
Virus-Encoded Complement Regulators: Current Status

Abstract
:1. Introduction
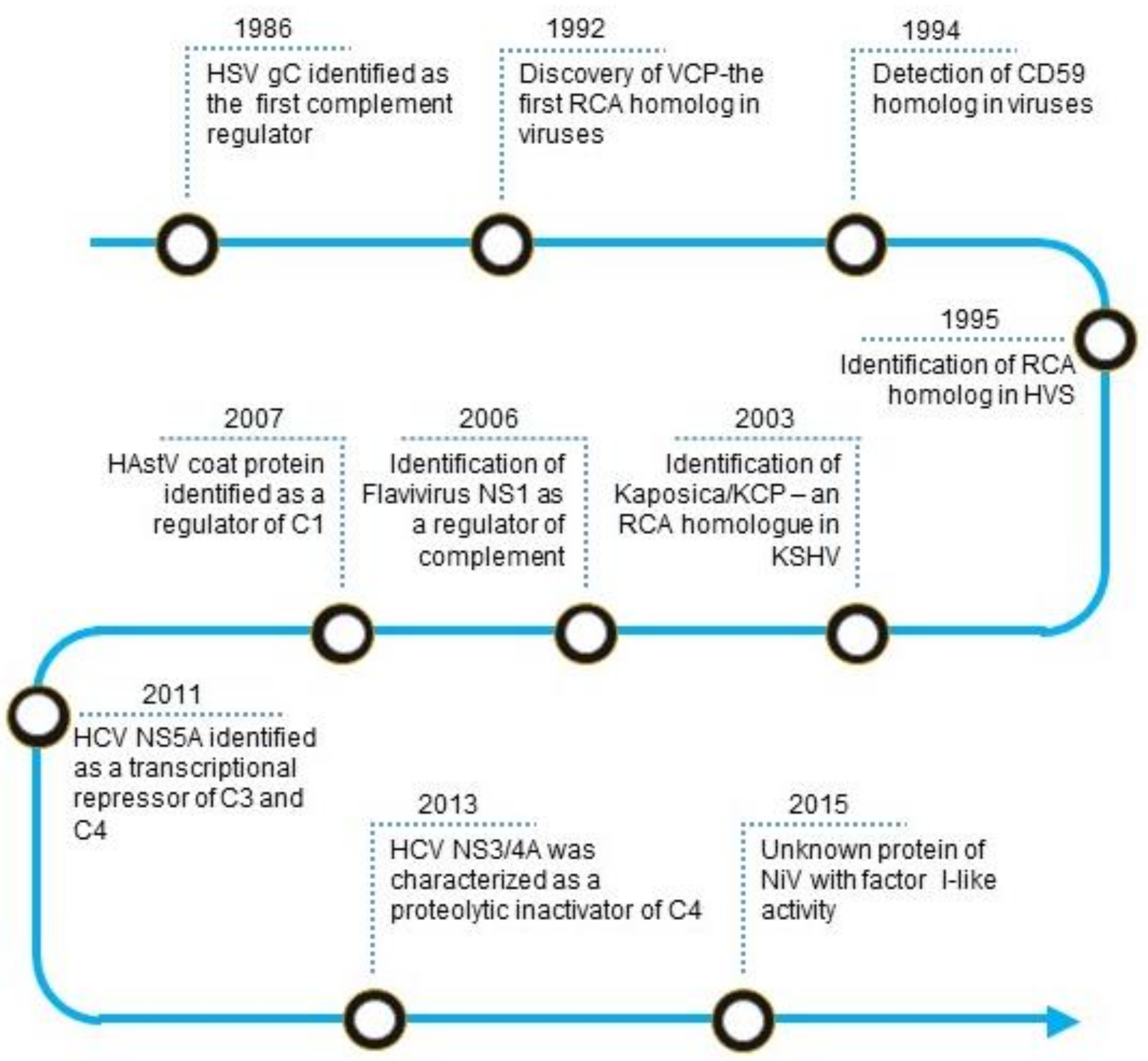
2. Historical Perspective
3. Viral Complement Regulators
3.1. Structural Proteins
3.1.1. Herpes Simplex Virus Glycoprotein C (gC)
3.1.2. Astrovirus Capsid Protein (CaPt)
3.1.3. Influenza Virus Matrix Protein M1
3.1.4. Nipah and Chikungunya Virus Proteins with Factor I-Like Activity
3.1.5. Hepatitis C Virus Core Protein (HCV-CP)
3.1.6. Zika Virus E Protein
3.2. Non-Structural Proteins
3.2.1. Hepatitis C Virus NS3/4A Protease Complex and NS5A Protein
3.2.2. Hepatitis B Virus HBx Protein
3.2.3. Flavivirus Non-Structural Protein 1 (NS1)
3.2.4. Vaccinia Virus Complement Control Protein (VCP) and Smallpox Inhibitor of Complement Enzyme (SPICE)
3.2.5. Cowpox Virus Inflammation Modulatory Protein (IMP)
3.2.6. Monkeypox Inhibitor of Complement Enzyme (MOPICE)
3.2.7. Ectromelia Virus Inhibitor of Complement Enzymes (EMICE)
3.2.8. Herpesvirus Saimiri Virus Complement Control Protein Homolog (HVS CCPH)
3.2.9. Kaposi’s Sarcoma-Associated Herpesvirus Inhibitor of Complement Activation (Kaposica)
3.2.10. Rhesus Rhadinovirus Complement Control Protein (RCP)
3.2.11. Murine Gammaherpesvirus 68 (γHV68) RCA Protein
3.2.12. Herpesvirus Saimiri CD59
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wasik, B.R.; Turner, P.E. On the Biological Success of Viruses. Annu. Rev Microbiol. 2013, 67, 519–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doherty, M.; Robertson, M.J. Some Early Trends in Immunology. Trends Immunol. 2004, 25, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, P. Complement before Molecular Biology. Mol. Immunol. 2006, 43, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Bordet, J.; Gengou, O. Sur l’Existences de Substances Sensibilisatrices dans la Plupart des Serums Anti-Microbiens. Ann. I'Institut Pasteur 1901, 15, 289–302. [Google Scholar]
- Ember, J.A.; Jagels, M.A.; Hugli, T.E. Characterization of Complement Anaphylatoxins and Their Biological Responses. In The Human Complement System in Health and Disease; Volanakis, J.E., Frank, M.M., Eds.; Marcel Dekker, Inc.: New York, NY, USA, 1998; Chapter 11. [Google Scholar]
- Hopken, U.E.; Lu, B.; Gerard, N.P.; Gerard, C. The C5a Chemoattractant Receptor Mediates Mucosal Defence to Infection. Nature 1996, 383, 86–89. [Google Scholar] [CrossRef]
- Fearon, D.T.; Carroll, M.C. Regulation of B Lymphocyte Responses to Foreign and Self-Antigens by the CD19/CD21 Complex. Annu. Rev Immunol. 2000, 18, 393–422. [Google Scholar] [CrossRef]
- West, E.E.; Kolev, M.; Kemper, C. Complement and the Regulation of T Cell Responses. Annu. Rev Immunol. 2018, 36, 309–338. [Google Scholar] [CrossRef]
- Tam, J.C.; Bidgood, S.R.; McEwan, W.A.; James, L.C. Intracellular Sensing of Complement C3 Activates Cell Autonomous Immunity. Science 2014, 345, 1256070. [Google Scholar] [CrossRef] [Green Version]
- Paterson, S.; Vogwill, T.; Buckling, A.; Benmayor, R.; Spiers, A.J.; Thomson, N.R.; Quail, M.; Smith, F.; Walker, D.; Libberton, B.; et al. Antagonistic Coevolution Accelerates Molecular Evolution. Nature 2010, 464, 275–278. [Google Scholar] [CrossRef] [Green Version]
- Cines, D.B.; Lyss, A.P.; Bina, M. Fc and C3 Receptors Induced by Herpes Simplex Virus on Cultured Human Endothelial Cells. J. Clin. Investig. 1982, 69, 123–128. [Google Scholar] [CrossRef] [Green Version]
- Friedman, H.M.; Glorioso, J.C.; Cohen, G.H.; Hasting, J.C.; Harris, S.L.; Eisenberg, R.J. Binding of Complement Component C3b to Glycoprotein GC of Herpes Simplex Virus Type 1: Mapping of GC-Binding Sites and Demonstration of Conserved C3b Binding in Low-Passage Clinical Isolates. J. Virol. 1986, 60, 470–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fries, L.F.; Friedman, H.M.; Cohen, G.H.; Eisenberg, R.J.; Hammer, C.H.; Frank, M.M. Glycoprotein C of Herpes Simplex Virus 1 Is an Inhibitor of the Complement Cascade. J. Immunol. 1986, 137, 1636–1641. [Google Scholar] [PubMed]
- McNearney, T.A.; Odell, C.; Holers, V.M.; Spear, P.G.; Atkinson, J.P. Herpes Simplex Virus Glycoproteins GC-1 and GC-2 Bind to the Third Component of Complement and Provide Protection Against Complement-Mediated Neutralization of Viral Infectivity. J. Exp. Med. 1987, 166, 1525–1535. [Google Scholar] [CrossRef] [PubMed]
- Panwar, H.S.; Ojha, H.; Ghosh, P.; Barage, S.H.; Raut, S.; Sahu, A. Molecular Engineering of an Efficient Four-Domain DAF-MCP Chimera Reveals the Presence of Functional Modularity in RCA Proteins. Proc. Natl. Acad. Sci. USA 2019, 116, 9953–9958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotwal, G.J.; Moss, B. Vaccinia Virus Encodes a Secretory Polypeptide Structurally Related to Complement Control Proteins. Nature 1988, 335, 176–178. [Google Scholar] [CrossRef]
- Kotwal, G.J.; Isaacs, S.N.; Mckenzie, R.; Frank, M.M.; Moss, B. Inhibition of the Complement Cascade by the Major Secretory Protein of Vaccinia Virus. Science 1990, 250, 827–830. [Google Scholar] [CrossRef]
- Moore, P.S.; Chang, Y. Kaposi’s Sarcoma-Associated Herpesvirus Immunoevasion and Tumorigenesis: Two Sides of the Same Coin? Annu. Rev. Microbiol. 2003, 57, 609–639. [Google Scholar] [CrossRef] [Green Version]
- Mullick, J.; Bernet, J.; Singh, A.K.; Lambris, J.D.; Sahu, A. Kaposi’s Sarcoma-Associated Herpesvirus (Human Herpesvirus-8) Open Reading Frame 4 Protein (Kaposica) Is a Functional Homolog of Complement Control Proteins. J. Virol. 2003, 77, 3878–3881. [Google Scholar] [CrossRef] [Green Version]
