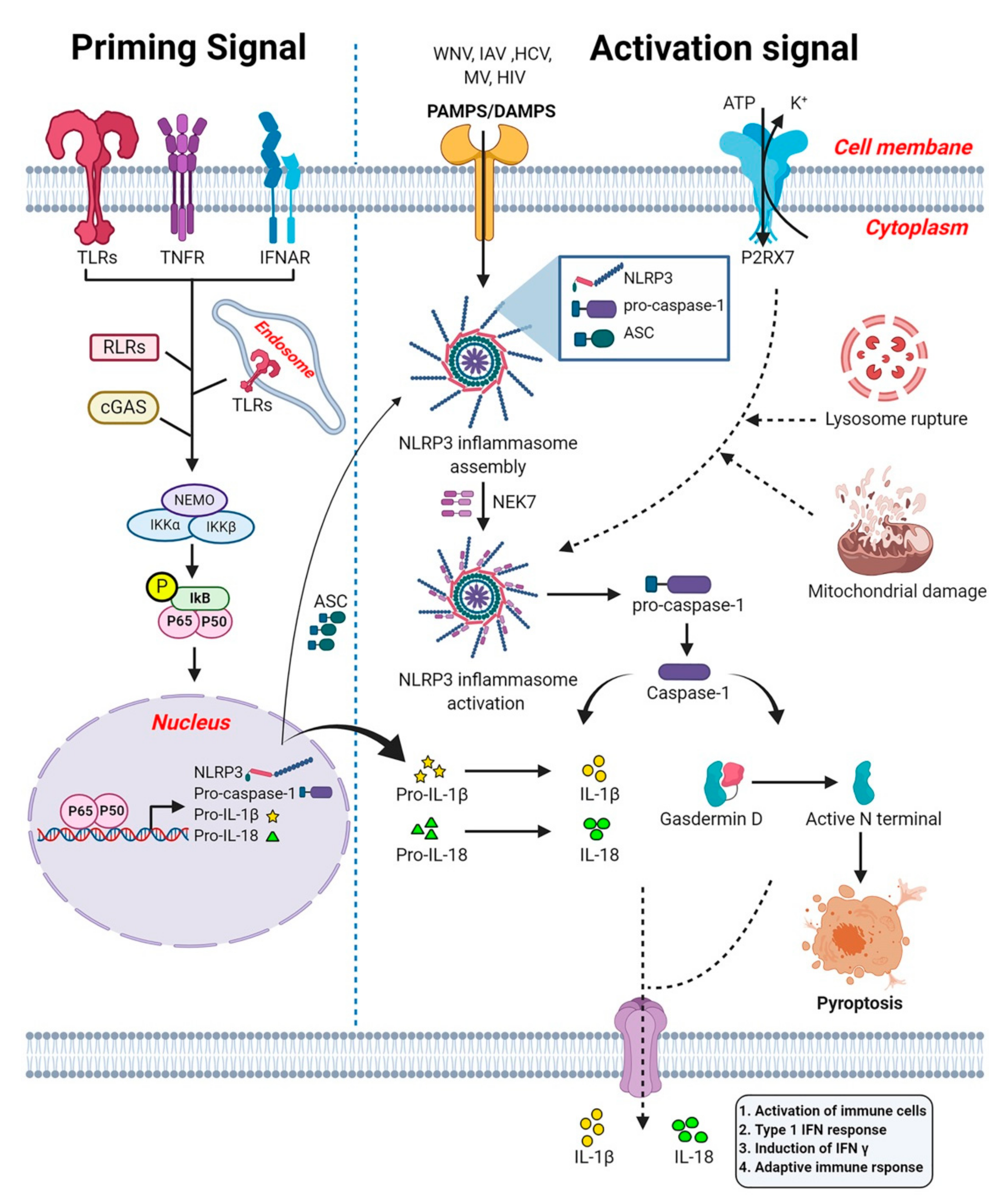
6. NOD-Like Receptors
7. The NLRP3 Inflammasome
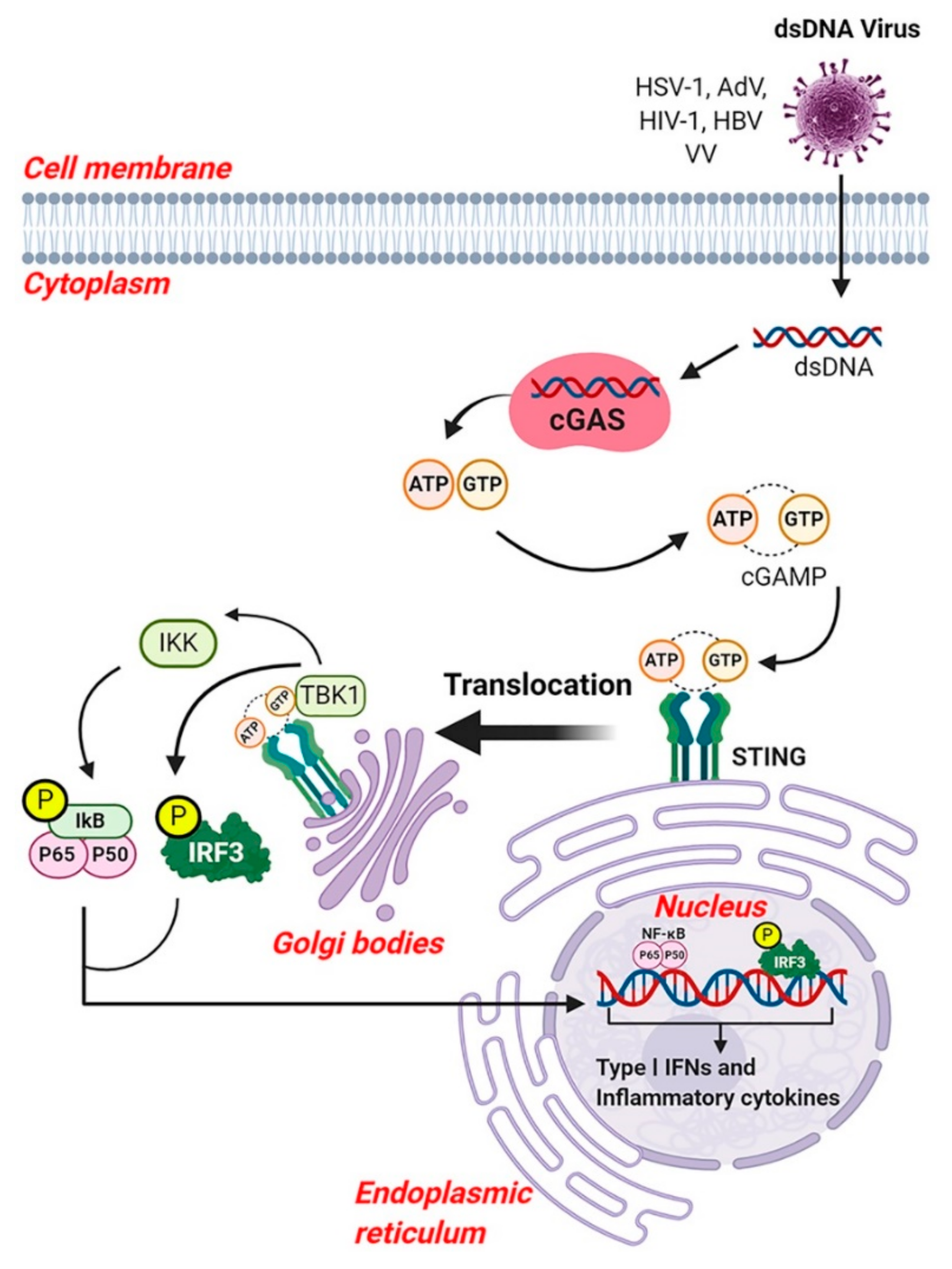
8. Cyclic GMP-AMP Synthase
9. Interferons during Viral Infection
10. PRR in Activation of Adaptive Immune Response
11. Consequences of the Innate Anti-Viral Immune Response in the CNS
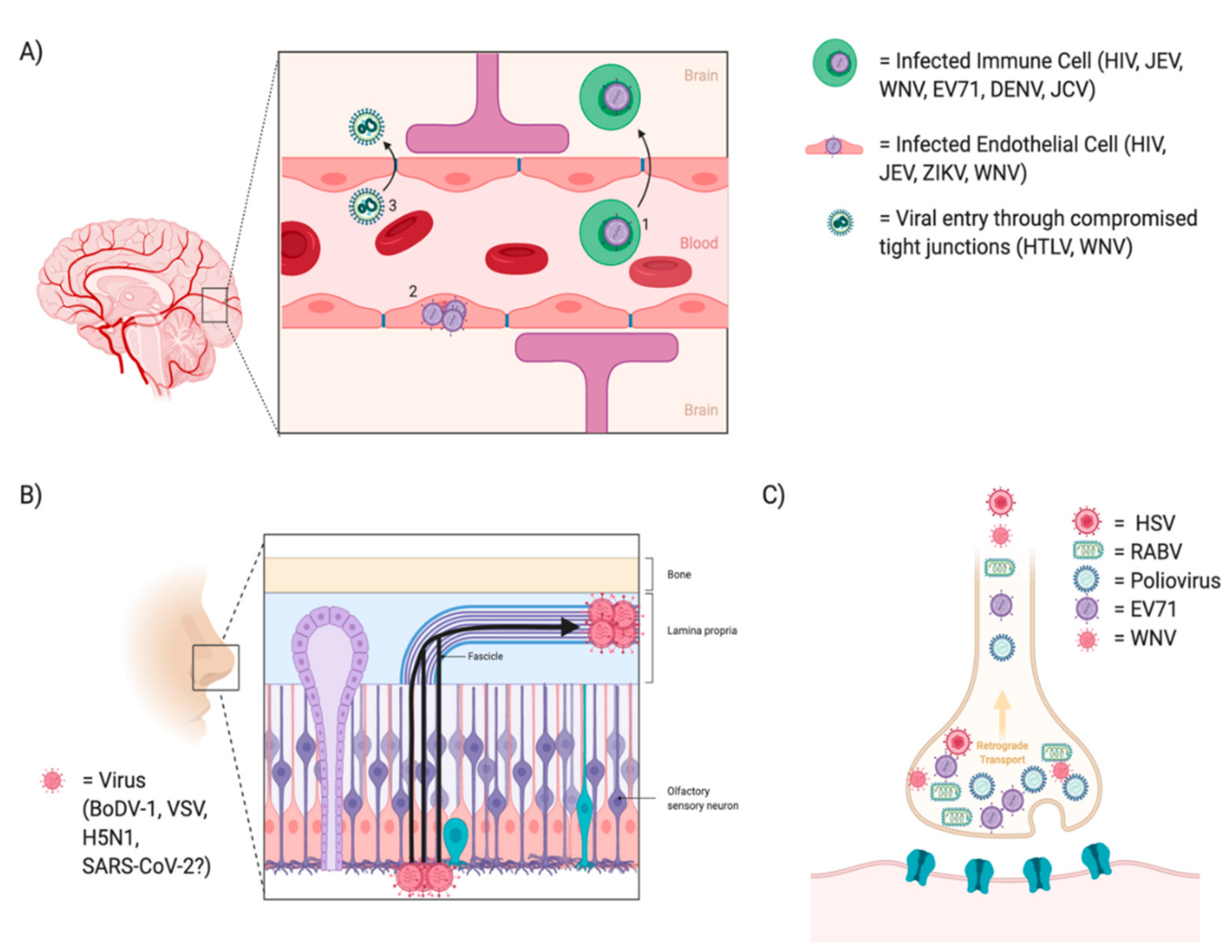
11.1. Viral Entry into the CNS
11.1.1. Infection through the Blood Brain Barrier
11.1.2. Infection through the Nasal Epithelium
11.1.3. Infection through Peripheral Nerves
11.2. Innate Immune Response in the CNS
11.2.1. Microglia and Astrocytes
11.2.2. Neurons
11.3. Cytokine and Chemokines Regulating the BBB
11.4. Human Inborn Errors of Innate Immune Pathways in the CNS
11.5. Viral Evasion of the Innate Immune Response
12. Therapeutic Approaches to Viral Infections of the CNS
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Morens, D.M.; Fauci, A.S. Emerging Pandemic Diseases: How We Got to COVID-19. Cell 2020, 182, 1077–1092. [Google Scholar] [CrossRef] [PubMed]
- Erbelding, E.J.; Post, D.J.; Stemmy, E.J.; Roberts, P.C.; Augustine, A.D.; Ferguson, S.; Paules, C.I.; Graham, B.S.; Fauci, A.S. A Universal Influenza Vaccine: The Strategic Plan for the National Institute of Allergy and Infectious Diseases. J. Infect. Dis. 2018, 218, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Marasini, B.; Chen, X.; Ding, L.; Wang, J.-J.; Xiao, P.; Villinger, F.; Spearman, P. A Bivalent, Spherical Virus-Like Particle Vaccine Enhances Breadth of Immune Responses against Pathogenic Ebola Viruses in Rhesus Macaques. J. Virol. 2020, 94, 1–19. [Google Scholar] [CrossRef]
- Mogensen, T.H. Pathogen Recognition and Inflammatory Signaling in Innate Immune Defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Goubau, D.; Deddouche, S.; Sousa, C.R.E. Cytosolic Sensing of Viruses. Immunity 2013, 38, 855–869. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, A.; Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004, 5, 987–995. [Google Scholar] [CrossRef]
- Le Bon, A.; Tough, D.F. Links between innate and adaptive immunity via type I interferon. Curr. Opin. Immunol. 2002, 14, 432–436. [Google Scholar] [CrossRef]
- Theofilopoulos, A.N.; Baccala, R.; Beutler, B.; Kono, D.H. Type I interferons (α/β) in immunity and autoimmunity. Annu. Rev. Immunol. 2005, 23, 307–335. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Zeisel, M.B.; Jilg, N.; Paranhos-Baccalà, G.; Stoll-Keller, F.; Wakita, T.; Hafkemeyer, P.; Blum, H.E.; Barth, H.; Henneke, P.; et al. Toll-Like Receptor 2 Senses Hepatitis C Virus Core Protein but Not Infectious Viral Particles. J. Innate Immun. 2009, 1, 446–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehme, K.W.; Guerrero, M.; Compton, T. Human Cytomegalovirus Envelope Glycoproteins B and H Are Necessary for TLR2 Activation in Permissive Cells. J. Immunol. 2006, 177, 7094–7102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Trippler, M.; Real, C.I.; Werner, M.; Luo, X.; Schefczyk, S.; Kemper, T.; Anastasiou, O.E.; Ladiges, Y.; Treckmann, J.; et al. Hepatitis B Virus Particles Activate Toll-Like Receptor 2 Signaling Initially Upon Infection of Primary Human Hepatocytes. Hepatology 2020, 72, 829–844. [Google Scholar] [CrossRef] [Green Version]
- Leoni, V.; Gianni, T.; Salvioli, S.; Campadelli-Fiume, G. Herpes Simplex Virus Glycoproteins gH/gL and gB Bind Toll-Like Receptor 2, and Soluble gH/gL Is Sufficient to Activate NF-B. J. Virol. 2012, 86, 6555–6562. [Google Scholar] [CrossRef] [Green Version]
- Kurt-Jones, E.A.; Chan, M.; Zhou, S.; Wang, J.P.; Reed, G.W.; Bronson, R.T.; Arnold, M.M.; Knipe, D.M.; Finberg, R.W. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc. Natl. Acad. Sci. USA 2004, 101, 1315–1320. [Google Scholar] [CrossRef] [Green Version]
- Cuevas, C.D.; Ross, S.R. Toll-like receptor 2-mediated innate immune responses against Junin virus in mice lead to an-tiviral adaptive immune responses during systemic infection and do not affect viral replication in the brain. J. Virol. 2014, 88, 7703–7714. [Google Scholar] [CrossRef] [Green Version]
- Georgel, P.; Jiang, Z.; Kunz, S.; Janssen, E.; Mols, J.; Hoebe, K.; Bahram, S.; Oldstone, M.B.; Beutler, B. Vesicular stomatitis virus glycoprotein G activates a specific antiviral Toll-like receptor 4-dependent pathway. Virology 2007, 362, 304–313. [Google Scholar] [CrossRef] [Green Version]
- Okumura, A.; Pitha, P.M.; Yoshimura, A.; Harty, R.N. Interaction between Ebola virus glycoprotein and host toll-like receptor 4 leads to induction of proin-flammatory cytokines and SOCS. J. Virol. 2010, 84, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Rassa, J.C.; Meyers, J.L.; Zhang, Y.; Kudaravalli, R.; Ross, S.R. Murine retroviruses activate B cells via interaction with toll-like receptor. Proc. Natl. Acad. Sci. USA 2002, 99, 2281–2286. [Google Scholar] [CrossRef] [Green Version]
- Del Cornò, M.; Cappon, A.; Donninelli, G.; Varano, B.; Marra, F.; Gessani, S. HIV-1 gp120 signaling through TLR4 modulates innate immune activation in human macrophages and the biology of hepatic stellate cells. J. Leukoc. Biol. 2016, 100, 599–606. [Google Scholar] [CrossRef]
- Lagos, D.; Vart, R.J.; Gratrix, F.; Westrop, S.J.; Emuss, V.; Wong, P.-P.; Robey, R.; Imami, N.; Bower, M.; Gotch, F.; et al. Toll-like Receptor 4 Mediates Innate Immunity to Kaposi Sarcoma Herpesvirus. Cell Host Microbe 2008, 4, 470–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Jude, B.; Pobezinskaya, Y.; Bishop, J.; Parke, S.; Medzhitov, R.M.; Chervonsky, A.V.; Golovkina, T.V. Subversion of the innate immune system by a retrovirus. Nat. Immunol. 2003, 4, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Burzyn, D.; Rassa, J.C.; Kim, D.; Nepomnaschy, I.; Ross, S.R.; Piazzon, I. Toll-Like Receptor 4-Dependent Activation of Dendritic Cells by a Retrovirus. J. Virol. 2004, 78, 576–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; Van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.C.; Wang, H.; et al. Identification of Oxidative Stress and Toll-like Receptor 4 Signaling as a Key Pathway of Acute Lung Injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Nhu, Q.M.; Shirey, K.; Teijaro, J.R.; Farber, D.L.; Netzel-Arnett, S.; Antalis, T.M.; Fasano, A.; Vogel, S.N. Novel signaling interactions between proteinase-activated receptor 2 and Toll-like receptors in vitro and in vivo. Mucosal Immunol. 2009, 3, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Piccinini, A.M.; Midwood, K.S. DAMPening inflammation by modulating TLR signalling. Mediat. Inflamm 2010, 2010, 672395. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, D.S.; A Chase, M.; Senft, A.P.; E Poynter, S.; Wong, H.R.; Page, K. Extracellular Hsp72, an endogenous DAMP, is released by virally infected airway epithelial cells and activates neutrophils via Toll-like receptor (TLR)-4. Respir. Res. 2009, 10, 31. [Google Scholar] [CrossRef] [Green Version]
- Borde, C.; Barnay-Verdier, S.; Gaillard, C.; Hocini, H.; Maréchal, V.; Gozlan, J. Stepwise Release of Biologically Active HMGB1 during HSV-2 Infection. PLoS ONE 2011, 6, e16145. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, D.; Zhou, L.; Samanta, M.; Matsumoto, M.; Ebihara, T.; Seya, T.; Imai, S.; Fujieda, M.; Kawa, K.; Takada, K. Epstein-Barr virus (EBV)–encoded small RNA is released from EBV-infected cells and activates signaling from toll-like receptor 3. J. Exp. Med. 2009, 206, 2091–2099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudd, B.D.; Burstein, E.; Duckett, C.S.; Li, X.; Lukacs, N.W. Differential Role for TLR3 in Respiratory Syncytial Virus-Induced Chemokine Expression. J. Virol. 2005, 79, 3350–3357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudd, B.D.; Smit, J.J.; Flavell, R.A.; Alexopoulou, L.; Schaller, M.A.; Gruber, A.; Berlin, A.A.; Lukacs, N.W. Deletion of TLR3 Alters the Pulmonary Immune Environment and Mucus Production during Respiratory Syncytial Virus Infection. J. Immunol. 2006, 176, 1937–1942. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Miller, D.J.; Bowman, E.R.; Nagarkar, D.R.; Schneider, D.; Zhao, Y.; Linn, M.J.; Goldsmith, A.M.; Bentley, J.K.; Sajjan, U.S.; et al. MDA5 and TLR3 initiate pro-inflammatory signaling pathways leading to rhinovirus-induced airways in-flammation and hyperresponsiveness. PLoS Pathog. 2011, 7, e1002070. [Google Scholar]
- Reinert, L.S.; Harder, L.; Holm, C.K.; Iversen, M.B.; Horan, K.A.; Dagnæs-Hansen, F.; Ulhøi, B.P.; Holm, T.H.; Mogensen, T.H.; Owens, T.; et al. TLR3 deficiency renders astrocytes permissive to herpes simplex virus infection and facilitates estab-lishment of CNS infection in mice. J. Clin. Investig. 2012, 122, 1368–1376. [Google Scholar] [CrossRef] [Green Version]
- Sancho-Shimizu, V.; De Diego, R.P.; Lorenzo, L.; Halwani, R.; Alangari, A.; Israelsson, E.; Fabrega, S.; Cardon, A.; Maluenda, J.; Tatematsu, M.; et al. Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J. Clin. Investig. 2011, 121, 4889–4902. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.-Y.; Jouanguy, E.; Ugolini, S.; Smahi, A.; Elain, G.; Romero, P.; Segal, D.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; et al. TLR3 Deficiency in Patients with Herpes Simplex Encephalitis. Science 2007, 317, 1522–1527. [Google Scholar] [CrossRef]
- Breckpot, K.; Escors, D.; Arce, F.; Lopes, L.; Karwacz, K.; Van Lint, S.; Keyaerts, M.; Collins, M. HIV-1 Lentiviral Vector Immunogenicity Is Mediated by Toll-Like Receptor 3 (TLR3) and TLR7. J. Virol. 2010, 84, 5627–5636. [Google Scholar] [CrossRef] [Green Version]
- Le Goffic, R.; Pothlichet, J.; Vitour, D.; Fujita, T.; Meurs, E.; Chignard, M.; Si-Tahar, M. Cutting Edge: Influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J. Immunol. 2007, 178, 3368–3372. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.-R.; Yu, C.-K.; Kung, S.-H.; Chen, S.-H.; Chang, C.-F.; Ho, T.-C.; Lee, Y.-P.; Chang, H.-C.; Kung, S.-H.; Lo, S.-Y.; et al. Toll-Like Receptor 3 Is Involved in Detection of Enterovirus A71 Infection and Targeted by Viral 2A Protease. Viruses 2018, 10, 689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbert, K.; Francois, S.; Sigmund, A.M.; Harper, M.S.; Barrett, B.S.; Kirschning, C.J.; Lu, M.; Santiago, M.L.; Dittmer, U. Friend retrovirus drives cytotoxic effectors through Toll-like receptor 3. Retrovirology 2014, 11, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Liang, Y.; Devaraj, S.; Wang, J.; Lemon, S.M.; Li, K. Toll-Like Receptor 3 Mediates Establishment of an Antiviral State against Hepatitis C Virus in Hepatoma Cells. J. Virol. 2009, 83, 9824–9834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazaleuskaya, L.; Veltrop, R.; Ikpeze, N.; Martín-García, J.; Navas-Martin, S. Protective Role of Toll-like Receptor 3-Induced Type I Interferon in Murine Coronavirus Infection of Macrophages. Viruses 2012, 4, 901–923. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Town, T.; Alexopoulou, L.; Anderson, J.F.; Fikrig, E.; A Flavell, R. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat. Med. 2004, 10, 1366–1373. [Google Scholar] [CrossRef]