- Spiller, O.B.; Blackbourn, D.J.; Mark, L.; Proctor, D.G.; Blom, A.M. Functional Activity of the Complement Regulator Encoded by Kaposi’s Sarcoma-Associated Herpesvirus. J. Biol. Chem. 2003, 278, 9283–9289. [Google Scholar] [CrossRef] [Green Version]
- Ojha, H.; Ghosh, P.; Singh, P.H.; Shende, R.; Gondane, A.; Mande, S.C.; Sahu, A. Spatially Conserved Motifs in Complement Control Protein Domains Determine Functionality in Regulators of Complement Activation-Family Proteins. Commun. Biol. 2019, 2, 290. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, J.C.; Fleckenstein, B. New Member of the Multigene Family of Complement Control Proteins in Herpesvirus Saimiri. J. Virol. 1992, 66, 3937–3940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rother, R.P.; Rollins, S.A.; Fodor, W.L.; Albrecht, J.C.; Setter, E.; Fleckenstein, B.; Squinto, S.P. Inhibition of Complement-Mediated Cytolysis by the Terminal Complement Inhibitor of Herpesvirus Saimiri. J. Virol. 1994, 68, 730–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bramley, J.C.; Davies, A.; Lachmann, P.J. Herpesvirus Saimiri CD59—Baculovirus Expression and Characterisation of Complement Inhibitory Activity. Biochem. Soc. Trans. 1997, 25, 354S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avirutnan, P.; Hauhart, R.E.; Somnuke, P.; Blom, A.M.; Diamond, M.S.; Atkinson, J.P. Binding of Flavivirus Nonstructural Protein NS1 to C4b Binding Protein Modulates Complement Activation. J. Immunol. 2011, 187, 424–433. [Google Scholar]
- Conde, J.N.; da Silva, E.M.; Allonso, D.; Coelho, D.R.; Andrade, I.D.; de Medeiros, L.N.; Menezes, J.L.; Barbosa, A.S.; Mohana-Borges, R. Inhibition of the Membrane Attack Complex by Dengue Virus NS1 Through Interaction with Vitronectin and Terminal Complement Proteins. J. Virol. 2016, 90, 9570–9581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avirutnan, P.; Fuchs, A.; Hauhart, R.E.; Somnuke, P.; Youn, S.; Diamond, M.S.; Atkinson, J.P. Antagonism of the Complement Component C4 by Flavivirus Nonstructural Protein NS1. J. Exp. Med. 2010, 207, 793–806. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Mazumdar, B.; Meyer, K.; Di Bisceglie, A.M.; Ray, R.B.; Ray, R. Transcriptional Repression of C4 Complement by Hepatitis C Virus Proteins. J. Virol. 2011, 85, 4157–4166. [Google Scholar] [CrossRef] [Green Version]
- Mazumdar, B.; Kim, H.; Meyer, K.; Bose, S.K.; Di Bisceglie, A.M.; Ray, R.B.; Diamond, M.S.; Atkinson, J.P.; Ray, R. Hepatitis C Virus Infection Upregulates CD55 Expression on the Hepatocyte Surface and Promotes Association with Virus Particles. J. Virol. 2013, 87, 7902–7910. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Meyer, K.; Di Bisceglie, A.M.; Ray, R. Hepatitis C Virus Suppresses C9 Complement Synthesis and Impairs Membrane Attack Complex Function. J. Virol. 2013, 87, 5858–5867. [Google Scholar] [CrossRef] [Green Version]
- Hair, P.S.; Gronemus, J.Q.; Crawford, K.B.; Salvi, V.P.; Cunnion, K.M.; Thielens, N.M.; Arlaud, G.J.; Rawal, N.; Krishna, N.K. Human Astrovirus Coat Protein Binds C1q and MBL and Inhibits the Classical and Lectin Pathways of Complement Activation. Mol. Immunol. 2010, 47, 792–798. [Google Scholar] [CrossRef]
- Zhang, J.; Li, G.; Liu, X.; Wang, Z.; Liu, W.; Ye, X. Influenza A Virus M1 Blocks the Classical Complement Pathway Through Interacting with C1qA. J. Gen. Virol. 2009, 90, 2751–2758. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.B.; Borisevich, V.; Rockx, B.; Parks, G.D. A Novel Factor I Activity in Nipah Virus Inhibits Human Complement Pathways through Cleavage of C3b. J. Virol. 2015, 89, 989–998. [Google Scholar] [PubMed] [Green Version]
- Agrawal, P.; Sharma, S.; Pal, P.; Ojha, H.; Mullick, J.; Sahu, A. The Imitation Game: A Viral Strategy to Subvert the Complement System. FEBS Lett. 2020, 594, 2518–2542. [Google Scholar] [CrossRef] [PubMed]
- Perrin, L.H.; Joseph, B.S.; Cooper, N.R.; Oldstone, M.B. Mechanism of Injury of Virus-Infected Cells by Antiviral Antibody and Complement: Participation of IgG, F(Ab’)2, and the Alternative Complement Pathway. J. Exp. Med. 1976, 143, 1027–1041. [Google Scholar] [CrossRef] [Green Version]
- Sissons, J.G.; Oldstone, M.B.; Schreiber, R.D. Antibody-Independent Activation of the Alternative Complement Pathway by Measles Virus-Infected Cells. Proc. Natl. Acad. Sci. USA 1980, 77, 559–562. [Google Scholar]
- Liu, J.; Ali, M.A.; Shi, Y.; Zhao, Y.; Luo, F.; Yu, J.; Xiang, T.; Tang, J.; Li, D.; Hu, Q.; et al. Specifically Binding of L-Ficolin to N-Glycans of HCV Envelope Glycoproteins E1 and E2 Leads to Complement Activation. Cell Mol. Immunol. 2009, 6, 235–244. [Google Scholar] [CrossRef] [Green Version]
- Kostavasili, I.; Sahu, A.; Friedman, H.M.; Eisenberg, R.J.; Cohen, G.H.; Lambris, J.D. Mechanism of Complement Inactivation by Glycoprotein C of Herpes Simplex Virus. J. Immunol. 1997, 158, 1763–1771. [Google Scholar]
- Fodor, W.L.; Rollins, S.A.; Biancocaron, S.; Rother, R.P.; Guilmette, E.R.; Burton, W.V.; Albrecht, J.C.; Fleckenstein, B.; Squinto, S.P. The Complement Control Protein Homolog of Herpesvirus Saimiri Regulates Serum Complement by Inhibiting C3 Convertase Activity. J. Virol. 1995, 69, 3889–3892. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.K.; Mullick, J.; Bernet, J.; Sahu, A. Functional Characterization of the Complement Control Protein Homolog of Herpesvirus Saimiri: R118 Is Critical for Factor I Cofactor Activities. J. Biol. Chem. 2006, 281, 23119–23128. [Google Scholar] [CrossRef] [Green Version]
- Mark, L.; Spiller, O.B.; Okroj, M.; Chanas, S.; Aitken, J.A.; Wong, S.W.; Damania, B.; Blom, A.M.; Blackbourn, D.J. Molecular Characterization of the Rhesus Rhadinovirus (RRV) ORF4 Gene and the RRV Complement Control Protein It Encodes. J. Virol. 2007, 81, 4166–4176. [Google Scholar]
- Okroj, M.; Mark, L.; Stokowska, A.; Wong, S.W.; Rose, N.; Blackbourn, D.J.; Villoutreix, B.O.; Spiller, O.B.; Blom, A.M. Characterization of the Complement Inhibitory Function of Rhesus Rhadinovirus Complement Control Protein (RCP). J. Biol. Chem. 2009, 284, 505–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapadia, S.B.; Molina, H.; van Berkel, V.; Speck, S.H.; Virgin, H.W. Murine Gammaherpesvirus 68 Encodes a Functional Regulator of Complement Activation. J. Virol. 1999, 73, 7658–7670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonaparte, R.S.; Hair, P.S.; Banthia, D.; Marshall, D.M.; Cunnion, K.M.; Krishna, N.K. Human Astrovirus Coat Protein Inhibits Serum Complement Activation Via C1, the First Component of the Classical Pathway. J. Virol. 2008, 82, 817–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nag, J.; Mukesh, R.K.; Suma, S.M.; Kunnakkadan, U.; Kumar, N.A.; Johnson, J.B. A Factor I-Like Activity Associated with Chikungunya Virus Contributes to Its Resistance to the Human Complement System. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mawatari, S.; Uto, H.; Ido, A.; Nakashima, K.; Suzuki, T.; Kanmura, S.; Kumagai, K.; Oda, K.; Tabu, K.; Tamai, T.; et al. Hepatitis C Virus NS3/4A Protease Inhibits Complement Activation by Cleaving Complement Component 4. PLoS ONE 2013, 8, e82094. [Google Scholar] [CrossRef]
- Mazumdar, B.; Kim, H.; Meyer, K.; Bose, S.K.; Di Bisceglie, A.M.; Ray, R.B.; Ray, R. Hepatitis C Virus Proteins Inhibit C3 Complement Production. J. Virol. 2012, 86, 2221–2228. [Google Scholar] [CrossRef] [Green Version]
- Malekshahi, Z.; Schiela, B.; Bernklau, S.; Banki, Z.; Wurzner, R.; Stoiber, H. Interference of the Zika Virus E-Protein with the Membrane Attack Complex of the Complement System. Front. Immunol. 2020, 11, 569549. [Google Scholar] [CrossRef]
- Shan, C.; Zhang, S.; Cui, W.; You, X.; Kong, G.; Du, Y.; Qiu, L.; Ye, L.; Zhang, X. Hepatitis B Virus X Protein Activates CD59 Involving DNA Binding and Let-7i in Protection of Hepatoma and Hepatic Cells from Complement Attack. Carcinogenesis 2011, 32, 1190–1197. [Google Scholar] [CrossRef] [Green Version]
- Feng, G.; Li, J.; Zheng, M.; Yang, Z.; Liu, Y.; Zhang, S.; Ye, L.; Zhang, W.; Zhang, X. Hepatitis B Virus X Protein Up-Regulates C4b-Binding Protein A through Activating Transcription Factor Sp1 in Protection of Hepatoma Cells from Complement Attack. Oncotarget 2016, 7, 28013–28026. [Google Scholar] [CrossRef] [Green Version]
- Rosengard, A.M.; Liu, Y.; Nie, Z.; Jimenez, R. Variola Virus Immune Evasion Design: Expression of a Highly Efficient Inhibitor of Human Complement. Proc. Natl. Acad. Sci. USA 2002, 99, 8808–8813. [Google Scholar] [CrossRef] [Green Version]
- Yadav, V.N.; Pyaram, K.; Mullick, J.; Sahu, A. Identification of Hot Spots in the Variola Virus Complement Inhibitor (SPICE) for Human Complement Regulation. J. Virol. 2008, 82, 3283–3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mckenzie, R.; Kotwal, G.J.; Moss, B.; Hammer, C.H.; Frank, M.M. Regulation of Complement Activity by Vaccinia Virus Complement-Control Protein. J. Infect. Dis. 1992, 166, 1245–1250. [Google Scholar] [CrossRef] [PubMed]
- Sahu, A.; Isaacs, S.N.; Soulika, A.M.; Lambris, J.D. Interaction of Vaccinia Virus Complement Control Protein with Human Complement Proteins: Factor I-Mediated Degradation of C3b to IC3b1 Inactivates the Alternative Complement Pathway. J. Immunol. 1998, 160, 5596–5604. [Google Scholar] [CrossRef]
- Miller, C.G.; Shchelkunov, S.N.; Kotwal, G.J. The Cowpox Virus-Encoded Homolog of the Vaccinia Virus Complement Control Protein Is an Inflammation Modulatory Protein. Virology 1997, 229, 126–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liszewski, M.K.; Leung, M.K.; Hauhart, R.; Buller, R.M.; Bertram, P.; Wang, X.; Rosengard, A.M.; Kotwal, G.J.; Atkinson, J.P. Structure and Regulatory Profile of the Monkeypox Inhibitor of Complement: Comparison to Homologs in Vaccinia and Variola and Evidence for Dimer Formation. J. Immunol. 2006, 176, 3725–3734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moulton, E.A.; Bertram, P.; Chen, N.; Buller, R.M.; Atkinson, J.P. Ectromelia Virus Inhibitor of Complement Enzymes Protects Intracellular Mature Virus and Infected Cells from Mouse Complement. J. Virol. 2010, 84, 9128–9139. [Google Scholar] [CrossRef] [Green Version]
- Herold, B.C.; Wudunn, D.; Soltys, N.; Spear, P.G. Glycoprotein-C of Herpes Simplex Virus Type-1 Plays a Principal Role in the Adsorption of Virus to Cells and in Infectivity. J. Virol. 1991, 65, 1090–1098. [Google Scholar] [CrossRef] [Green Version]
- Spear, P.G. Antigenic Structure of Herpes Simplex Viruses. In Immunochemistry of Viruses. The Basis for Serodiagnosis and Vaccines; van Regenmortel, M.V.H., Neurath, A.R., Eds.; Elsevier Science Publishers, B.V.: Amsterdam, The Netherlands, 1985. [Google Scholar]
- Wudunn, D.; Spear, P.G. Initial Interaction of Herpes-Simplex Virus with Cells Is Binding to Heparan-Sulfate. J. Virol. 1989, 63, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Zezulak, K.M.; Spear, P.G. Mapping of the Structural Gene for the Herpes Simplex Virus Type 2 Counterpart of Herpes Simplex Virus Type 1 Glycoprotein C and Identification of a Type 2 Mutant Which Does Not Express This Glycoprotein. J. Virol. 1984, 49, 741–747. [Google Scholar] [CrossRef] [Green Version]