- Hutchens, M.; Luker, K.E.; Sottile, P.; Sonstein, J.; Lukacs, N.W.; Núñez, G.; Curtis, J.L.; Luker, G.D. TLR3 Increases Disease Morbidity and Mortality from Vaccinia Infection. J. Immunol. 2008, 180, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-J. IPC: Professional Type I Interferon-Producing Cells and Plasmacytoid Dendritic Cell Precursors. Annu. Rev. Immunol. 2005, 23, 275–306. [Google Scholar] [CrossRef]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-Specific Recognition of Single-Stranded RNA via Toll-like Receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [Green Version]
- Triantafilou, K.; Orthopoulos, G.; Vakakis, E.; Ahmed, M.A.E.; Golenbock, D.T.; Lepper, P.M.; Triantafilou, M. Human cardiac inflammatory responses triggered by Coxsackie B viruses are mainly Toll-like receptor (TLR) 8-dependent. Cell. Microbiol. 2005, 7, 1117–1126. [Google Scholar] [CrossRef]
- Melchjorsen, J.; Jensen, S.B.; Malmgaard, L.; Rasmussen, S.B.; Weber, F.; Bowie, A.G.; Matikainen, S.; Paludan, S.R. Activation of Innate Defense against a Paramyxovirus Is Mediated by RIG-I and TLR7 and TLR8 in a Cell-Type-Specific Manner. J. Virol. 2005, 79, 12944–12951. [Google Scholar] [CrossRef] [Green Version]
- Alter, G.; Suscovich, T.J.; Teigen, N.; Meier, A.; Streeck, H.; Brander, C.; Altfeld, M. Single-Stranded RNA Derived from HIV-1 Serves as a Potent Activator of NK Cells. J. Immunol. 2007, 178, 7658–7666. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.M.; Alexopoulou, L.; Sato, A.; Karow, M.; Adams, N.C.; Gale, N.W.; Iwasaki, A.; Flavell, R.A. Recognition of single-stranded RNA viruses by Toll-like receptor 3. Proc. Natl. Acad. Sci. USA 2004, 101, 5598–5603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; El-Far, M.; Dupuy, F.P.; Abdel-Hakeem, M.S.; He, Z.; Procopio, F.A.; Shi, Y.; Haddad, E.K.; Ancuta, P.; Sekaly, R.-P.; et al. HCV RNA Activates APCs via TLR7/TLR8 While Virus Selectively Stimulates Macrophages Without Inducing Antiviral Responses. Sci. Rep. 2016, 6, 29447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Z.; Li, Y.; Zhou, M.; Lv, L.; Wu, Q.; Chen, C.; Zhang, Y.; Sui, B.; Tu, C.; Cui, M.; et al. Toll-Like Receptor 7 Enhances Rabies Virus-Induced Humoral Immunity by Facilitating the Formation of Germinal Centers. Front. Immunol. 2019, 10, 429. [Google Scholar] [CrossRef] [PubMed]
- Pi, R.; Iwasaki, A.; Sewald, X.; Mothes, W.; Uchil, P.D. Murine Leukemia Virus Exploits Innate Sensing by Toll-Like Receptor 7 in B-1 Cells to Establish Infection and Locally Spread in Mice. J. Virol. 2019, 93, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awais, M.; Wang, K.; Lin, X.; Qian, W.; Zhang, N.; Wang, C.; Wang, K.; Zhao, L.; Fu, Z.F.; Cui, M. TLR7 Deficiency Leads to TLR8 Compensative Regulation of Immune Response against JEV in Mice. Front. Immunol. 2017, 8, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeMaria, O.; Pagni, P.P.; Traub, S.; De Gassart, A.; Branzk, N.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Flavell, R.A.; Alexopoulou, L. TLR8 deficiency leads to autoimmunity in mice. J. Clin. Investig. 2010, 120, 3651–3662. [Google Scholar] [CrossRef]
- De Marcken, M.; Dhaliwal, K.; Danielsen, A.C.; Gautron, A.S.; Dominguez-Villar, M. TLR7 and TLR8 activate distinct pathways in monocytes during RNA virus infection. Sci. Signal 2019, 12, 1–18. [Google Scholar] [CrossRef]
- Varani, S.; Cederarv, M.; Feld, S.; Tammik, C.; Frascaroli, G.; Landini, M.P.; Söderberg-Nauclér, C. Human Cytomegalovirus Differentially Controls B Cell and T Cell Responses through Effects on Plasmacytoid Dendritic Cells. J. Immunol. 2007, 179, 7767–7776. [Google Scholar] [CrossRef]
- Fiola, S.; Gosselin, D.; Takada, K.; Gosselin, J. TLR9 Contributes to the Recognition of EBV by Primary Monocytes and Plasmacytoid Dendritic Cells. J. Immunol. 2010, 185, 3620–3631. [Google Scholar] [CrossRef] [Green Version]
- Lim, W.H.; Kireta, S.; Russ, G.R.; Coates, P.T.H. Human plasmacytoid dendritic cells regulate immune responses to Epstein-Barr virus (EBV) infection and delay EBV-related mortality in humanized NOD-SCID mice. Blood 2006, 109, 1043–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.-R.; Huang, H.-C.; Kuo, H.-C.; Sheen, J.-M.; Ou, C.-Y.; Hsu, T.-Y.; Yang, K.D. IFN-α production by human mononuclear cells infected with varicella-zoster virus through TLR9-dependent and -independent pathways. Cell. Mol. Immunol. 2011, 8, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.; Sato, A.; Akira, S.; Medzhitov, R.; Iwasaki, A. Toll-like Receptor 9–mediated Recognition of Herpes Simplex Virus-2 by Plasmacytoid Dendritic Cells. J. Exp. Med. 2003, 198, 513–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochrein, H.; Schlatter, B.; O’Keeffe, M.; Wagner, C.; Schmitz, F.; Schiemann, M.; Bauer, S.; Suter, M.; Wagner, H. Herpes simplex virus type-1 induces IFN- production via Toll-like receptor 9-dependent and -independent pathways. Proc. Natl. Acad. Sci. USA 2004, 101, 11416–11421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, A.; Punke, E.B.; Mehmetoglu-Gurbuz, T.; Peralta, D.P.; Garg, H. TLR9 polymorphism correlates with immune activation, CD4 decline and plasma IP10 levels in HIV patients. BMC Infect. Dis. 2019, 19, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Vincent, I.E.; Zannetti, C.; Lucifora, J.; Norder, H.; Protzer, U.; Hainaut, P.; Zoulim, F.; Tommasino, M.; Trepo, C.; Hasan, U.A.; et al. Hepatitis B Virus Impairs TLR9 Expression and Function in Plasmacytoid Dendritic Cells. PLoS ONE 2011, 6, e26315. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, H.B.; Chou, A.H.; Lin, S.I.; Chen, I.H.; Lien, S.P.; Liu, C.C.; Chong, P.; Liu, S.J. Toll-like receptor 9-mediated protection of enterovirus 71 infection in mice is due to the release of dan-ger-associated molecular patterns. J. Virol. 2014, 88, 11658–11670. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.M.Y.; Kok, K.-H.; Jaume, M.; Cheung, T.K.W.; Yip, T.-F.; Lai, J.C.C.; Guan, Y.; Webster, R.G.; Jin, D.-Y.; Peiris, M. Toll-like receptor 10 is involved in induction of innate immune responses to influenza virus infection. Proc. Natl. Acad. Sci. USA 2014, 111, 3793–3798. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Li, X.; Hess, N.J.; Guan, Y.; Tapping, R.I. TLR10 Is a Negative Regulator of Both MyD88-Dependent and -Independent TLR Signaling. J. Immunol. 2016, 196, 3834–3841. [Google Scholar] [CrossRef] [Green Version]
- Hess, N.J.; Jiang, S.; Li, X.; Guan, Y.; Tapping, R.I. TLR10 Is a B Cell Intrinsic Suppressor of Adaptive Immune Responses. J. Immunol. 2017, 198, 699–707. [Google Scholar] [CrossRef] [Green Version]
- Bieback, K.; Lien, E.; Klagge, I.M.; Avota, E.; Schneider-Schaulies, J.; Duprex, W.P.; Wagner, H.; Kirschning, C.J.; Ter Meulen, V.; Schneider-Schaulies, S. Hemagglutinin Protein of Wild-Type Measles Virus Activates Toll-Like Receptor 2 Signaling. J. Virol. 2002, 76, 8729–8736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.P.; Kurt-Jones, E.A.; Shin, O.S.; Manchak, M.D.; Levin, M.J.; Finberg, R.W. Varicella-Zoster Virus Activates Inflammatory Cytokines in Human Monocytes and Macrophages via Toll-Like Receptor 2. J. Virol. 2005, 79, 12658–12666. [Google Scholar] [CrossRef] [Green Version]
- Gaudreault, E.; Fiola, S.; Olivier, M.; Gosselin, J. Epstein-Barr Virus Induces MCP-1 Secretion by Human Monocytes via TLR2. J. Virol. 2007, 81, 8016–8024. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Martinez, J.; Huang, X.; Yang, Y. Innate immunity against vaccinia virus is mediated by TLR2 and requires TLR-independent production of IFN-β. Blood 2007, 109, 619–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triantafilou, K.; Triantafilou, M. Coxsackievirus B4-Induced Cytokine Production in Pancreatic Cells Is Mediated through Toll-Like Receptor 4. J. Virol. 2004, 78, 11313–11320. [Google Scholar] [CrossRef] [Green Version]
- Kurt-Jones, E.A.; Popova, L.; A Kwinn, L.; Haynes, L.M.; Jones, L.P.; Tripp, R.A.; Walsh, E.E.; Freeman, M.W.; Golenbock, D.T.; Anderson, L.J.; et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 2000, 1, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Henrick, B.M.; Yao, X.-D.; Zahoor, M.A.; Abimiku, A.; Osawe, S.; Rosenthal, K.L. TLR10 Senses HIV-1 Proteins and Significantly Enhances HIV-1 Infection. Front. Immunol. 2019, 10, 482. [Google Scholar] [CrossRef]
- Lester, S.N.; Li, K. Toll-Like Receptors in Antiviral Innate Immunity. J. Mol. Biol. 2014, 426, 1246–1264. [Google Scholar] [CrossRef]
- Lin, S.C.; Lo, Y.C.; Wu, H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 2010, 465, 885–890. [Google Scholar] [CrossRef] [Green Version]
- Kollewe, C.; Mackensen, A.-C.; Neumann, D.; Knop, J.; Cao, P.; Li, S.; Wesche, H.; Martin, M.U. Sequential Autophosphorylation Steps in the Interleukin-1 Receptor-associated Kinase-1 Regulate its Availability as an Adapter in Interleukin-1 Signaling. J. Biol. Chem. 2004, 279, 5227–5236. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Ninomiya-Tsuji, J.; Qian, Y.; Matsumoto, K.; Li, X. Interleukin-1 (IL-1) Receptor-Associated Kinase-Dependent IL-1-Induced Signaling Complexes Phosphorylate TAK1 and TAB2 at the Plasma Membrane and Activate TAK1 in the Cytosol. Mol. Cell. Biol. 2002, 22, 7158–7167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z. Ubiquitination in signaling to and activation of IKK. Immunol. Rev. 2012, 246, 95–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uematsu, S.; Sato, S.; Yamamoto, M.; Hirotani, T.; Kato, H.; Takeshita, F.; Matsuda, M.; Coban, C.; Ishii, K.J.; Kawai, T.; et al. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-{alpha} induction. J. Exp. Med. 2005, 201, 915–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, K.; Yanai, H.; Mizutani, T.; Negishi, H.; Shimada, N.; Suzuki, N.; Ohba, Y.; Takaoka, A.; Yeh, W.-C.; Taniguchi, T. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 15416–15421. [Google Scholar] [CrossRef] [Green Version]
- Cushing, L.; Winkler, A.; Jelinsky, S.A.; Lee, K.; Korver, W.; Hawtin, R.; Rao, V.R.; Fleming, M.; Lin, L.-L. IRAK4 kinase activity controls Toll-like receptor–induced inflammation through the transcription factor IRF5 in primary human monocytes. J. Biol. Chem. 2017, 292, 18689–18698. [Google Scholar] [CrossRef] [Green Version]