- Swain, M.A.; Peet, R.W.; Galloway, D.A. Characterization of the Gene Encoding Herpes Simplex Virus Type 2 Glycoprotein C and Comparison with the Type 1 Counterpart. J. Virol. 1985, 53, 561–569. [Google Scholar] [CrossRef] [Green Version]
- Frink, R.J.; Eisenberg, R.; Cohen, G.; Wagner, E.K. Detailed Analysis of the Portion of the Herpes Simplex Virus Type 1 Genome Encoding Glycoprotein C. J. Virol. 1983, 45, 634–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, D.C.; Spear, P.G. O-Linked Oligosaccharides Are Acquired by Herpes Simplex Virus Glycoproteins in the Golgi Apparatus. Cell 1983, 32, 987–997. [Google Scholar] [CrossRef]
- Dall’Olio, F.; Malagolini, N.; Speziali, V.; Campadelli-Fiume, G.; Serafini-Cessi, F. Sialylated Oligosaccharides O-Glycosidically Linked to Glycoprotein C from Herpes Simplex Virus Type 1. J. Virol. 1985, 56, 127–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, H.M.; Wang, L.; Fishman, N.O.; Lambris, J.D.; Eisenberg, R.J.; Cohen, G.H.; Lubinsky, J. Immune Evasion Properties of Herpes Simplex Virus Type 1 Glycoprotein GC. J. Virol. 1996, 70, 4253–4260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenberg, R.J.; Ponce de Leon, P.; Friedman, H.M.; Fries, L.F.; Frank, M.M.; Hastings, J.C.; Cohen, G.H. Complement Component C3b Binds Directly to Purified Glycoprotein C of Herpes Simplex Virus Types 1 and 2. Microb. Path. 1987, 3, 423–435. [Google Scholar] [CrossRef]
- Hung, S.L.; Peng, C.; Kostavasili, I.; Friedman, H.M.; Lambris, J.D.; Eisenberg, R.J.; Cohen, G.H. The Interaction of Glycoprotein C of Herpes Simplex Virus Types 1 and 2 with the Alternative Complement Pathway. Virology 1994, 203, 299–312. [Google Scholar] [CrossRef]
- Lubinski, J.; Wang, L.; Mastellos, D.; Sahu, A.; Lambris, J.D.; Friedman, H.M. In Vivo Role of Complement-Interacting Domains of Herpes Simplex Virus Type 1 Glycoprotein GC. J. Exp. Med. 1999, 190, 1637–1646. [Google Scholar] [CrossRef] [Green Version]
- Rux, A.H.; Lou, H.; Lambris, J.D.; Friedman, H.M.; Eisenberg, R.J.; Cohen, G.H. Kinetic Analysis of Glycoprotein C of Herpes Simplex Virus Types 1 and 2 Binding to Heparin, Heparan Sulfate, and Complement Component C3b. Virology 2002, 294, 324–332. [Google Scholar] [CrossRef] [Green Version]
- Patel, C.D.; Taylor, S.A.; Mehrbach, J.; Awasthi, S.; Friedman, H.M.; Leib, D.A. Trivalent Glycoprotein Subunit Vaccine Prevents Neonatal Herpes Simplex Virus Mortality and Morbidity. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Madeley, C.R.; Cosgrove, B.P. Letter: Viruses in Infantile Gastroenteritis. Lancet 1975, 2, 124. [Google Scholar] [CrossRef]
- Toh, Y.; Harper, J.; Dryden, K.A.; Yeager, M.; Arias, C.F.; Mendez, E.; Tao, Y.J. Crystal Structure of the Human Astrovirus Capsid Protein. J. Virol. 2016, 90, 9008–9017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, T.L.; Greenberg, H.B.; Herrmann, J.E.; Smith, L.S.; Matsui, S.M. Analysis of Astrovirus Serotype 1 RNA, Identification of the Viral RNA-Dependent RNA Polymerase Motif, and Expression of a Viral Structural Protein. J. Virol. 1994, 68, 77–83. [Google Scholar] [PubMed]
- Willcocks, M.M.; Carter, M.J. Identification and Sequence Determination of the Capsid Protein Gene of Human Astrovirus Serotype 1. FEMS Microbiol. Lett. 1993, 114, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Teillet, F.; Lacroix, M.; Thiel, S.; Weilguny, D.; Agger, T.; Arlaud, G.J.; Thielens, N.M. Identification of the Site of Human Mannan-Binding Lectin Involved in the Interaction with Its Partner Serine Proteases: The Essential Role of Lys55. J. Immunol. 2007, 178, 5710–5716. [Google Scholar] [CrossRef] [Green Version]
- Groeneveld, T.W.; Ramwadhdoebe, T.H.; Trouw, L.A.; van den Ham, D.L.; van der Borden, V.; Drijfhout, J.W.; Hiemstra, P.S.; Daha, M.R.; Roos, A. Human Neutrophil Peptide-1 Inhibits Both the Classical and the Lectin Pathway of Complement Activation. Mol. Immunol. 2007, 44, 3608–3614. [Google Scholar] [CrossRef]
- Van den Berg, R.H.; Faber-Krol, M.C.; van Wetering, S.; Hiemstra, P.S.; Daha, M.R. Inhibition of Activation of the Classical Pathway of Complement by Human Neutrophil Defensins. Blood 1998, 92, 3898–3903. [Google Scholar] [CrossRef]
- Gronemus, J.Q.; Hair, P.S.; Crawford, K.B.; Nyalwidhe, J.O.; Cunnion, K.M.; Krishna, N.K. Potent Inhibition of the Classical Pathway of Complement by a Novel C1q-Binding Peptide Derived from the Human Astrovirus Coat Protein. Mol. Immunol. 2010, 48, 305–313. [Google Scholar] [CrossRef]
- Sharp, J.A.; Whitley, P.H.; Cunnion, K.M.; Krishna, N.K. Peptide Inhibitor of Complement C1, a Novel Suppressor of Classical Pathway Activation: Mechanistic Studies and Clinical Potential. Front. Immunol. 2014, 5, 406. [Google Scholar] [CrossRef] [Green Version]
- Sharp, J.A.; Hair, P.S.; Pallera, H.K.; Kumar, P.S.; Mauriello, C.T.; Nyalwidhe, J.O.; Phelps, C.A.; Park, D.; Thielens, N.M.; Pascal, S.M.; et al. Peptide Inhibitor of Complement C1 (PIC1) Rapidly Inhibits Complement Activation After Intravascular Injection in Rats. PLoS ONE 2015, 10, e0132446. [Google Scholar] [CrossRef]
- Lambert, L.C.; Fauci, A.S. Influenza Vaccines for the Future. N. Engl. J. Med. 2010, 363, 2036–2044. [Google Scholar] [CrossRef]
- Shtykova, E.V.; Baratova, L.A.; Fedorova, N.V.; Radyukhin, V.A.; Ksenofontov, A.L.; Volkov, V.V.; Shishkov, A.V.; Dolgov, A.A.; Shilova, L.A.; Batishchev, O.V.; et al. Structural Analysis of Influenza A Virus Matrix Protein M1 and Its Self-Assemblies at Low PH. PLoS ONE 2013, 8, e82431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akarsu, H.; Burmeister, W.P.; Petosa, C.; Petit, I.; Muller, C.W.; Ruigrok, R.W.; Baudin, F. Crystal Structure of the M1 Protein-Binding Domain of the Influenza A Virus Nuclear Export Protein (NEP/NS2). EMBO J. 2003, 22, 4646–4655. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Handa, H.; Mizumoto, K.; Nagata, K. Mechanism for Inhibition of Influenza Virus RNA Polymerase Activity by Matrix Protein. J. Virol. 1996, 70, 241–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poynard, T.; Ratziu, V.; Benhamou, Y.; Opolon, P.; Cacoub, P.; Bedossa, P. Natural History of HCV Infection. Baillieres Best. Pract. Res. Clin. Gastroenterol. 2000, 14, 211–228. [Google Scholar]
- McLauchlan, J. Properties of the Hepatitis C Virus Core Protein: A Structural Protein That Modulates Cellular Processes. J. Viral Hepat. 2000, 7, 2–14. [Google Scholar] [CrossRef]
- Yao, Z.Q.; Nguyen, D.T.; Hiotellis, A.I.; Hahn, Y.S. Hepatitis C Virus Core Protein Inhibits Human T Lymphocyte Responses by a Complement-Dependent Regulatory Pathway. J. Immunol. 2001, 167, 5264–5272. [Google Scholar] [CrossRef] [Green Version]
- Yao, Z.Q.; Ray, S.; Eisen-Vandervelde, A.; Waggoner, S.; Hahn, Y.S. Hepatitis C Virus: Immunosuppression by Complement Regulatory Pathway. Viral Immunol. 2001, 14, 277–295. [Google Scholar] [CrossRef]
- Zhang, X.; Jia, R.; Shen, H.; Wang, M.; Yin, Z.; Cheng, A. Structures and Functions of the Envelope Glycoprotein in Flavivirus Infections. Viruses 2017, 9, 338. [Google Scholar]
- Schiela, B.; Bernklau, S.; Malekshahi, Z.; Deutschmann, D.; Koske, I.; Banki, Z.; Thielens, N.M.; Wurzner, R.; Speth, C.; Weiss, G.; et al. Active Human Complement Reduces the Zika Virus Load Via Formation of the Membrane-Attack Complex. Front. Immunol. 2018, 9, 2177. [Google Scholar]
- Belda, O.; Targett-Adams, P. Small Molecule Inhibitors of the Hepatitis C Virus-Encoded NS5A Protein. Virus Res. 2012, 170, 1–14. [Google Scholar] [CrossRef]
- Hughes, M.; Griffin, S.; Harris, M. Domain III of NS5A Contributes to Both RNA Replication and Assembly of Hepatitis C Virus Particles. J. Gen. Virol. 2009, 90, 1329–1334. [Google Scholar] [CrossRef] [PubMed]
- Appel, N.; Zayas, M.; Miller, S.; Krijnse-Locker, J.; Schaller, T.; Friebe, P.; Kallis, S.; Engel, U.; Bartenschlager, R. Essential Role of Domain III of Nonstructural Protein 5A for Hepatitis C Virus Infectious Particle Assembly. PLoS Pathog. 2008, 4, e1000035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, N.P.; Easterbrook, P.J.; McMahon, B.J. Epidemiology of Hepatitis B Virus Infection and Impact of Vaccination on Disease. Clin. Liver Dis. 2016, 20, 607–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchard, M.J.; Schneider, R.J. The Enigmatic X Gene of Hepatitis B Virus. J. Virol. 2004, 78, 12725–12734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanda, T.; Yokosuka, O.; Nagao, K.; Saisho, H. State of Hepatitis C Viral Replication Enhances Activation of NF-KB- and AP-1-Signaling Induced by Hepatitis B Virus, X. Cancer Lett. 2006, 234, 143–148. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, H.; Ye, L. Effects of Hepatitis B Virus X Protein on the Development of Liver Cancer. J. Lab. Clin. Med. 2006, 147, 58–66. [Google Scholar] [CrossRef]
- Akey, D.L.; Brown, W.C.; Dutta, S.; Konwerski, J.; Jose, J.; Jurkiw, T.J.; DelProposto, J.; Ogata, C.M.; Skiniotis, G.; Kuhn, R.J.; et al. Flavivirus NS1 Structures Reveal Surfaces for Associations with Membranes and the Immune System. Science 2014, 343, 881–885. [Google Scholar] [CrossRef] [Green Version]
- Akey, D.L.; Brown, W.C.; Jose, J.; Kuhn, R.J.; Smith, J.L. Structure-Guided Insights on the Role of NS1 in Flavivirus Infection. Bioessays 2015, 37, 489–494. [Google Scholar] [CrossRef] [Green Version]
- Wallis, T.P.; Huang, C.Y.; Nimkar, S.B.; Young, P.R.; Gorman, J.J. Determination of the Disulfide Bond Arrangement of Dengue Virus NS1 Protein. J. Biol. Chem. 2004, 279, 20729–20741. [Google Scholar] [CrossRef] [Green Version]
- Crooks, A.J.; Lee, J.M.; Easterbrook, L.M.; Timofeev, A.V.; Stephenson, J.R. The NS1 Protein of Tick-Borne Encephalitis Virus Forms Multimeric Species Upon Secretion from the Host Cell. J. Gen. Virol. 1994, 75, 3453–3460. [Google Scholar] [CrossRef]
- Flamand, M.; Megret, F.; Mathieu, M.; Lepault, J.; Rey, F.A.; Deubel, V. Dengue Virus Type 1 Nonstructural Glycoprotein NS1 Is Secreted from Mammalian Cells as a Soluble Hexamer in a Glycosylation-Dependent Fashion. J. Virol. 1999, 73, 6104–6110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, G.; Randolph, V.B.; Cleaves, G.R.; Ryan, T.E.; Stollar, V. Evidence That the Mature Form of the Flavivirus Nonstructural Protein NS1 Is a Dimer. Virology 1988, 162, 187–196. [Google Scholar] [CrossRef]
- Winkler, G.; Maxwell, S.E.; Ruemmler, C.; Stollar, V. Newly Synthesized Dengue-2 Virus Nonstructural Protein NS1 Is a Soluble Protein but Becomes Partially Hydrophobic and Membrane-Associated after Dimerization. Virology 1989, 171, 302–305. [Google Scholar] [CrossRef]
- Khromykh, A.A.; Sedlak, P.L.; Guyatt, K.J.; Hall, R.A.; Westaway, E.G. Efficient Trans-Complementation of the Flavivirus Kunjin NS5 Protein but Not of the NS1 Protein Requires Its Coexpression with Other Components of the Viral Replicase. J. Virol. 1999, 73, 10272–10280. [Google Scholar]