- Lomaga, M.A.; Yeh, W.-C.; Sarosi, I.; Duncan, G.S.; Furlonger, C.; Ho, A.; Morony, S.; Capparelli, C.; Van, G.; Kaufman, S.; et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999, 13, 1015–1024. [Google Scholar] [CrossRef] [Green Version]
- Walsh, M.C.; Lee, J.; Choi, Y. Tumor necrosis factor receptor- associated factor 6 (TRAF6) regulation of development, function, and homeostasis of the immune system. Immunol. Rev. 2015, 266, 72–92. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Yamaguchi, M.; Zhou, M.; Nishio, M.; Itoh, M.; Gotoh, B. Human Parainfluenza Virus Type 2 V Protein Inhibits TRAF6-Mediated Ubiquitination of IRF7 To Prevent TLR7- and TLR9-Dependent Interferon Induction. J. Virol. 2013, 87, 7966–7976. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Sanjo, H.; Takeda, K.; Ninomiya-Tsuji, J.; Yamamoto, M.; Kawai, T.; Matsumoto, K.; Takeuchi, O.; Akira, S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 2005, 6, 1087–1095. [Google Scholar] [CrossRef]
- Wan, Y.Y.; Chi, H.; Xie, M.; Schneider, M.D.; Flavell, R.A. The kinase TAK1 integrates antigen and cytokine receptor signaling for T cell development, survival and function. Nat. Immunol. 2006, 7, 851–858. [Google Scholar] [CrossRef]
- Ajibade, A.A.; Wang, H.Y.; Wang, R.-F. Cell type-specific function of TAK1 in innate immune signaling. Trends Immunol. 2013, 34, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.-H.; Xiao, C.; Paschal, A.E.; Bailey, S.T.; Rao, P.; Hayden, M.S.; Lee, K.-Y.; Bussey, C.; Steckel, M.; Tanaka, N.; et al. TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 2005, 19, 2668–2681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ermolaeva, M.A.; Michallet, M.-C.; Papadopoulou, N.; Utermöhlen, O.; Kranidioti, K.; Kollias, G.; Tschopp, J.; Pasparakis, M. Function of TRADD in tumor necrosis factor receptor 1 signaling and in TRIF-dependent inflammatory responses. Nat. Immunol. 2008, 9, 1037–1046. [Google Scholar] [CrossRef]
- Chang, M.; Jin, W.; Sun, S.-C. Peli1 facilitates TRIF-dependent Toll-like receptor signaling and proinflammatory cytokine production. Nat. Immunol. 2009, 10, 1089–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oganesyan, G.; Saha, S.K.; Guo, B.; He, J.Q.; Shahangian, A.; Zarnegar, B.; Perry, A.K.; Cheng, G. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nat. Cell Biol. 2005, 439, 208–211. [Google Scholar] [CrossRef]
- Häcker, H.; Redecke, V.; Blagoev, B.; Kratchmarova, I.; Hsu, L.-C.; Wang, G.G.; Kamps, M.P.; Raz, E.; Wagner, H.; Häcker, G.; et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nat. Cell Biol. 2005, 439, 204–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nat. Cell Biol. 2005, 434, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Mamane, Y.; Hiscott, J. Multiple Regulatory Domains Control IRF-7 Activity in Response to Virus Infection. J. Biol. Chem. 2000, 275, 34320–34327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wathelet, M.G.; Lin, C.H.; Parekh, B.S.; Ronco, L.V.; Howley, P.M.; Maniatis, T. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-beta enhancer in vivo. Mol. Cell 1998, 1, 507–518. [Google Scholar] [CrossRef]
- Zelensky, A.N.; Gready, J.E. The C-type lectin-like domain superfamily. FEBS J. 2005, 272, 6179–6217. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, B.; Smed-Sörensen, A.; Cohn, L.; Chalouni, C.; Vandlen, R.; Lee, B.-C.; Widger, J.; Keler, T.; Delamarre, L.; Mellman, I. Internalization and endosomal degradation of receptor-bound antigens regulate the efficiency of cross presentation by human dendritic cells. Blood 2012, 120, 2011–2020. [Google Scholar] [CrossRef]
- Ribeiro, C.M.S.; Sarrami-Forooshani, R.; Setiawan, L.C.; Zijlstra-Willems, E.M.; Van Hamme, J.L.; Tigchelaar, W.; Van Der Wel, N.N.; Kootstra, N.A.; Gringhuis, S.I.; Geijtenbeek, T.B.H. Receptor usage dictates HIV-1 restriction by human TRIM5α in dendritic cell subsets. Nat. Cell Biol. 2016, 540, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Bermejo-Jambrina, M.; Eder, J.; Helgers, L.C.; Hertoghs, N.; Nijmeijer, B.M.; Stunnenberg, M.; Geijtenbeek, T.B. C-Type Lectin Receptors in Antiviral Immunity and Viral Escape. Front. Immunol. 2018, 9, 590. [Google Scholar] [CrossRef] [PubMed]
- Chiffoleau, E. C-Type Lectin-Like Receptors as Emerging Orchestrators of Sterile Inflammation Represent Potential Ther-apeutic Targets. Front. Immunol. 2018, 9, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moris, A.; Nobile, C.; Buseyne, F.; Porrot, F.; Abastado, J.-P.; Schwartz, O. DC-SIGN promotes exogenous MHC-I–restricted HIV-1 antigen presentation. Blood 2004, 103, 2648–2654. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; Dewet, B.J.; Martinez-Pomares, L.; Radcliffe, C.M.; Dwek, R.A.; Rudd, P.M.; Gordon, S. The mannose receptor mediates dengue virus infection of macrophages. PLoS Pathog. 2008, 4, e17. [Google Scholar] [CrossRef]
- Geijtenbeek, T.B.; Kwon, D.S.; Torensma, R.; Van Vliet, S.J.; Van Duijnhoven, G.C.; Middel, J.; Cornelissen, I.L.; Nottet, H.S.; KewalRamani, V.N.; Littman, D.R.; et al. DC-SIGN, a Dendritic Cell–Specific HIV-1-Binding Protein that Enhances trans-Infection of T Cells. Cell 2000, 100, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Hoving, J.C.; Wilson, G.J.; Brown, G.D. Signalling C-Type lectin receptors, microbial recognition and immunity. Cell. Microbiol. 2014, 16, 185–194. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, J.T.; Lepenies, B. Myeloid C-Type Lectin Receptors in Viral Recognition and Antiviral Immunity. Viruses 2017, 9, 59. [Google Scholar] [CrossRef]
- Ludwig, I.S.; Lekkerkerker, A.N.; Depla, E.; Bosman, F.; Musters, R.J.P.; Depraetere, S.; Van Kooyk, Y.; Geijtenbeek, T.B.H. Hepatitis C Virus Targets DC-SIGN and L-SIGN to Escape Lysosomal Degradation. J. Virol. 2004, 78, 8322–8332. [Google Scholar] [CrossRef] [Green Version]
- Simmons, G.; Reeves, J.D.; Grogan, C.C.; Vandenberghe, L.H.; Baribaud, F.; Whitbeck, J.C.; Burke, E.; Buchmeier, M.J.; Soilleux, E.J.; Riley, J.L.; et al. DC-SIGN and DC-SIGNR Bind Ebola Glycoproteins and Enhance Infection of Macrophages and Endothelial Cells. Virology 2003, 305, 115–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klimstra, W.B.; Nangle, E.M.; Smith, M.S.; Yurochko, A.D.; Ryman, K.D. DC-SIGN and L-SIGN can act as attachment receptors for alphaviruses and distinguish between mosquito cell- and mammalian cell-derived viruses. J. Virol. 2003, 77, 12022–12032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouw, M.L.O.D.; De Jong, M.A.W.P.; Ludwig, I.S.; Van Der Molen, R.G.; Janssen, H.L.A.; Geijtenbeek, T.B.H.; Woltman, A.M. Branched oligosaccharide structures on HBV prevent interaction with both DC-SIGN and L-SIGN. J. Viral Hepat. 2008, 15, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Han, D.P.; Lohani, M.; Cho, M.W. Specific Asparagine-Linked Glycosylation Sites Are Critical for DC-SIGN- and L-SIGN-Mediated Severe Acute Respiratory Syndrome Coronavirus Entry. J. Virol. 2007, 81, 12029–12039. [Google Scholar] [CrossRef] [Green Version]
- Gramberg, T.; Hofmann, H.; Möller, P.; Lalor, P.F.; Marzi, A.; Geier, M.; Krumbiegel, M.; Winkler, T.; Kirchhoff, F.; Adams, D.H.; et al. LSECtin interacts with filovirus glycoproteins and the spike protein of SARS coronavirus. Virology 2005, 340, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Gramberg, T.; Hofmann, H.; Möller, P.; Lalor, P.F.; Marzi, A.; Geier, M.; Krumbiegel, M.; Winkler, T.; Kirchhoff, F.; Adams, D.H.; et al. CLEC5A regulates Japanese encephalitis virus-induced neuroinflammation and lethality. PLoS Pathog. 2012, 8, e1002655. [Google Scholar]
- Chen, S.-T.; Lin, Y.-L.; Huang, M.-T.; Wu, M.-F.; Cheng, S.-C.; Lei, H.-Y.; Lee, C.-K.; Chiou, T.-W.; Wong, C.-H.; Hsieh, S.-L. CLEC5A is critical for dengue-virus-induced lethal disease. Nat. Cell Biol. 2008, 453, 672–676. [Google Scholar] [CrossRef]
- Teng, O.; Chen, S.-T.; Hsu, T.-L.; Sia, S.F.; Cole, S.; Valkenburg, S.A.; Hsu, T.-Y.; Zheng, J.T.; Tu, W.; Bruzzone, R.; et al. CLEC5A-Mediated Enhancement of the Inflammatory Response in Myeloid Cells Contributes to Influenza Virus Pathogenicity In Vivo. J. Virol. 2016, 91, e01813-16. [Google Scholar] [CrossRef] [Green Version]
- De Witte, L.; Nabatov, A.; Pion, M.; Fluitsma, D.; De Jong, M.A.; de Gruijl, T.; Piguet, V.; van Kooyk, Y.; Geijtenbeek, T.B. Langerin is a natural barrier to HIV-1 transmission by Langerhans cells. Nat. Med. 2007, 13, 367–371. [Google Scholar] [CrossRef]
- Ng, W.C.; Londrigan, S.L.; Nasr, N.; Cunningham, A.L.; Turville, S.; Brooks, A.G.; Reading, P.C. The C-type Lectin Langerin Functions as a Receptor for Attachment and Infectious Entry of Influenza A Virus. J. Virol. 2015, 90, 206–221. [Google Scholar] [CrossRef] [Green Version]
- Lambert, A.A.; Gilbert, C.; Richard, M.; Beaulieu, A.D.; Tremblay, M.J. The C-type lectin surface receptor DCIR acts as a new attachment factor for HIV-1 in dendritic cells and contributes to trans- and cis-infection pathways. Blood 2008, 112, 1299–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iborra, S.; Izquierdo, H.M.; Martínez-López, M.; Blanco-Menéndez, N.; Sousa, C.R.E.; Sancho, D. The DC receptor DNGR-1 mediates cross-priming of CTLs during vaccinia virus infection in mice. J. Clin. Investig. 2012, 122, 1628–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteiro, J.T.; Schön, K.; Ebbecke, T.; Goethe, R.; Ruland, J.; Baumgärtner, W.; Becker, S.C.; Lepenies, B. The CARD9-Associated C-Type Lectin, Mincle, Recognizes La Crosse Virus (LACV) but Plays a Limited Role in Early Antiviral Responses against LACV. Viruses 2019, 11, 303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahim, M.M.A.; Chen, P.; Mottashed, A.N.; Mahmoud, A.B.; Thomas, M.J.; Zhu, Q.; Brooks, C.G.; Kartsogiannis, V.; Gillespie, M.T.; Carlyle, J.R.; et al. The mouse NKR-P1B:Clr-b recognition system is a negative regulator of innate immune responses. Blood 2015, 125, 2217–2227. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, O.A.; Sampaio, I.S.; Rahim, M.M.A.; Samaniego, J.D.; Tilahun, M.E.; Krishnamoorthy, M.; Popović, B.; Babić, M.; Krmpotić, A.; Treanor, B.; et al. Mouse Cytomegalovirus m153 Protein Stabilizes Expression of the Inhibitory NKR-P1B Ligand Clr-b. J. Virol. 2019, 94, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, T.; Aratanib, S.; Nakajimab, T.; Carlsonc, M.; Matsumiyad, T.; Tanjie, K.; Ookawaf, K.; Yoshidaa, H.; Tsuchidaf, S.; McIntyre, T.M.; et al. Retinoic Acid-Inducible Gene-I Is Induced in Endothelial Cells by LPS and Regulates Expression of COX-2. Biochem. Biophys. Res. Commun. 2002, 292, 274–279. [Google Scholar] [CrossRef]