- Lindenbach, B.D.; Rice, C.M. Trans-Complementation of Yellow Fever Virus NS1 Reveals a Role in Early RNA Replication. J. Virol. 1997, 71, 9608–9617. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, J.M.; Jones, M.K.; Young, P.R. Immunolocalization of the Dengue Virus Nonstructural Glycoprotein NS1 Suggests a Role in Viral RNA Replication. Virology 1996, 220, 232–240. [Google Scholar] [CrossRef] [Green Version]
- Brandt, W.E.; Chiewslip, D.; Harris, D.L.; Russell, P.K. Partial Purification and Characterization of a Dengue Virus Soluble Complement-Fixing Antigen. J. Immunol. 1970, 105, 1565–1568. [Google Scholar]
- Kurosu, T.; Chaichana, P.; Yamate, M.; Anantapreecha, S.; Ikuta, K. Secreted Complement Regulatory Protein Clusterin Interacts with Dengue Virus Nonstructural Protein 1. Biochem. Biophys. Res. Commun. 2007, 362, 1051–1056. [Google Scholar] [CrossRef]
- Chung, K.M.; Liszewski, M.K.; Nybakken, G.; Davis, A.E.; Townsend, R.R.; Fremont, D.H.; Atkinson, J.P.; Diamond, M.S. West Nile Virus Nonstructural Protein NS1 Inhibits Complement Activation by Binding the Regulatory Protein Factor, H. Proc. Natl. Acad. Sci. USA 2006, 103, 19111–19116. [Google Scholar]
- Thiemmeca, S.; Tamdet, C.; Punyadee, N.; Prommool, T.; Songjaeng, A.; Noisakran, S.; Puttikhunt, C.; Atkinson, J.P.; Diamond, M.S.; Ponlawat, A.; et al. Secreted NS1 Protects Dengue Virus from Mannose-Binding Lectin-Mediated Neutralization. J. Immunol. 2016, 197, 4053–4065. [Google Scholar] [CrossRef]
- Gurav, Y.K.; Raut, C.G.; Yadav, P.D.; Tandale, B.V.; Sivaram, A.; Pore, M.D.; Basu, A.; Mourya, D.T.; Mishra, A.C. Buffalopox Outbreak in Humans and Animals in Western Maharashtra, India. Prev. Vet. Med. 2011, 100, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Trindade, G.S.; Lobato, Z.I.; Drumond, B.P.; Leite, J.A.; Trigueiro, R.C.; Guedes, M.I.; da Fonseca, F.G.; Dos, S., Jr.; Bonjardim, C.A.; Ferreira, P.C.; et al. Short Report: Isolation of Two Vaccinia Virus Strains from a Single Bovine Vaccinia Outbreak in Rural Area from Brazil: Implications on the Emergence of Zoonotic Orthopoxviruses. Am. J. Trop. Med. Hyg. 2006, 75, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Bernet, J.; Mullick, J.; Panse, Y.; Parab, P.B.; Sahu, A. Kinetic Analysis of the Interactions Between Vaccinia Virus Complement Control Protein and Human Complement Proteins C3b and C4b. J. Virol. 2004, 78, 9446–9457. [Google Scholar] [PubMed] [Green Version]
- Mullick, J.; Bernet, J.; Panse, Y.; Hallihosur, S.; Singh, A.K.; Sahu, A. Identification of Complement Regulatory Domains in Vaccinia Virus Complement Control Protein. J. Virol. 2005, 79, 12382–12393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, M.; Raut, S.; Pyaram, K.; Kamble, A.; Mullick, J.; Sahu, A. Domain Swapping Reveals Complement Control Protein Modules Critical for Imparting Cofactor and Decay-Accelerating Activities in Vaccinia Virus Complement Control Protein. J. Immunol. 2010, 185, 6128–6137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaacs, S.N.; Kotwal, G.J.; Moss, B. Vaccinia Virus Complement-Control Protein Prevents Antibody- Dependent Complement-Enhanced Neutralization of Infectivity and Contributes to Virulence. Proc. Natl. Acad. Sci. USA 1992, 89, 628–632. [Google Scholar] [CrossRef] [Green Version]
- Girgis, N.M.; Dehaven, B.C.; Xiao, Y.; Alexander, E.; Viner, K.M.; Isaacs, S.N. The Vaccinia Virus Complement Control Protein Modulates Adaptive Immune Responses During Infection. J. Virol. 2010, 85, 2547–2556. [Google Scholar]
- Bernet, J.; Ahmad, M.; Mullick, J.; Panse, Y.; Singh, A.K.; Parab, P.B.; Sahu, A. Disabling Complement Regulatory Activities of Vaccinia Virus Complement Control Protein Reduces Vaccinia Virus Pathogenicity. Vaccine 2011, 29, 7435–7443. [Google Scholar] [CrossRef] [Green Version]
- Kotwal, G.J.; Fernando, N.; Zhou, J.; Valter, K. Exploring the Potential Benefits of Vaccinia Virus Complement Control Protein in Controlling Complement Activation in Pathogenesis of the Central Nervous System Diseases. Mol. Immunol. 2014, 61, 204–209. [Google Scholar] [CrossRef]
- Fernando, N.; Natoli, R.; Racic, T.; Wooff, Y.; Provis, J.; Valter, K. The Use of the Vaccinia Virus Complement Control Protein (VCP) in the Rat Retina. PLoS ONE 2018, 13, e0193740. [Google Scholar] [CrossRef] [Green Version]
- Sfyroera, G.; Katragadda, M.; Morikis, D.; Isaacs, S.N.; Lambris, J.D. Electrostatic Modeling Predicts the Activities of Orthopoxvirus Complement Control Proteins. J. Immunol. 2005, 174, 2143–2151. [Google Scholar] [CrossRef] [PubMed]
- Liszewski, M.K.; Bertram, P.; Leung, M.K.; Hauhart, R.; Zhang, L.; Atkinson, J.P. Smallpox Inhibitor of Complement Enzymes (SPICE): Regulation of Complement Activation on Cells and Mechanism of Its Cellular Attachment. J. Immunol. 2008, 181, 4199–4207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, V.N.; Pyaram, K.; Ahmad, M.; Sahu, A. Species Selectivity in Poxviral Complement Regulators Is Dictated by the Charge Reversal in the Central Complement Control Protein Modules. J. Immunol. 2012, 189, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Yadav, V.N.; Phulera, S.; Kamble, A.; Gautam, A.K.; Panwar, H.S.; Sahu, A. Species Specificity of Vaccinia Virus Complement Control Protein for the Bovine Classical Pathway Is Governed Primarily by Direct Interaction of Its Acidic Residues with Factor I. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFadden, G. Poxvirus Tropism. Nat. Rev. Microbiol. 2005, 3, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Andreani, J.; Arnault, J.P.; Bou Khalil, J.Y.; Abrahao, J.; Tomei, E.; Vial, E.; Le Bideau, M.; Raoult, D.; La Scola, B. Atypical Cowpox Virus Infection in Smallpox-Vaccinated Patient, France. Emerg. Infect. Dis. 2019, 25, 212–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shchelkunov, S.N.; Safronov, P.F.; Totmenin, A.V.; Petrov, N.A.; Ryazankina, O.I.; Gutorov, V.V.; Kotwal, G.J. The Genomic Sequence Analysis of the Left and Right Species-Specific Terminal Region of a Cowpox Virus Strain Reveals Unique Sequences and a Cluster of Intact ORFs for Immunomodulatory and Host Range Proteins. Virology 1998, 243, 432–460. [Google Scholar]
- Reed, K.D.; Melski, J.W.; Graham, M.B.; Regnery, R.L.; Sotir, M.J.; Wegner, M.V.; Kazmierczak, J.J.; Stratman, E.J.; Li, Y.; Fairley, J.A.; et al. The Detection of Monkeypox in Humans in the Western Hemisphere. N. Engl. J. Med. 2004, 350, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Li, G.; Liszewski, M.K.; Atkinson, J.P.; Jahrling, P.B.; Feng, Z.; Schriewer, J.; Buck, C.; Wang, C.; Lefkowitz, E.J.; et al. Virulence Differences between Monkeypox Virus Isolates from West Africa and the Congo Basin. Virology 2005, 340, 46–63. [Google Scholar] [CrossRef] [Green Version]
- Estep, R.D.; Messaoudi, I.; O’Connor, M.A.; Li, H.; Sprague, J.; Barron, A.; Engelmann, F.; Yen, B.; Powers, M.F.; Jones, J.M.; et al. Deletion of the Monkeypox Virus Inhibitor of Complement Enzymes Locus Impacts the Adaptive Immune Response to Monkeypox Virus in a Nonhuman Primate Model of Infection. J. Virol. 2011, 85, 9527–9542. [Google Scholar] [CrossRef] [Green Version]
- Hudson, P.N.; Self, J.; Weiss, S.; Braden, Z.; Xiao, Y.; Girgis, N.M.; Emerson, G.; Hughes, C.; Sammons, S.A.; Isaacs, S.N.; et al. Elucidating the Role of the Complement Control Protein in Monkeypox Pathogenicity. PLoS ONE 2012, 7, e35086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteban, D.J.; Buller, R.M.L. Ectromelia Virus: The Causative Agent of Mousepox. J. Gen. Virol. 2005, 86, 2645–2659. [Google Scholar] [CrossRef] [PubMed]
- Desrosiers, R.C.; Falk, L.A. Herpesvirus Saimiri Strain Variability. J. Virol. 1982, 43, 352–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desrosiers, R.C.; Bakker, A.; Kamine, J.; Falk, L.A.; Hunt, R.D.; King, N.W. A Region of the Herpesvirus Saimiri Genome Required for Oncogenicity. Science 1985, 228, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Biesinger, B.; Trimble, J.J.; Desrosiers, R.C.; Fleckenstein, B. The Divergence Between Two Oncogenic Herpesvirus Saimiri Strains in a Genomic Region Related to the Transforming Phenotype. Virology 1990, 176, 505–514. [Google Scholar] [CrossRef]
- Cabanillas, J.A.; Cambronero, R.; Pacheco-Castro, A.; Garcia-Rodriguez, M.C.; Martin-Fernandez, J.M.; Fontan, G.; Regueiro, J.R. Characterization of Herpesvirus Saimiri-Transformed T Lymphocytes from Common Variable Immunodeficiency Patients. Clin. Exp. Immunol. 2002, 127, 366–373. [Google Scholar] [CrossRef]
- Albrecht, J.C.; Nicholas, J.; Biller, D.; Cameron, K.R.; Biesinger, B.; Newman, C.; Wittmann, S.; Craxton, M.A.; Coleman, H.; Fleckenstein, B. Primary Structure of the Herpesvirus Saimiri Genome. J. Virol. 1992, 66, 5047–5058. [Google Scholar] [CrossRef] [Green Version]
- Means, R.E.; Choi, J.K.; Nakamura, H.; Chung, Y.H.; Ishido, S.; Jung, J.U. Immune Evasion Strategies of Kaposi’s Sarcoma-Associated Herpesvirus. Curr. Top. Microbiol. Immunol. 2002, 269, 187–201. [Google Scholar]
- Singh, A.K.; Yadav, V.N.; Pyaram, K.; Mullick, J.; Sahu, A. Mapping of Functional Domains in Herpesvirus Saimiri Complement Control Protein Homolog: Complement Control Protein Domain 2 Is the Smallest Structural Unit Displaying Cofactor and Decay-Accelerating Activities. J. Virol. 2009, 83, 10299–10304. [Google Scholar]
- Reza, M.J.; Kamble, A.; Ahmad, M.; Krishnasastry, M.V.; Sahu, A. Dissection of Functional Sites in Herpesvirus Saimiri Complement Control Protein Homolog. J. Virol. 2013, 87, 282–295. [Google Scholar] [CrossRef] [Green Version]
- Mullick, J.; Singh, A.K.; Panse, Y.; Yadav, V.; Bernet, J.; Sahu, A. Identification of Functional Domains in Kaposica, the Complement Control Protein Homolog of Kaposi’s Sarcoma-Associated Herpesvirus (Human Herpesvirus-8). J. Virol. 2005, 79, 5850–5856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiller, O.B.; Mark, L.; Blue, C.E.; Proctor, D.G.; Aitken, J.A.; Blom, A.M.; Blackbourn, D.J. Dissecting the Regions of Virion-Associated Kaposi’s Sarcoma-Associated Herpesvirus Complement Control Protein Required for Complement Regulation and Cell Binding. J. Virol. 2006, 80, 4068–4078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyaram, K.; Kieslich, C.A.; Yadav, V.N.; Morikis, D.; Sahu, A. Influence of Electrostatics on the Complement Regulatory Functions of Kaposica, the Complement Inhibitor of Kaposi’s Sarcoma-Associated Herpesvirus. J. Immunol. 2010, 184, 1956–1967. [Google Scholar] [CrossRef] [PubMed]
- Gautam, A.K.; Panse, Y.; Ghosh, P.; Reza, M.J.; Mullick, J.; Sahu, A. Mutational Analysis of Kaposica Reveals That Bridging of MG2 and CUB Domains of Target Protein Is Crucial for the Cofactor Activity of RCA Proteins. Proc. Natl. Acad. Sci. USA 2015, 112, 12794–12799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estep, R.D.; Rawlings, S.D.; Li, H.; Manoharan, M.; Blaine, E.T.; O’Connor, M.A.; Messaoudi, I.; Axthelm, M.K.; Wong, S.W. The Rhesus Rhadinovirus CD200 Homologue Affects Immune Responses and Viral Loads During In Vivo Infection. J. Virol. 2014, 88, 10635–10654. [Google Scholar] [CrossRef] [Green Version]