- Brisse, M.; Ly, H. Comparative Structure and Function Analysis of the RIG-I-Like Receptors: RIG-I and MDA5. Front. Immunol. 2019, 10, 1586. [Google Scholar] [CrossRef]
- Kang, D.-C.; Gopalkrishnan, R.V.; Wu, Q.; Jankowsky, E.; Pyle, A.M.; Fisher, P.B. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc. Natl. Acad. Sci. USA 2002, 99, 637–642. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Li, M.; Walton, K.D.; Sun, K.; Hanover, J.A.; Furth, P.A.; Hennighausen, L. The Stat3/5 locus encodes novel endoplasmic reticulum and helicase-like proteins that are preferentially ex-pressed in normal and neoplastic mammary tissue. Genomics 2001, 78, 129–134. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Loo, Y.-M.; Gale, M. Immune Signaling by RIG-I-like Receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruns, A.M.; Horvath, C.M. Antiviral RNA recognition and assembly by RLR family innate immune sensors. Cytokine Growth Factor Rev. 2014, 25, 507–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahasi, K.; Kumeta, H.; Tsuduki, N.; Narita, R.; Shigemoto, T.; Hirai, R.; Yoneyama, M.; Horiuchi, M.; Ogura, K.; Fujita, T.; et al. Solution structures of cytosolic RNA sensor MDA5 and LGP2 C-terminal domains: Identification of the RNA recognition loop in RIG-I-like receptors. J. Biol. Chem. 2009, 284, 17465–17474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, T.; Kato, H.; Kumagai, Y.; Yoneyama, M.; Sato, S.; Matsushita, K.; Tsujimura, T.; Fujita, T.; Akira, S.; Takeuchi, O. LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc. Natl. Acad. Sci. USA 2010, 107, 1512–1517. [Google Scholar] [CrossRef] [Green Version]
- Uchikawa, E.; Lethier, M.; Malet, H.; Brunel, J.; Gerlier, D.; Cusack, S. Structural Analysis of dsRNA Binding to Anti-viral Pattern Recognition Receptors LGP2 and MDA5. Mol. Cell 2016, 62, 586–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Xu, H.; Ranjith-Kumar, C.T.; Brooks, M.T.; Hou, T.Y.; Hu, F.; Herr, A.B.; Strong, R.K.; Kao, C.C.; Li, P. The Structural Basis of 5′ Triphosphate Double-Stranded RNA Recognition by RIG-I C-Terminal Domain. Structure 2010, 18, 1032–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Ludwig, J.; Schuberth, C.; Goldeck, M.; Schlee, M.; Li, H.; Juranek, S.; Sheng, G.; Micura, R.; Tuschl, T.; et al. Structural and functional insights into 5′-ppp RNA pattern recognition by the innate immune receptor RIG-I. Nat. Struct. Mol. Biol. 2010, 17, 781–787. [Google Scholar] [CrossRef] [Green Version]
- Goubau, D.; Schlee, M.; Deddouche, S.; Pruijssers, A.J.; Zillinger, T.; Goldeck, M.; Schuberth, C.; Van Der Veen, A.G.; Fujimura, T.; Rehwinkel, J.; et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nat. Cell Biol. 2014, 514, 372–375. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.G.; Kim, M.V.; Chen, X.; Batista, P.J.; Aoyama, S.; Wilusz, J.E.; Iwasaki, A.; Chang, H.Y. Sensing Self and Foreign Circular RNAs by Intron Identity. Mol. Cell 2017, 67, 228–238.e5. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Karijolich, J. Know Thyself: RIG-I-Like Receptor Sensing of DNA Virus Infection. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nat. Cell Biol. 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Uematsu, S.; Matsui, K.; Tsujimura, T.; Takeda, K.; Fujita, T.; Takeuchi, O.; et al. Cell Type-Specific Involvement of RIG-I in Antiviral Response. Immunity 2005, 23, 19–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minamitani, T.; Iwakiri, D.; Takada, K. Adenovirus Virus-Associated RNAs Induce Type I Interferon Expression through a RIG-I-Mediated Pathway. J. Virol. 2011, 85, 4035–4040. [Google Scholar] [CrossRef] [Green Version]
- Samanta, M.; Iwakiri, D.; Kanda, T.; Imaizumi, T.; Takada, K. EB virus-encoded RNAs are recognized by RIG-I and activate signaling to induce type I IFN. EMBO J. 2006, 25, 4207–4214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samanta, M.; Iwakiri, D.; Takada, K. Epstein–Barr virus-encoded small RNA induces IL-10 through RIG-I-mediated IRF-3 signaling. Oncogene 2008, 27, 4150–4160. [Google Scholar] [CrossRef] [Green Version]
- Myskiw, C.; Arsenio, J.; Booy, E.P.; Hammett, C.; Deschambault, Y.; Gibson, S.B.; Cao, J. RNA species generated in vaccinia virus infected cells activate cell type-specific MDA5 or RIG-I dependent interferon gene transcription and PKR dependent apoptosis. Virology 2011, 413, 183–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaloye, J.; Roger, T.; Steiner-Tardivel, Q.-G.; Le Roy, D.; Reymond, M.K.; Akira, S.; Petrilli, V.; Gomez, C.E.; Perdiguero, B.; Tschopp, J.; et al. Innate Immune Sensing of Modified Vaccinia Virus Ankara (MVA) Is Mediated by TLR2-TLR6, MDA-5 and the NALP3 Inflammasome. PLoS Pathog. 2009, 5, e1000480. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Zhong, J.; Chung, J.; Chisari, F.V. Double-stranded DNA and double-stranded RNA induce a common antiviral signaling pathway in human cells. Proc. Natl. Acad. Sci. USA 2007, 104, 9035–9040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funabiki, M.; Kato, H.; Miyachi, Y.; Toki, H.; Motegi, H.; Inoue, M.; Minowa, O.; Yoshida, A.; Deguchi, K.; Sato, H.; et al. Autoimmune Disorders Associated with Gain of Function of the Intracellular Sensor MDA5. Immunity 2014, 40, 199–212. [Google Scholar] [CrossRef] [Green Version]
- Smyth, D.J.; Cooper, J.D.; Bailey, R.; Field, S.; Burren, O.; Smink, L.J.; Guja, C.; Ionescu-Tirgoviste, C.; Widmer, B.; Dunger, D.B.; et al. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the inter-feron-induced helicase (IFIH1) region. Nat. Genet. 2006, 38, 617–619. [Google Scholar] [CrossRef]
- Sheng, Y.; Jin, X.; Xu, J.; Gao, J.; Du, X.; Duan, D.; Li, B.; Zhao, J.; Zhan, W.; Tang, H.; et al. Sequencing-based approach identified three new susceptibility loci for psoriasis. Nat. Commun. 2014, 5, 4331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, A.; Varade, J.; Lamas, J.R.; Fernandez-Arquero, M.; Jover, J.A.; De La Concha, E.G.; Fernandez-Gutierrez, B.; Urcelay, E. Association of the IFIH1-GCA-KCNH7 chromosomal region with rheumatoid arthritis. Ann. Rheum. Dis. 2008, 67, 137–138. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Birlea, S.A.; Fain, P.R.; Ferrara, T.M.; Ben, S.; Riccardi, S.L.; Cole, J.B.; Gowan, K.; Holland, P.J.; Bennett, D.C.; et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat. Genet. 2012, 44, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Enevold, C.; Oturai, A.B.; Sørensen, P.S.; Ryder, L.P.; Koch-Henriksen, N.; Bendtzen, K. Multiple sclerosis and polymorphisms of innate pattern recognition receptors TLR1-10, NOD1-2, DDX58, and IFIH. J. Neuroimmunol. 2009, 212, 125–131. [Google Scholar] [CrossRef]
- Graham, D.S.C.; Morris, D.L.; Bhangale, T.R.; Criswell, L.A.; Syvänen, A.-C.; Rönnblom, L.; Behrens, T.W.; Graham, R.R.; Vyse, T.J. Association of NCF2, IKZF1, IRF8, IFIH1, and TYK2 with Systemic Lupus Erythematosus. PLoS Genet. 2011, 7, e1002341. [Google Scholar] [CrossRef]
- Rutsch, F.; MacDougall, M.; Lu, C.; Buers, I.; Mamaeva, O.; Nitschke, Y.; Rice, G.I.; Erlandsen, H.; Kehl, H.G.; Thiele, H.; et al. A Specific IFIH1 Gain-of-Function Mutation Causes Singleton-Merten Syndrome. Am. J. Hum. Genet. 2015, 96, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Rice, G.I.; del Toro Duany, Y.; Jenkinson, E.M.; Forte, G.M.; Anderson, B.H.; Ariaudo, G.; Bader-Meunier, B.; Baildam, E.M.; Battini, R.; Beresford, M.W.; et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upreg-ulated type I interferon signaling. Nat. Genet. 2014, 46, 503–509. [Google Scholar] [CrossRef]
- Barral, P.M.; Morrison, J.M.; Drahos, J.; Gupta, P.; Sarkar, D.; Fisher, P.B.; Racaniello, V.R. MDA-5 Is Cleaved in Poliovirus-Infected Cells. J. Virol. 2007, 81, 3677–3684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gitlin, L.; Barchet, W.; Gilfillan, S.; Cella, M.; Beutler, B.; Flavell, R.A.; Diamond, M.S.; Colonna, M. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalo-myocarditis picornavirus. Proc. Natl. Acad. Sci. USA 2006, 103, 8459–8464. [Google Scholar] [CrossRef] [Green Version]
- McCartney, S.A.; Thackray, L.B.; Gitlin, L.; Gilfillan, S.; Virgin IV, H.W.; Colonna, M. MDA-5 recognition of a murine norovirus. PLoS Pathog. 2008, 4, e1000108. [Google Scholar] [CrossRef]
- Zaki, M.; Thoenes, M.; Kawalia, A.; Nürnberg, P.; Kaiser, R.; Heller, R.; Bolz, H.J. Recurrent and Prolonged Infections in a Child with a Homozygous IFIH1 Nonsense Mutation. Front. Genet. 2017, 8, 130. [Google Scholar] [CrossRef]
- Asgari, S.; Schlapbach, L.J.; Anchisi, S.; Hammer, C.; Bartha, I.; Junier, T.; Mottet-Osman, G.; Posfay-Barbe, K.M.; Longchamp, D.; Stocker, M.; et al. Severe viral respiratory infections in children with IFIH1 loss-of-function mutations. Proc. Natl. Acad. Sci. USA 2017, 114, 8342–8347. [Google Scholar] [CrossRef] [Green Version]
- Lamborn, I.T.; Jing, H.; Zhang, Y.; Drutman, S.; Abbott, J.; Munir, S.; Bade, S.; Murdock, H.M.; Santos, C.P.; Brock, L.G.; et al. Recurrent rhinovirus infections in a child with inherited MDA5 deficiency. J. Exp. Med. 2017, 214, 1949–1972. [Google Scholar] [CrossRef]
- Fredericksen, B.L.; Keller, B.C.; Fornek, J.; Katze, M.G.; Gale, M. Establishment and Maintenance of the Innate Antiviral Response to West Nile Virus Involves both RIG-I and MDA5 Signaling through IPS-1. J. Virol. 2007, 82, 609–616. [Google Scholar] [CrossRef] [Green Version]
- Uno, N.; Ross, T.M. Dengue virus and the host innate immune response. Emerg. Microbes Infect. 2018, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid–inducible gene-I and melanoma differentiation–associated gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.M.; Gale, M.; Akira, S.; et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 2005, 175, 2851–2858. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Zhuang, M.W.; Han, L.; Zhang, J.; Nan, M.L.; Zhan, P.; Kang, D.; Liu, X.; Gao, C.; Wang, P.H. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) membrane (M) protein inhibits type I and III interferon production by targeting RIG-I/MDA-5 signaling. Signal Transduct. Target Ther. 2020, 5, 299. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Hill, T.E.; Yoshikawa, N.; Popov, V.L.; Galindo, C.L.; Garner, H.R.; Peters, C.J.; Tseng, C.K. Dynamic Innate Immune Responses of Human Bronchial Epithelial Cells to Severe Acute Respiratory Syndrome-Associated Coronavirus Infection. PLoS ONE 2010, 5, e8729. [Google Scholar] [CrossRef]
- Roth-Cross, J.K.; Bender, S.J.; Weiss, S.R. Murine Coronavirus Mouse Hepatitis Virus Is Recognized by MDA5 and Induces Type I Interferon in Brain Macrophages/Microglia. J. Virol. 2008, 82, 9829–9838. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, Y.; Zhang, X. Murine Coronavirus Induces Type I Interferon in Oligodendrocytes through Recognition by RIG-I and MDA5. J. Virol. 2010, 84, 6472–6482. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zhao, J.; Van Rooijen, N.; Perlman, S. Evasion by stealth: Inefficient immune activation underlies poor T cell response and severe disease in SARS-CoV-infected mice. PLoS Pathog. 2009, 5, e1000636. [Google Scholar] [CrossRef]
- Totura, A.L.; Baric, R. SARS coronavirus pathogenesis: Host innate immune responses and viral antagonism of interferon. Curr. Opin. Virol. 2012, 2, 264–275. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Kato, H.; Sato, S.; Takahashi, K.; Coban, C.; Yamamoto, M.; Uematsu, S.; Ishii, K.J.; Takeuchi, O.; et al. Essential role of IPS-1 in innate immune responses against RNA viruses. J. Exp. Med. 2006, 203, 1795–1803. [Google Scholar] [CrossRef] [Green Version]
- Xing, F.; Matsumiya, T.; Hayakari, R.; Yoshida, H.; Kawaguchi, S.; Takahashi, I.; Nakaji, S.; Imaizumi, T. Alteration of Antiviral Signalling by Single Nucleotide Polymorphisms (SNPs) of Mitochondrial Antiviral Signalling Protein (MAVS). PLoS ONE 2016, 11, e0151173. [Google Scholar] [CrossRef]
- Simpson, J.; Lynch, J.P.; Loh, Z.; Zhang, V.; Werder, R.B.; Spann, K.; Phipps, S. The Absence of Interferon-beta Promotor Stimulator-1 (IPS-1) Predisposes to Bronchiolitis and Asthma-like Pathology in Response to Pneumoviral Infection in Mice. Sci. Rep. 2017, 7, 2353. [Google Scholar] [CrossRef] [Green Version]
- Suthar, M.S.; Ma, D.Y.; Thomas, S.; Lund, J.M.; Zhang, N.; Daffis, S.; Rudensky, A.Y.; Bevan, M.J.; Clark, E.A.; Kaja, M.-K.; et al. IPS-1 Is Essential for the Control of West Nile Virus Infection and Immunity. PLoS Pathog. 2010, 6, e1000757. [Google Scholar] [CrossRef]
- Roe, K.; Giordano, D.; Young, L.B.; Draves, K.E.; Holder, U.; Suthar, M.S.; Gale, M.; Clark, E.A. Dendritic cell-associated MAVS is required to control West Nile virus replication and ensuing humoral immune responses. PLoS ONE 2019, 14, e0218928. [Google Scholar] [CrossRef]
- Takaki, H.; Takeda, M.; Tahara, M.; Shingai, M.; Oshiumi, H.; Matsumoto, M.; Seya, T. The MyD88 Pathway in Plasmacytoid and CD4+Dendritic Cells Primarily Triggers Type I IFN Production against Measles Virus in a Mouse Infection Model. J. Immunol. 2013, 191, 4740–4747. [Google Scholar] [CrossRef] [Green Version]
- Faul, E.J.; Wanjalla, C.N.; Suthar, M.S.; Gale, M.; Wirblich, C.; Schnell, M.J. Rabies Virus Infection Induces Type I Interferon Production in an IPS-1 Dependent Manner While Dendritic Cell Activation Relies on IFNAR Signaling. PLoS Pathog. 2010, 6, e1001016. [Google Scholar] [CrossRef]
- Cárdenas, W.; Loo, Y.-M.; Gale, M.; Hartman, A.L.; Kimberlin, C.R.; Martínez-Sobrido, L.; Saphire, E.O.; Basler, C.F. Ebola Virus VP35 Protein Binds Double-Stranded RNA and Inhibits Alpha/Beta Interferon Production Induced by RIG-I Signaling. J. Virol. 2006, 80, 5168–5178. [Google Scholar] [CrossRef] [Green Version]
- Habjan, M.; Andersson, I.; Klingström, J.; Schümann, M.; Martin, A.; Zimmermann, P.; Wagner, V.; Pichlmair, A.; Schneider, U.; Mühlberger, E.; et al. Processing of Genome 5′ Termini as a Strategy of Negative-Strand RNA Viruses to Avoid RIG-I-Dependent Interferon Induction. PLoS ONE 2008, 3, e2032. [Google Scholar] [CrossRef]
- Sen, A.; Pruijssers, A.J.; Dermody, T.S.; García-Sastre, A.; Greenberg, H.B. The Early Interferon Response to Rotavirus Is Regulated by PKR and Depends on MAVS/IPS-1, RIG-I, MDA-5, and IRF3. J. Virol. 2011, 85, 3717–3732. [Google Scholar] [CrossRef] [Green Version]
- Ikegame, S.; Takeda, M.; Ohno, S.; Nakatsu, Y.; Nakanishi, Y.; Yanagi, Y. Both RIG-I and MDA5 RNA helicases contribute to the induction of alpha/beta interferon in measles vi-rus-infected human cells. J. Virol. 2010, 84, 372–379. [Google Scholar] [CrossRef] [Green Version]
- Solis, M.; Nakhaei, P.; Jalalirad, M.; Lacoste, J.; Douville, R.; Arguello, M.; Zhao, T.; Laughrea, M.; Wainberg, M.A.; Hiscott, J. RIG-I-Mediated Antiviral Signaling Is Inhibited in HIV-1 Infection by a Protease-Mediated Sequestration of RIG-I. J. Virol. 2010, 85, 1224–1236. [Google Scholar] [CrossRef] [Green Version]
- Sabbah, A.; Bose, S. Retinoic acid inducible gene I Activates innate antiviral response against human parainfluenza virus type 3. J. Virol. 2009, 6, 200. [Google Scholar] [CrossRef]
- Sumpter, R.; Loo, Y.-M.; Foy, E.; Li, K.; Yoneyama, M.; Fujita, T.; Lemon, S.M.; Gale, M. Regulating Intracellular Antiviral Defense and Permissiveness to Hepatitis C Virus RNA Replication through a Cellular RNA Helicase, RIG-I. J. Virol. 2005, 79, 2689–2699. [Google Scholar] [CrossRef] [Green Version]
- Holm, G.H.; Zurney, J.; Tumilasci, V.; Leveille, S.; Danthi, P.; Hiscott, J.; Sherry, B.; Dermody, T.S. Retinoic acid-inducible gene-I and interferon-beta promoter stimulator-1 augment proapoptotic responses following mammalian reovirus infection via interferon regulatory factor-3. J. Biol. Chem. 2007, 282, 21953–21961. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Cerny, A.M.; Zacharia, A.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Finberg, R.W. Induction and inhibition of type I interferon responses by distinct components of lymphocytic choriomen-ingitis virus. J. Virol. 2010, 84, 9452–9462. [Google Scholar] [CrossRef] [Green Version]
- Gitlin, L.; Benoit, L.; Song, C.; Cella, M.; Gilfillan, S.; Holtzman, M.J.; Colonna, M. Melanoma differentiation-associated gene 5 (MDA5) is involved in the innate immune response to Para-myxoviridae infection in vivo. PLoS Pathog. 2010, 6, e1000734. [Google Scholar] [CrossRef]
- Wang, J.P.; Cerny, A.; Asher, D.R.; Kurt-Jones, E.A.; Bronson, R.T.; Finberg, R.W. MDA5 and MAVS Mediate Type I Interferon Responses to Coxsackie B Virus. J. Virol. 2009, 84, 254–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melchjorsen, J.; Rintahaka, J.; Søby, S.; Horan, K.A.; Poltajainen, A.; Østergaard, L.; Paludan, S.R.; Matikainen, S. Early innate recognition of herpes simplex virus in human primary macrophages is mediated via the MDA5/MAVS-dependent and MDA5/MAVS/RNA polymerase III-independent pathways. J. Virol. 2010, 84, 11350–11358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayagaki, N.; Phung, Q.; Chan, S.; Chaudhari, R.; Quan, C.; O’Rourke, K.M.; Eby, M.; Pietras, E.; Cheng, G.; Bazan, J.F.; et al. DUBA: A Deubiquitinase That Regulates Type I Interferon Production. Science 2007, 318, 1628–1632. [Google Scholar] [CrossRef]
- Li, S.; Zheng, H.; Mao, A.-P.; Zhong, B.; Li, Y.; Liu, Y.; Gao, Y.; Ran, Y.; Tien, P.; Shu, H.-B. Regulation of Virus-triggered Signaling by OTUB1- and OTUB2-mediated Deubiquitination of TRAF3 and TRAF6. J. Biol. Chem. 2010, 285, 4291–4297. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Xu, R.; Zheng, X. HSCARG Negatively Regulates the Cellular Antiviral RIG-I Like Receptor Signaling Pathway by Inhibiting TRAF3 Ubiquitination via Recruiting OTUB1. PLoS Pathog. 2014, 10, e1004041. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Jin, J.; Zhu, L.; Jie, Z.; Li, Y.; Zhao, B.; Cheng, X.; Li, P.; Sun, S.-C. Cell type-specific function of TRAF2 and TRAF3 in regulating type I IFN induction. Cell Biosci. 2019, 9, 5. [Google Scholar] [CrossRef]
- Hatesuer, B.; Hoang, H.T.T.; Riese, P.; Trittel, S.; Gerhauser, I.; Elbahesh, H.; Geffers, R.; Wilk, E.; Schughart, K. Deletion of Irf3 and Irf7 Genes in Mice Results in Altered Interferon Pathway Activation and Granulo-cyte-Dominated Inflammatory Responses to Influenza a Infection. J. Innate. Immunol. 2017, 9, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-W.; King, K.; Tu, J.; Sanchez, M.; Luster, A.D.; Shresta, S. The roles of IRF-3 and IRF-7 in innate antiviral immunity against dengue virus. J. Immunol. 2013, 191, 4194–4201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Honda, K.; Taniguchi, T. IRFs: Master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006, 6, 644–658. [Google Scholar] [CrossRef]
- Balachandran, S.; Thomas, E.; Barber, G.N. A FADD-dependent innate immune mechanism in mammalian cells. Nat. Cell Biol. 2004, 432, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Komuro, A.; Horvath, C.M. RNA- and Virus-Independent Inhibition of Antiviral Signaling by RNA Helicase LGP2. J. Virol. 2006, 80, 12332–12342. [Google Scholar] [CrossRef] [Green Version]
- Childs, K.S.; Randall, R.E.; Goodbourn, S. LGP2 Plays a Critical Role in Sensitizing mda-5 to Activation by Double-Stranded RNA. PLoS ONE 2013, 8, e64202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruns, A.M.; Pollpeter, D.; Hadizadeh, N.; Myong, S.; Marko, J.F.; Horvath, C.M. ATP Hydrolysis Enhances RNA Recognition and Antiviral Signal Transduction by the Innate Immune Sensor, Laboratory of Genetics and Physiology 2 (LGP2). J. Biol. Chem. 2013, 288, 938–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deddouche, S.; Goubau, D.; Rehwinkel, J.; Chakravarty, P.; Begum, S.; Maillard, P.V.; Borg, A.; Matthews, N.; Feng, Q.; Van Kuppeveld, F.J.M.; et al. Identification of an LGP2-associated MDA5 agonist in picornavirus-infected cells. eLife 2014, 3, e01535. [Google Scholar] [CrossRef] [PubMed]
- Hei, L.; Zhong, J. Laboratory of genetics and physiology 2 (LGP2) plays an essential role in hepatitis C virus infec-tion-induced interferon responses. Hepatology 2017, 65, 1478–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franchi, L.; Warner, N.; Viani, K.; Nuñez, G. Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev. 2008, 227, 106–128. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.K.; Shin, J.-S.; Nahm, M.H. NOD-Like Receptors in Infection, Immunity, and Diseases. Yonsei Med. J. 2016, 57, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, M.H.; Reimer, T.; Kim, Y.-G.; Núñez, G. NOD-like receptors (NLRs): Bona fide intracellular microbial sensors. Curr. Opin. Immunol. 2008, 20, 377–382. [Google Scholar] [CrossRef] [Green Version]
- Kanneganti, T.D.; Body-Malapel, M.; Amer, A.; Park, J.H.; Whitfield, J.; Franchi, L.; Taraporewala, Z.F.; Miller, D.; Patton, J.T.; Inohara, N.; et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and dou-ble-stranded RNA. J. Biol. Chem. 2006, 281, 36560–36568. [Google Scholar] [CrossRef] [Green Version]
- Muruve, D.A.; Pétrilli, V.; Zaiss, A.K.; White, L.R.; Clark, S.A.; Ross, P.J.; Parks, R.J.; Tschopp, J. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune re-sponse. Nature 2008, 452, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Ramos, H.J.; Lanteri, M.C.; Blahnik, G.; Negash, A.; Suthar, M.S.; Brassil, M.M.; Sodhi, K.; Treuting, P.M.; Busch, M.P.; Norris, P.J.; et al. IL-1beta signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLoS Pathog. 2012, 8, e1003039. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhao, W. NLRP3 Inflammasome—A Key Player in Antiviral Responses. Front. Immunol. 2020, 11, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and Functions of Inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [Green Version]