- Alexander, L.; Denekamp, L.; Knapp, A.; Auerbach, M.R.; Damania, B.; Desrosiers, R.C. The Primary Sequence of Rhesus Monkey Rhadinovirus Isolate 26-95: Sequence Similarities to Kaposi’s Sarcoma-Associated Herpesvirus and Rhesus Monkey Rhadinovirus Isolate 17577. J. Virol. 2000, 74, 3388–3398. [Google Scholar]
- O’Connor, C.M.; Kedes, D.H. Mass Spectrometric Analyses of Purified Rhesus Monkey Rhadinovirus Reveal 33 Virion-Associated Proteins. J. Virol. 2006, 80, 1574–1583. [Google Scholar]
- Efstathiou, S.; Ho, Y.M.; Minson, A.C. Cloning and Molecular Characterization of the Murine Herpesvirus 68 Genome. J. Gen. Virol. 1990, 71, 1355–1364. [Google Scholar] [CrossRef]
- Virgin, H.W.; Latreille, P.; Wamsley, P.; Hallsworth, K.; Weck, K.E.; DalCanto, A.J.; Speck, S.H. Complete Sequence and Genomic Analysis of Murine Gammaherpesvirus 68. J. Virol. 1997, 71, 5894–5904. [Google Scholar] [CrossRef] [Green Version]
- Kapadia, S.; Levine, B.; Speck, S.; Virgin, H. Critical Role of Complement and Viral Evasion of Complement in Acute, Persistent, and Latent Gamma-Herpesvirus Infection. Immunity 2002, 17, 143–155. [Google Scholar] [CrossRef] [Green Version]
- Meri, S.; Morgan, B.P.; Davies, A.; Daniels, R.H.; Olavesen, M.G.; Waldmann, H.; Lachmann, P.J. Human Protectin (CD59), an 18,000–20,000 MW Complement Lysis Restricting Factor, Inhibits C5b-8 Catalysed Insertion of C9 into Lipid Bilayers. Immunology 1990, 71, 1–9. [Google Scholar] [PubMed]
- Hahn, W.C.; Menu, E.; Bothwell, A.L.; Sims, P.J.; Bierer, B.E. Overlapping but Nonidentical Binding Sites on CD2 for CD58 and a Second Ligand CD59. Science 1992, 256, 1805–1807. [Google Scholar] [CrossRef] [PubMed]
- Bratke, K.A.; McLysaght, A. Identification of Multiple Independent Horizontal Gene Transfers into Poxviruses Using a Comparative Genomics Approach. BMC Evol. Biol. 2008, 8, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewald, P.W. The Evolution of Virulence and Emerging Diseases. J. Urban Health 1998, 75, 480–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullen, B.R. MicroRNAs As Mediators of Viral Evasion of the Immune System. Nat. Immunol. 2013, 14, 205–210. [Google Scholar] [CrossRef] [Green Version]
- Rajsbaum, R.; Garcia-Sastre, A. Viral Evasion Mechanisms of Early Antiviral Responses Involving Regulation of Ubiquitin Pathways. Trends Microbiol. 2013, 21, 421–429. [Google Scholar] [CrossRef]
- Arbore, G.; Kemper, C.; Kolev, M. Intracellular Complement—The Complosome—In Immune Cell Regulation. Mol. Immunol. 2017, 89, 2–9. [Google Scholar] [CrossRef]
- Arbore, G.; West, E.E.; Spolski, R.; Robertson, A.A.; Klos, A.; Rheinheimer, C.; Dutow, P.; Woodruff, T.M.; Yu, Z.X.; O’Neill, L.A.; et al. T Helper 1 Immunity Requires Complement-Driven NLRP3 Inflammasome Activity in CD4(+) T Cells. Science 2016, 352, aad1210. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Virus Family | Virus | Complement Evasion Protein | Target | Reference |
|---|---|---|---|---|
| Herpesviridae | Herpes simplex virus | Glycoprotein C-1 | AP C3 convertase and C3b | [13,38] |
| Herpesvirus saimiri | CD59 homolog | C5b-8 and C5b-9 | [23,24] | |
| HVS CCPH | CP/LP and AP C3 convertase | [39,40] | ||
| Kaposi sarcoma-associated herpesvirus | Kaposica/KCP | CP/LP and AP C3 convertase | [19,20] | |
| Rhesus rhadinovirus | RCP-H and RCP-1 | CP/LP and AP C3 convertase | [41,42] | |
| Murine gammaherpesvirus 68 | γHV68 RCA protein | CP/LP and AP C3 convertase | [43] | |
| Astroviridae | Astrovirus | CaPt | C1q and MBL | [31,44] |
| Orthomyxoviridae | Influenza virus | M1 | C1q | [32] |
| Paramyxoviridae | Nipah virus | Unknown protein with factor I like activity | C3b | [33] |
| Togaviridae | Chikungunya virus | Unknown protein with factor I like activity | C3b | [45] |
| Flaviviridae | Hepatitis C virus | Core protein | C3, C4 and C9 genes | [28,29,30] |
| NS3/4A | C4 | [46] | ||
| NS5A | C3 and C4 genes | [28,47] | ||
| West Nile virus and Dengue virus | NS1 | C4 and C9 | [26,27] | |
| Zika virus | E protein | C5b-6, C8 and C9 | [48] | |
| Hepadnaviridae | Hepatitis B virus | HBx protein | CD59 and C4BP genes | [49,50] |
| Poxviridae | Variola virus | SPICE | CP/LP and AP C3/C5 convertase | [51,52] |
| Vaccinia virus | VCP | CP/LP and AP C3/C5 convertase | [17,53,54] | |
| Cowpox virus | IMP | Unknown | [55] | |
| Monkeypox virus | MOPICE | CP/LP C3 convertase | [56] | |
| Ectromelia virus | EMICE | CP/LP C3 convertase | [57] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinha, A.; Singh, A.K.; Kadni, T.S.; Mullick, J.; Sahu, A. Virus-Encoded Complement Regulators: Current Status. Viruses 2021, 13, 208. https://doi.org/10.3390/v13020208
Sinha A, Singh AK, Kadni TS, Mullick J, Sahu A. Virus-Encoded Complement Regulators: Current Status. Viruses. 2021; 13(2):208. https://doi.org/10.3390/v13020208
Chicago/Turabian StyleSinha, Anwesha, Anup Kumar Singh, Trupti Satish Kadni, Jayati Mullick, and Arvind Sahu. 2021. "Virus-Encoded Complement Regulators: Current Status" Viruses 13, no. 2: 208. https://doi.org/10.3390/v13020208
APA StyleSinha, A., Singh, A. K., Kadni, T. S., Mullick, J., & Sahu, A. (2021). Virus-Encoded Complement Regulators: Current Status. Viruses, 13(2), 208. https://doi.org/10.3390/v13020208