- Gram, A.M.; Frenkel, J.; Ressing, M.E. Inflammasomes and viruses: Cellular defence versus viral offence. J. Gen. Virol. 2012, 93, 2063–2075. [Google Scholar] [CrossRef]
- Sharif, H.; Wang, L.; Wang, W.L.; Magupalli, V.G.; Andreeva, L.; Qiao, Q.; Hauenstein, A.V.; Wu, Z.; Núñez, G.; Mao, Y.; et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nat. Cell Biol. 2019, 570, 338–343. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Rathinam, V.A.K.; Fitzgerald, K.A. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell 2016, 165, 792–800. [Google Scholar] [CrossRef] [Green Version]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Joosten, L.A.; Netea, M.G.; Dinarello, C.A. Interleukin-1beta in innate inflammation, autophagy and immunity. Semin. Immunol. 2013, 25, 416–424. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Novick, D.; Kim, S.; Kaplanski, G. Interleukin-18 and IL-18 Binding Protein. Front. Immunol. 2013, 4, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuriakose, T.; Kanneganti, T.-D. Regulation and functions of NLRP3 inflammasome during influenza virus infection. Mol. Immunol. 2017, 86, 56–64. [Google Scholar] [CrossRef] [PubMed]
- De Castro-Jorge, L.A.; de Carvalho, R.V.; Klein, T.M.; Hiroki, C.H.; Lopes, A.H.; Guimarães, R.M.; Fumagalli, M.J.; Floriano, V.G.; Agostinho, M.R.; Slhessarenko, R.D.; et al. The NLRP3 inflammasome is involved with the pathogenesis of Mayaro virus. PLoS Pathog. 2019, 15, e1007934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negash, A.A.; Ramos, H.J.; Crochet, N.; Lau, D.T.; Doehle, B.; Papic, N.; Delker, D.A.; Jo, J.; Bertoletti, A.; Hagedorn, C.H.; et al. IL-1beta production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 2013, 9, e1003330. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Xiong, M.; Wang, S.; Wang, Y.-Y.; Ran, Y. The Regulation of cGAS. Virol. Sin. 2018, 33, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Dai, P.; Wang, W.; Cao, H.; Avogadri, F.; Dai, L.; Drexler, I.; Joyce, J.A.; Li, X.D.; Chen, Z.; Merghoub, T.; et al. Modified vaccinia virus Ankara triggers type I IFN production in murine conventional dendritic cells via a cGAS/STING-mediated cytosolic DNA-sensing pathway. PLoS Pathog. 2014, 10, e1003989. [Google Scholar] [CrossRef] [Green Version]
- Lam, E.; Stein, S.; Falck-Pedersen, E. Adenovirus Detection by the cGAS/STING/TBK1 DNA Sensing Cascade. J. Virol. 2014, 88, 974–981. [Google Scholar] [CrossRef] [Green Version]
- El-Jesr, M.; Teir, M.; De Motes, C.M. Vaccinia Virus Activation and Antagonism of Cytosolic DNA Sensing. Front. Immunol. 2020, 11, 568412. [Google Scholar] [CrossRef]
- Muntjewerff, E.M.; Meesters, L.D.; Bogaart, G.V.D. Antigen Cross-Presentation by Macrophages. Front. Immunol. 2020, 11, 1276. [Google Scholar] [CrossRef]
- Haag, S.M.; Gulen, M.F.; Reymond, L.; Gibelin, A.; Abrami, L.; Decout, A.; Heymann, M.; Van Der Goot, F.G.; Turcatti, G.; Behrendt, R.; et al. Targeting STING with covalent small-molecule inhibitors. Nat. Cell Biol. 2018, 559, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Georgana, I.; Sumner, R.P.; Towers, G.J.; De Motes, C.M. Virulent Poxviruses Inhibit DNA Sensing by Preventing STING Activation. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Röhl, I.; Hopfner, K.-P.; Ludwig, J.; Hornung, V. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nat. Cell Biol. 2013, 498, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Schoggins, J.W.; MacDuff, D.A.; Imanaka, N.; Gainey, M.D.; Shrestha, B.; Eitson, J.L.; Mar, K.B.; Richardson, R.B.; Ratushny, A.V.; Litvak, V.; et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 2014, 505, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Wu, J.; Wu, Y.-T.; Du, F.; Aroh, C.; Yan, N.; Sun, L.; Chen, Z. Cyclic GMP-AMP Synthase Is an Innate Immune Sensor of HIV and Other Retroviruses. Science 2013, 341, 903–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzner, A.-M.; Hagmann, C.A.; Goldeck, M.; Wolter, S.; Kübler, K.; Wittmann, S.; Gramberg, T.; Andreeva, L.; Hopfner, K.-P.; Mertens, C.; et al. Sequence-specific activation of the DNA sensor cGAS by Y-form DNA structures as found in primary HIV-1 cDNA. Nat. Immunol. 2015, 16, 1025–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre, S.; Luthra, P.; Sanchez-Aparicio, M.T.; Maestre, A.M.; Patel, J.; Lamothe, F.; Fredericks, A.C.; Tripathi, S.; Zhu, T.; Pintado-Silva, J.; et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat. Microbiol. 2017, 2, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; MacDuff, D.A.; Imanaka, N.; Gainey, M.D.; Shrestha, B.; Eitson, J.L.; Mar, K.B.; Richardson, R.B.; Ratushny, A.V.; Litvak, V.; et al. Corrigendum: Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate im-munity. Nature 2015, 525, 144. [Google Scholar] [CrossRef] [Green Version]
- Verrier, E.R.; Yim, S.A.; Heydmann, L.; El Saghire, H.; Bach, C.; Turon-Lagot, V.; Mailly, L.; Durand, S.C.; Lucifora, J.; Durantel, D.; et al. Hepatitis B Virus Evasion from Cyclic Guanosine Monophosphate-Adenosine Monophosphate Synthase Sensing in Human Hepatocytes. Hepatology 2018, 68, 1695–1709. [Google Scholar] [CrossRef] [Green Version]
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. Proc. R. Soc. London. Ser. B Biol. Sci. 1957, 147, 258–267. [Google Scholar] [CrossRef]
- Parkin, J.; Cohen, B. An overview of the immune system. Lancet 2001, 357, 1777–1789. [Google Scholar] [CrossRef]
- Thaney, V.E.; Kaul, M. Type I Interferons in NeuroHIV. Viral Immunol. 2019, 32, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Innate immunity to virus infection. Immunol. Rev. 2008, 227, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Chelbi-Alix, M.K.; Wietzerbin, J. Interferon, a growing cytokine family: 50 years of interferon research. Biochimie 2007, 89, 713–718. [Google Scholar] [CrossRef]
- Mantegazza, A.R.; Magalhaes, J.G.; Amigorena, S.; Marks, M.S. Presentation of Phagocytosed Antigens by MHC Class I and II. Traffic 2013, 14, 135–152. [Google Scholar] [CrossRef] [Green Version]
- Thomas, R.; Yang, X. NK-DC Crosstalk in Immunity to Microbial Infection. J. Immunol. Res. 2016, 2016, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dionisio, D.; Valassina, M.; Ciufolini, M.G.; Vivarelli, A.; Esperti, F.; Cusi, M.G.; Marchi, A.; Mazzoli, F.; Lupi, C. Encephalitis without Meningitis Due to Sandfly Fever Virus Serotype Toscana. Clin. Infect. Dis. 2001, 32, 1241–1243. [Google Scholar] [CrossRef] [Green Version]
- Ojeda-Juárez, D.; Shah, R.; Fields, J.A.; Harahap-Carrillo, I.S.; Koury, J.; Maung, R.; Gelman, B.B.; Baaten, B.J.; Roberts, A.J.; Kaul, M. Lipocalin-2 mediates HIV-1 induced neuronal injury and behavioral deficits by overriding CCR5-dependent protection. Brain Behav. Immun. 2020, 89, 184–199. [Google Scholar] [CrossRef]
- Kincaid, O.; Lipton, H.L. Viral myelitis: An update. Curr. Neurol. Neurosci. Rep. 2006, 6, 469–474. [Google Scholar] [CrossRef]
- Koyuncu, O.O.; Hogue, I.B.; Enquist, L.W. Virus Infections in the Nervous System. Cell Host Microbe 2013, 13, 379–393. [Google Scholar] [CrossRef] [Green Version]
- Spindler, K.R.; Hsu, T.-H. Viral disruption of the blood–brain barrier. Trends Microbiol. 2012, 20, 282–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludlow, M.; Kortekaas, J.; Herden, C.; Hoffmann, B.; Tappe, D.; Trebst, C.; Griffin, D.E.; Brindle, H.E.H.; Solomon, T.; Brown, A.S.A.; et al. Neurotropic virus infections as the cause of immediate and delayed neuropathology. Acta Neuropathol. 2016, 131, 159–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muoio, V.; Persson, P.B.; Sendeski, M.M. The neurovascular unit-concept review. Acta Physiol. 2014, 210, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Welte, T.; McGargill, M.; Town, T.; Thompson, J.; Anderson, J.F.; Flavell, R.A.; Fikrig, E.; Hedrick, S.M.; Wang, T. Drak2 Contributes to West Nile Virus Entry into the Brain and Lethal Encephalitis. J. Immunol. 2008, 181, 2084–2091. [Google Scholar] [CrossRef] [Green Version]
- Kaul, M.; Garden, G.A.; Lipton, S.A. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nat. Cell Biol. 2001, 410, 988–994. [Google Scholar] [CrossRef]
- Karim, Q.A. Current status of the HIV epidemic & challenges in prevention. Indian J. Med. Res. 2017, 146, 673–676. [Google Scholar] [CrossRef]
- Sacktor, N.; Skolasky, R.L.; Seaberg, E.; Munro, C.; Becker, J.T.; Martin, E.; Ragin, A.; Levine, A.; Miller, E. Prevalence of HIV-associated neurocognitive disorders in the Multicenter AIDS Cohort Study. Neurology 2016, 86, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Lutgen, V.; Narasipura, S.D.; Barbian, H.J.; Richards, M.; Wallace, J.; Razmpour, R.; Buzhdygan, T.; Ramirez, S.H.; Prevedel, L.; Eugenin, E.A.; et al. HIV infects astrocytes in vivo and egresses from the brain to the periphery. PLoS Pathog. 2020, 16, e1008381. [Google Scholar] [CrossRef]
- Aleyas, A.G.; George, J.A.; Han, Y.W.; Rahman, M.; Kim, S.J.; Han, S.B.; Kim, B.S.; Kim, K.; Eo, S.K. Functional Modulation of Dendritic Cells and Macrophages by Japanese Encephalitis Virus through MyD88 Adaptor Molecule-Dependent and -Independent Pathways. J. Immunol. 2009, 183, 2462–2474. [Google Scholar] [CrossRef] [Green Version]
- Dutta, K.; Kumawat, K.L.; Nazmi, A.; Mishra, M.K.; Basu, A. Minocycline Differentially Modulates Viral Infection and Persistence in an Experimental Model of Japanese Encephalitis. J. Neuroimmune Pharmacol. 2010, 5, 553–565. [Google Scholar] [CrossRef]
- Yang, K.D.; Yeh, W.T.; Chen, R.F.; Chuon, H.L.; Tsai, H.P.; Yao, C.W.; Shaio, M.F. A model to study neurotropism and persistency of Japanese encephalitis virus infection in human neuro-blastoma cells and leukocytes. J. Gen. Virol. 2004, 85 Pt 3, 635–642. [Google Scholar] [CrossRef]
- Eugenin, E.A.; Osiecki, K.; Lopez, L.; Goldstein, H.; Calderon, T.M.; Berman, J.W. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodefi-ciency virus (HIV)-infected leukocytes across the blood-brain barrier: A potential mechanism of HIV-CNS invasion and NeuroAIDS. J. Neurosci. 2006, 26, 1098–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, S.; Lo, Y.; Chapagain, M.; Lum, S.; Kumar, M.; Gurjav, U.; Luo, H.; Nakatsuka, A.; Nerurkar, V.R. West Nile virus infection modulates human brain microvascular endothelial cells tight junction proteins and cell adhesion molecules: Transmigration across the in vitro blood-brain barrier. Virology 2009, 385, 425–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slavuljica, I.; Kveštak, D.; Huszthy, P.C.; Kosmac, K.; Britt, W.J.; Jonjić, S. Immunobiology of congenital cytomegalovirus infection of the central nervous system—The murine cytomegalovirus model. Cell. Mol. Immunol. 2014, 12, 180–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lokensgard, J.R.; Cheeran, M.C.; Gekker, G.; Hu, S.; Chao, C.C.; Peterson, P.K. Human cytomegalovirus replication and modulation of apoptosis in astrocytes. J. Hum. Virol. 1999, 2, 91–101. [Google Scholar]
- Solomon, T.; Lewthwaite, P.; Perera, D.; Cardosa, M.J.; McMINN, P.; Ooi, M.-H. Virology, epidemiology, pathogenesis, and control of enterovirus. Lancet Infect Dis. 2010, 10, 778–790. [Google Scholar] [CrossRef]
- Calderón-Peláez, M.-A.; Velandia-Romero, M.L.; Bastidas-Legarda, L.Y.; Beltrán, E.O.; Camacho-Ortega, S.J.; Castellanos, J.E. Dengue Virus Infection of Blood–Brain Barrier Cells: Consequences of Severe Disease. Front. Microbiol. 2019, 10, 1435. [Google Scholar] [CrossRef] [Green Version]
- Boothpur, R.; Brennan, D.C. Human polyoma viruses and disease with emphasis on clinical BK and JC. J. Clin. Virol. 2010, 47, 306–312. [Google Scholar] [CrossRef] [Green Version]
- Argyris, E.G.; Acheampong, E.; Nunnari, G.; Mukhtar, M.; Williams, K.J.; Pomerantz, R.J. Human Immunodeficiency Virus Type 1 Enters Primary Human Brain Microvascular Endothelial Cells by a Mechanism Involving Cell Surface Proteoglycans Independent of Lipid Rafts. J. Virol. 2003, 77, 12140–12151. [Google Scholar] [CrossRef] [Green Version]
- Liou, M.-L.; Hsu, C.Y. Japanese encephalitis virus is transported across the cerebral blood vessels by endocytosis in mouse brain. Cell Tissue Res. 1998, 293, 389–394. [Google Scholar] [CrossRef]
- Chien, Y.-J.; Chen, W.; Hsu, W.-L.; Chiou, S.-S. Bovine lactoferrin inhibits Japanese encephalitis virus by binding to heparan sulfate and receptor for low density lipoprotein. Virology 2008, 379, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Bayer, A.; Lennemann, N.J.; Ouyang, Y.; Bramley, J.C.; Morosky, S.; Marques, E.T.D.A., Jr.; Cherry, S.; Sadovsky, Y.; Coyne, C.B. Type III Interferons Produced by Human Placental Trophoblasts Confer Protection against Zika Virus In-fection. Cell Host Microbe 2016, 19, 705–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mladinich, M.C.; Schwedes, J.; Mackow, E.R. Zika Virus Persistently Infects and Is Basolaterally Released from Primary Human Brain Microvascular Endothelial Cells. mBio 2017, 8, e00952-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papa, M.P.; Meuren, L.M.; Coelho, S.V.A.; Lucas, C.G.D.O.; Mustafá, Y.M.; Matassoli, F.L.; Silveira, P.P.; Frost, P.S.; Pezzuto, P.; Ribeiro, M.R.; et al. Zika Virus Infects, Activates, and Crosses Brain Microvascular Endothelial Cells, without Barrier Disruption. Front. Microbiol. 2017, 8, 2557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afonso, P.V.; Ozden, S.; Cumont, M.-C.; Seilhean, D.; Cartier, L.; Rezaie, P.; Mason, S.; Lambert, S.; Huerre, M.; Gessain, A.; et al. Alteration of Blood–Brain Barrier Integrity by Retroviral Infection. PLoS Pathog. 2008, 4, e1000205. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Dai, J.; Bai, F.; Kong, K.-F.; Wong, S.J.; Montgomery, R.R.; Madri, J.A.; Fikrig, E. Matrix Metalloproteinase 9 Facilitates West Nile Virus Entry into the Brain. J. Virol. 2008, 82, 8978–8985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniels, B.P.; Holman, D.W.; Cruz-Orengo, L.; Jujjavarapu, H.; Durrant, D.M.; Klein, R.S. Viral Pathogen-Associated Molecular Patterns Regulate Blood-Brain Barrier Integrity via Competing Innate Cytokine Signals. mBio 2014, 5, e01476-14. [Google Scholar] [CrossRef] [Green Version]
- Mori, I.; Nishiyama, Y.; Yokochi, T.; Kimura, Y. Olfactory transmission of neurotropic viruses. J. Neurovirology 2005, 11, 129–137. [Google Scholar] [CrossRef]
- Kupke, A.; Becker, S.; Wewetzer, K.; Ahlemeyer, B.; Eickmann, M.; Herden, C. Intranasal Borna Disease Virus (BoDV-1) Infection: Insights into Initial Steps and Potential Contagiosity. Int. J. Mol. Sci. 2019, 20, 1318. [Google Scholar] [CrossRef] [Green Version]
- Van Riel, D.; Leijten, L.M.; Verdijk, R.M.; GeurtsvanKessel, C.; van der Vries, E.; van Rossum, A.M.; Osterhaus, A.D.; Kuiken, T. Evidence for Influenza Virus CNS Invasion Along the Olfactory Route in an Immunocompromised Infant. J. Infect. Dis. 2014, 210, 419–423. [Google Scholar] [CrossRef]
- Plakhov, I.V.; Arlund, E.E.; Aoki, C.; Reiss, C.S. The Earliest Events in Vesicular Stomatitis Virus Infection of the Murine Olfactory Neuroepithelium and Entry of the Central Nervous System. Virology 1995, 209, 257–262. [Google Scholar] [CrossRef]
- Mao, L.; Jin, H.; Wang, M.; Hu, Y.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D.; et al. Neurologic Manifestations of Hospitalized Patients with Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol. 2020, 77, 683. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.H.; Jiang, D.; Huang, J.T. SARS-CoV-2 Detected in Cerebrospinal Fluid by PCR in a Case of COVID-19 En-cephalitis. Brain Behav. Immun. 2020, 87, 149. [Google Scholar] [CrossRef] [PubMed]
- Baig, A.M. Neurological manifestations in COVID-19 caused by SARS-CoV-CNS. Neurosci. Ther. 2020, 26, 499–501. [Google Scholar] [CrossRef] [Green Version]
- Bullen, C.K.; Hogberg, H.T.; Bahadirli-Talbott, A.; Bishai, W.R.; Hartung, T.; Keuthan, C.; Looney, M.M.; Pekosz, A.; Romero, J.C.; Sillé, F.C.; et al. Infectability of human BrainSphere neurons suggests neurotropism of SARS-CoV-2. ALTEX 2020, 37, 665–671. [Google Scholar] [PubMed]
- Davies, J.; Randeva, H.S.; Chatha, K.; Hall, M.; Spandidos, D.A.; Karteris, E.; Kyrou, I. Neuropilin-1 as a new potential SARS-CoV-2 infection mediator implicated in the neurologic features and central nervous system involvement of COVID-19. Mol. Med. Rep. 2020, 22, 4221–4226. [Google Scholar] [CrossRef] [PubMed]
- Sungnak, W.; Huang, N.; Bécavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousaf, M.Z.; Qasim, M.; Zia, S.; Khan, M.U.R.; Ashfaq, U.A.; Khan, S. Rabies molecular virology, diagnosis, prevention and treatment. Virol. J. 2012, 9, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafon, M. Rabies virus receptors. J. NeuroVirol. 2005, 11, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Gluska, S.; Zahavi, E.E.; Chein, M.; Gradus, T.; Bauer, A.; Finke, S.; Perlson, E. Rabies Virus Hijacks and Accelerates the p75NTR Retrograde Axonal Transport Machinery. PLoS Pathog. 2014, 10, e1004348. [Google Scholar] [CrossRef] [Green Version]
- Ugolini, G. Rabies Virus as a Transneuronal Tracer of Neuronal Connections. Adv. Virus Res. 2011, 79, 165–202. [Google Scholar] [CrossRef] [PubMed]
- Mehndiratta, M.M.; Mehndiratta, P.; Pande, R. Poliomyelitis: Historical facts, epidemiology, and current challenges in eradication. Neurohospitalist 2014, 4, 223–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racaniello, V. One hundred years of poliovirus pathogenesis. Virology 2006, 344, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, R.; Racaniello, V.R. Human poliovirus receptor gene expression and poliovirus tissue tropism in transgenic mice. J. Virol. 1992, 66, 296–304. [Google Scholar] [CrossRef] [Green Version]
- Ohka, S.; Nihei, C.-I.; Yamazaki, M.; Nomoto, A. Poliovirus trafficking toward central nervous system via human poliovirus receptor-dependent and -independent pathway. Front. Microbiol. 2012, 3, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, M.A.; Wang, H.; Siddharthan, V.; Morrey, J.D.; Diamond, M.S. Axonal transport mediates West Nile virus entry into the central nervous system and induces acute flaccid paralysis. Proc. Natl. Acad. Sci. USA 2007, 104, 17140–17145. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.T.; Munisamy, B.; Ong, K.C.; Kojima, H.; Noriyo, N.; Chua, K.B.; Ong, B.B.; Nagashima, K. The Distribution of Inflammation and Virus in Human Enterovirus 71 Encephalomyelitis Suggests Possible Viral Spread by Neural Pathways. J. Neuropathol. Exp. Neurol. 2008, 67, 162–169. [Google Scholar] [CrossRef] [Green Version]
- Diefenbach, R.J.; Miranda-Saksena, M.; Douglas, M.W.; Cunningham, A.L. Transport and egress of herpes simplex virus in neurons. Rev. Med. Virol. 2007, 18, 35–51. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Brown, M.A. Innate immunity in the central nervous system. J. Clin. Investig. 2012, 122, 1164–1171. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Perdiguero, E.G.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; De Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nat. Cell Biol. 2015, 518, 547–551. [Google Scholar] [CrossRef]
- Illes, P.; Ribeiro, J.A. Molecular physiology of P2 receptors in the central nervous system. Eur. J. Pharmacol. 2004, 483, 5–17. [Google Scholar] [CrossRef]
- Fekete, R.; Cserép, C.; Lénárt, N.; Tóth, K.; Orsolits, B.; Martinecz, B.; Méhes, E.; Szabó, B.; Németh, V.; Gönci, B.; et al. Microglia control the spread of neurotropic virus infection via P2Y12 signalling and recruit monocytes through P2Y12-independent mechanisms. Acta Neuropathol. 2018, 136, 461–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Detje, C.N.; Meyer, T.; Schmidt, H.; Kreuz, D.; Rose, J.K.; Bechmann, I.; Prinz, M.; Kalinke, U. Local Type I IFN Receptor Signaling Protects against Virus Spread within the Central Nervous System. J. Immunol. 2009, 182, 2297–2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Detje, C.N.; Lienenklaus, S.; Chhatbar, C.; Spanier, J.; Prajeeth, C.K.; Soldner, C.; Tovey, M.G.; Schlüter, D.; Weiss, S.; Stangel, M.; et al. Upon Intranasal Vesicular Stomatitis Virus Infection, Astrocytes in the Olfactory Bulb Are Important Interferon Beta Producers That Protect from Lethal Encephalitis. J. Virol. 2014, 89, 2731–2738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drokhlyansky, E.; Aytürk, D.G.; Soh, T.K.; Chrenek, R.; O’Loughlin, E.; Madore, C.; Butovsky, O.; Cepko, C.L. The brain parenchyma has a type I interferon response that can limit virus spread. Proc. Natl. Acad. Sci. USA 2017, 114, E95–E104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuertz, K.M.; Treuting, P.M.; Hemann, E.A.; Esser-Nobis, K.; Snyder, A.G.; Graham, J.B.; Daniels, B.P.; Wilkins, C.; Snyder, J.M.; Voss, K.M.; et al. STING is required for host defense against neuropathological West Nile virus infection. PLoS Pathog. 2019, 15, e1007899. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Proll, S.C.; Szretter, K.J.; Katze, M.G.; Gale, M.; Diamond, M.S. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat. Med. 2013, 19, 458–464. [Google Scholar] [CrossRef]
- Durrant, D.M.; Daniels, B.P.; Klein, R.S. IL-1R1 signaling regulates CXCL12-mediated T cell localization and fate within the central nervous system during West Nile Virus encephalitis. J. Immunol. 2014, 193, 4095–4106. [Google Scholar] [CrossRef] [Green Version]
- Suthar, M.S.; Diamond, M.S.; Gale, M., Jr. West Nile virus infection and immunity. Nat. Rev. Genet. 2013, 11, 115–128. [Google Scholar] [CrossRef]
- Chopy, D.; Pothlichet, J.; Lafage, M.; Mégret, F.; Fiette, L.; Si-Tahar, M.; Lafon, M. Ambivalent Role of the Innate Immune Response in Rabies Virus Pathogenesis. J. Virol. 2011, 85, 6657–6668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Hammack, C.; Ogden, S.C.; Wen, Z.; Qian, X.; Li, Y.; Yao, B.; Shin, J.; Zhang, F.; Lee, E.M.; et al. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell Stem Cell 2016, 18, 587–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, J.; Tiwari, S.K.; Lichinchi, G.; Qin, Y.; Patil, V.S.; Eroshkin, A.M.; Rana, T.M. Zika Virus Depletes Neural Progenitors in Human Cerebral Organoids through Activation of the Innate Immune Receptor TLR3. Cell Stem Cell 2016, 19, 258–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serman, T.M.; Gack, M.U. Evasion of Innate and Intrinsic Antiviral Pathways by the Zika Virus. Viruses 2019, 11, 970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, J.; Issacson, R.S.; Koppel, B.S. Subacute sclerosing panencephalitis: An update. Dev. Med. Child Neurol. 2010, 52, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, R.; Bonthius, D.J. Measles Virus and Associated Central Nervous System Sequelae. Semin. Pediatr. Neurol. 2012, 19, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Patterson, C.E.; Daley, J.K.; Echols, L.A.; Lane, T.E.; Rall, G.F. Measles Virus Infection Induces Chemokine Synthesis by Neurons. J. Immunol. 2003, 171, 3102–3109. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Yan, C.; Gieling, R.G.; Kida, Y.; Garner, W.L.; Li, W.; Han, Y.-P. Tumor necrosis factor-alpha induced expression of matrix metalloproteinase-9 through p21-activated Kinase-1. BMC Immunol. 2009, 10, 15. [Google Scholar] [CrossRef] [Green Version]
- Daniels, B.P.; Jujjavarapu, H.; Durrant, D.M.; Williams, J.L.; Green, R.R.; White, J.P.; LaZear, H.M.; Gale, M.; Diamond, M.S.; Klein, R.S. Regional astrocyte IFN signaling restricts pathogenesis during neurotropic viral infection. J. Clin. Investig. 2017, 127, 843–856. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.; Luo, H.; Shan, C.; Muruato, A.E.; Nunes, B.T.D.; Medeiros, D.B.A.; Zou, J.; Xie, X.; Giraldo, M.I.; Vasconcelos, P.F.C.; et al. An evolutionary NS1 mutation enhances Zika virus evasion of host interferon induction. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Fredericksen, B.L.; Gale, M. West Nile Virus Evades Activation of Interferon Regulatory Factor 3 through RIG-I-Dependent and -Independent Pathways without Antagonizing Host Defense Signaling. J. Virol. 2006, 80, 2913–2923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelemans, T.; Kikkert, M. Viral Innate Immune Evasion and the Pathogenesis of Emerging RNA Virus Infections. Viruses 2019, 11, 961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beachboard, D.C.; Horner, S.M. Innate immune evasion strategies of DNA and RNA viruses. Curr. Opin. Microbiol. 2016, 32, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Waheed, A.A.; Zhang, Z.-R.; Freed, E.O.; Bonifacino, J.S. HIV-1 Vpu Accessory Protein Induces Caspase-mediated Cleavage of IRF3 Transcription Factor. J. Biol. Chem. 2014, 289, 35102–35110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirois, M.; Robitaille, L.; Allary, R.; Shah, M.; Woelk, C.H.; Estaquier, J.; Corbeil, J. TRAF6 and IRF7 Control HIV Replication in Macrophages. PLoS ONE 2011, 6, e28125. [Google Scholar] [CrossRef] [PubMed]
- Kitai, R.; Zhao, M.L.; Zhang, N.; Hua, L.L.; Lee, S.C. Role of MIP-1beta and RANTES in HIV-1 infection of microglia: Inhibition of infection and induction by IFNβ. J. Neuroimmunol. 2000, 110, 230–239. [Google Scholar] [CrossRef]
- Chan, Y.K.; Gack, M.U. Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Genet. 2016, 14, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Suh, H.-S.; Zhao, M.-L.; Choi, N.; Belbin, T.J.; Brosnan, C.F.; Lee, S.C. TLR3 and TLR4 are innate antiviral immune receptors in human microglia: Role of IRF3 in modulating antiviral and inflammatory response in the CNS. Virology 2009, 392, 246–259. [Google Scholar] [CrossRef] [Green Version]
- Kelesidis, T.; Mastoris, I.; Metsini, A.; Tsiodras, S. How to approach and treat viral infections in ICU patients. BMC Infect. Dis. 2014, 14, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Saylor, D.; Dickens, A.M.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-associated neurocognitive disorder—Pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016, 12, 234–248. [Google Scholar] [CrossRef]
- Anderson, A.M.; Muñoz-Moreno, J.A.; McClernon, D.R.; Ellis, R.J.; Cookson, D.; Clifford, D.B.; Collier, A.C.; Gelman, B.B.; Marra, C.M.; McArthur, J.C.; et al. Prevalence and Correlates of Persistent HIV-1 RNA in Cerebrospinal Fluid During Antiretroviral Therapy. J. Infect. Dis. 2017, 215, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.; Tyler, K.L. Novel approaches and challenges to treatment of central nervous system viral infections. Ann. Neurol. 2013, 74, 412–422. [Google Scholar] [CrossRef] [PubMed]
- The IFNB Multiple Sclerosis Study Group. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. Neurology 1993, 43, 655. [Google Scholar] [CrossRef]
- Stone, L.A.; Frank, J.A.; Albert, P.S.; Bash, C.; Smith, M.E.; Maloni, H.; McFarland, H.F. The effect of interferon-beta on blood-brain barrier disruptions demonstrated by contrast-enhanced magnetic resonance imaging in relapsing-remitting multiple sclerosis. Ann. Neurol. 1995, 37, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Kuruganti, P.A.; Hinojoza, J.R.; Eaton, M.J.; Ehmann, U.K.; Sobel, R.A. Interferon-beta counteracts inflammatory mediator-induced effects on brain endothelial cell tight junction molecules-implications for multiple sclerosis. J. Neuropathol. Exp. Neurol. 2002, 61, 710–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madsen, C. The innovative development in interferon beta treatments of relapsing-remitting multiple sclerosis. Brain Behav. 2017, 7, e00696. [Google Scholar] [CrossRef] [Green Version]
- Thaney, V.E.; O’Neill, A.M.; Hoefer, M.M.; Maung, R.; Sanchez, A.B.; Kaul, M. IFNβ Protects Neurons from Damage in a Murine Model of HIV-1 Associated Brain Injury. Sci. Rep. 2017, 7, srep46514. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-O.; Si, Q.; Zhou, J.N.; Pestell, R.G.; Brosnan, C.F.; Locker, J.; Lee, S.C. Interferon-β activates multiple signaling cascades in primary human microglia. J. Neurochem. 2002, 81, 1361–1371. [Google Scholar] [CrossRef]
- Kaul, M.; Ma, Q.; E Medders, K.; Desai, M.K.; Lipton, S.A. HIV-1 coreceptors CCR5 and CXCR4 both mediate neuronal cell death but CCR5 paradoxically can also contribute to protection. Cell Death Differ. 2006, 14, 296–305. [Google Scholar] [CrossRef] [Green Version]
- Wintergerst, U.; Gangemi, J.D.; Whitley, R.J.; Chatterjee, S.; Kern, E.R. Effect of recombinant human interferon α B/D (rHu-IFN-α B/D) in combination with acyclovir in experimental HSV-1 encephalitis. Antivir. Res. 1999, 44, 75–78. [Google Scholar] [CrossRef]
- Boivin, N.; Sergerie, Y.; Rivest, S.; Boivin, G. Effect of Pretreatment with Toll-like Receptor Agonists in a Mouse Model of Herpes Simplex Virus Type 1 Encephalitis. J. Infect. Dis. 2008, 198, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Boivin, N.; Menasria, R.; Piret, J.; Boivin, G. Modulation of TLR9 response in a mouse model of herpes simplex virus encephalitis. Antivir. Res. 2012, 96, 414–421. [Google Scholar] [CrossRef] [PubMed]







| PRRs/Receptor | Localization | Adapter | Viruses PAMPS | Viruses | Reference |
|---|---|---|---|---|---|
| TLR2 | Cell membrane | MyD88 | Envelope glycoprotein, core protein | Cytomegalovirus | [14] |
| Hepatitis C virus | [13] | ||||
| Hepatitis B virus | [15] | ||||
| Herpes simplex virus | [16] | ||||
| Measles virus | [71] | ||||
| Varicella-zoster virus | [72] | ||||
| Epstein-Barr virus | [73] | ||||
| Vaccinia virus | [74] | ||||
| Junin virus | [18] | ||||
| TLR3 | Endosome | TRIF | dsRNA, RNA, siRNAs, self RNA, polyI:C | Respiratory syncytial virus | [33] |
| Rhinovirus | [35] | ||||
| Reovirus | [31] | ||||
| Herpes simplex virus-2 | [36] | ||||
| Influenza A virus | [40] | ||||
| Enterovirus A71 | [41] | ||||
| Friend retrovirus | [42] | ||||
| West Nile virus | [45] | ||||
| Murine coronavirus | [44] | ||||
| Human immunodeficiency virus | [39] | ||||
| Epstein-Barr virus | [32] | ||||
| Hepatitis C virus | [43] | ||||
| TLR4 | Cell membrane | MyD88 and TRIF | Glycoprotein, envelop protein, fusion protein, | Vesicular stomatitis virus | [19] |
| Ebola virus | [20] | ||||
| Mouse mammary tumor virus | [21] | ||||
| Moloney murine leukemia virus | [21] | ||||
| Coxsackie B virus | [75] | ||||
| Respiratory syncytial virus | [76] | ||||
| Human immunodeficiency virus | [22] | ||||
| TLR7/8 | Endosome | MyD88 | ssRNA | Vesicular stomatitis virus | [52] |
| Human immunodeficiency virus | [51] | ||||
| Coxsackie B virus | [49] | ||||
| Hepatitis C virus | [53] | ||||
| Sendai virus | [50] | ||||
| Influenza virus | [52] | ||||
| Rabies virus | [54] | ||||
| Friend murine leukemia virus | [55] | ||||
| TLR9 | Endosome | MyD88 | Viral DNA | Herpes simplex virus 1 | [64] |
| Herpes simplex virus 2 | [63] | ||||
| Varicella-zoster virus | [62] | ||||
| Cytomegalovirus | [59] | ||||
| Epstein-Barr virus | [61] | ||||
| Human immunodeficiency virus | [65] | ||||
| Hepatitis B virus | [66] | ||||
| Enterovirus 71 | [67] | ||||
| TLR10 | Cell membrane | Unknown | Envelope protein, RNA | Influenza A virus | [68] |
| Human immunodeficiency virus | [77] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, H.; Koury, J.; Kaul, M. Innate Immune Sensing of Viruses and Its Consequences for the Central Nervous System. Viruses 2021, 13, 170. https://doi.org/10.3390/v13020170
Singh H, Koury J, Kaul M. Innate Immune Sensing of Viruses and Its Consequences for the Central Nervous System. Viruses. 2021; 13(2):170. https://doi.org/10.3390/v13020170
Chicago/Turabian StyleSingh, Hina, Jeffrey Koury, and Marcus Kaul. 2021. "Innate Immune Sensing of Viruses and Its Consequences for the Central Nervous System" Viruses 13, no. 2: 170. https://doi.org/10.3390/v13020170
APA StyleSingh, H., Koury, J., & Kaul, M. (2021). Innate Immune Sensing of Viruses and Its Consequences for the Central Nervous System. Viruses, 13(2), 170. https://doi.org/10.3390/v13020170